

Abstract
Enzyme replacement therapy (ERT) has become the standard of care for several lysosomal storage disorders (LSDs). Despite ERT's undisputed efficacy, the requirement for multiple and costly administrations as well as ERT's limited improvement of some LSD manifestations prompts the search for better therapies. Using a mouse model of mucopolysaccharidosis VI, we compared the efficacy of a single intravascular administration of an adeno-associated viral vector targeting liver to weekly infusions of human recombinant enzyme at the same doses used in mucopolysaccharidosis VI patients. While gene therapy results in increased and stable levels of circulating enzyme up to 1 year after vector administration, ERT has typical peak-and-drop serum kinetics. Both therapies similarly reduced glycosaminoglycan levels in urine and tissues including heart valves and myocardium, with gene therapy improving skeletal skull abnormalities slightly better, although not significantly, than ERT. Both therapies seem to similarly improve animal motor performance, with gene therapy possibly associated with less animal distress. Thus, a single vector administration that converts liver into a factory organ for systemic secretion of therapeutic proteins is at least as effective as ERT in a mouse model of LSD, potentially eliminating problems with compliance and costs. Only testing in humans will prove whether this holds true in a clinical setting.
Introduction
Lysosomal storage disorders (LSDs) are a heterogeneous group of rare inborn errors of metabolism encompassing more than 70 distinct conditions that are characterized by progressive storage of undigested or partially digested materials within the lysosomes, which lead to multisystemic cellular and organ dysfunction (Neufeld, 1991; Neufeld and Muenzer, 2010). The majority of LSDs are caused by a deficiency in soluble lysosomal enzymes. Lysosomal enzymes are targeted to the endosomal system via binding to the mannose 6-phosphate receptor (Man6PR). Lysosomal enzymes can be secreted, taken up by distal cells via the Man6PRs located on the plasma membrane, and then trafficked to the lysosome (Sands and Davidson, 2006). This is the basis for the cross-correction of deficient cells through enzyme replacement therapy (ERT), which is currently the standard of care for several types of LSD, including Fabry disease, mucopolysaccharidoses (MPS) I, II, and VI, and Pompe disease. Enzyme replacement therapies for LSDs have been demonstrated to slow disease progression and significantly improve patient outcomes. However, ERT shows limited therapeutic efficacy in bone, cartilage, and brain, presumably because of the poor biodistribution of recombinant enzymes to these regions (Desnick and Schuchman, 2012; Wyatt et al., 2012). In addition, the requirement of repeated (weekly or biweekly) intravenous infusions, as a result of the short plasma half-life of recombinant enzymes that are taken up by peripheral tissues, results in limited compliance to the therapy. Finally, ERTs are extremely expensive, which constitutes a barrier for their widespread use in less developed countries. Thus, alternative strategies with a comparable or better efficacy, but without the inconvenience of multiple infusions, would be desirable.
Gene therapy has the potential to convert a target organ like liver into a factory for the stable systemic production and secretion of therapeutic proteins after a single vector administration (Mingozzi and High, 2011). This could be particularly advantageous for LSDs given the potential cross-correction of enzyme-deficient cells by enzyme from transduced cells, that is, hepatocytes (Sands and Davidson, 2006; Byrne et al., 2012).
Adeno-associated viral vectors (AAVs) are the most frequently used viral vectors for in vivo gene transfer because of their safety profile and their ability to transduce a variety of tissues and provide long-term expression of the therapeutic gene (Brunetti-Pierri and Auricchio, 2010). AAV-mediated gene therapy has been successful in both small- and large-animal models of LSDs, including Pompe disease, Fabry disease, and MPS (Hartung et al., 2004; Sun et al., 2005; Ziegler et al., 2007; Tessitore et al., 2008; Cotugno et al., 2010, 2011; Wolf et al., 2011; Ferla et al., 2012; Sorrentino et al., 2013). Importantly, the safety and efficacy of intravascular administrations of AAV vectors based on serotype 8 (AAV2/8) and targeting liver (Thomas et al., 2004; Nathwani et al., 2011a) has been recently demonstrated in patients with hemophilia B (Nathwani et al., 2011b).
In the present study, we compare the efficacy of gene therapy and ERT in a murine model of MPS VI, a rare lysosomal storage disease caused by a deficiency in arylsulfatase B (ARSB), which leads to lysosomal accumulation and the excretion of elevated amounts of the glycosaminoglycan (GAG) dermatan sulfate in the urine (Neufeld and Muenzer, 2010). MPS VI is an attractive LSD for this comparison for two reasons: (1) MPS VI presents systemic life-threatening symptoms without central nervous system (CNS) involvement, and therefore the efficacy of potential therapies is not limited by the presence of the blood–brain barrier; (2) ERT is available as a current treatment for MPS VI patients, though its efficacy has been limited to reduction of both visceromegaly and urinary GAG excretion and improvement in endurance (Harmatz et al., 2004, 2005a,b, 2006, 2008). We used a murine model of MPS VI with a targeted disruption of the ARSB locus (Evers et al., 1996) and made it immune tolerant to human ARSB (hARSB) through transgenic insertion of the C91S hARSB mutant, resulting in the production of inactive hARSB (Brooks et al., 1995). Therefore, in this model a side-by-side comparison of ERT using the same enzyme, recombinant hARSB (rhARSB), used in treating MPS VI patients with gene therapy using an AAV2/8 vector expressing hARSB is possible without the interference of potential immune responses to the therapeutic product.
We show here that a single systemic administration of an AAV vector targeting liver provides similar therapeutic efficacy to weekly administrations of rhARSB with possibly less animal distress.
Materials and Methods
Animal colony
MPS VI mice, a kind gift from Prof. C. Peters (Institute of Molecular Medicine and Cell Research, University of Freidburg), were maintained at the Cardarelli Hospital Animal House (Naples, Italy). Animals were raised in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines for the care and use of animals in research. This mouse model carries a targeted disruption of the ARSB locus (Evers et al., 1996) and is made immune tolerant to human ARSB by transgenic insertion of the C91S hARSB mutant, resulting in the production of inactive hARSB (Brooks et al., 1995). Genotype analysis was performed by polymerase chain reaction (PCR) on genomic DNA obtained from tail using the DNeasy Blood and Tissue Extraction kit (Qiagen, Hilden, Germany). Three different PCRs were performed using the following forward (Fw) and reverse (Rev) primers: (1) Fw 5′-TGGGCAGACTAGGTCTGG-3′ and Rev 5′-TGTCTTCCACATGTTGAAGC-3′ to discriminate affected from not affected mice; (2) Fw 5′-TCTGGAGGCAACAACTGGC 3′ and Rev 5′-CGCGTCACCTTAATATGCGA 3′ to discriminate wild type from heterozygous mice; (3) Fw 5′ TTAAGAAGCTGATAAAATCTGCAACAC-3′ and Rev 5′-AACAATCAAGGGTCCCCAAAC 3′ to check for the presence of the C91S hARSB transgene.
Plasmid and vector production
The plasmid pAAV2.1-thyroxine binding globulin (TBG)-hARSB encoding the hARSB protein was generated as described previously (Tessitore et al., 2008). The therapeutic AAV2/8-TBG-hARSB and the control AAV2/8-TBG-eGFP (enhanced green fluorescent protein) vectors were produced by the AAV Vector Core of the Telethon Institute of Genetics and Medicine (Telethon Institute of Genetics and Medicine [TIGEM], Naples, Italy), as previously described (Cotugno et al., 2011).
Treatment administration
MPS VI mice treated with gene therapy received a single intravenous retro-orbital injection of 2×1012 genome copies (gc)/kg of AAV2/8-TBG-hARSB at postnatal day 30 (p30). As control, MPS VI mice received 2×1012 gc/kg of AAV2/8-TBG-eGFP or were left untreated. MPS VI mice treated with ERT received weekly retro-orbital injections of 1 mg/kg of rhARSB protein (Naglazyme; BioMarin Europe, London, UK), appropriately diluted in phosphate buffered saline (PBS), starting from p30. As control, MPS VI mice received weekly administrations of PBS only. Mice were injected retro-orbitally because of technical issues caused by the tail skin thickness of MPS VI mice. Please note that retro-orbital injections are routinely used in mice for intravenous delivery of several agents, including viral vectors and are extraocular. Thus, unless the needle is erroneously stuck intraocularly, which did not happen in this study, there is no possibility that retro-orbital injections damage vision. This was clearly demonstrated by Li et al., who performed electroretinograms to rule out any retinal damage after retro-orbital delivery and found similar AAV biodistribution between the animals administered through tail vein and those administered retro-orbitally (Li et al., 2013). Nonetheless, we have compared AAV2/8 biodistribution after either retro-orbital sinus or tail vein injection and found that this is similar in liver, brain, kidney, and spleen of adult mice (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/hum).
Blood, urine, and tissue collection
Blood was collected every month from treated and control mice via eye bleeding and centrifuged at 10,000×g in a microcentrifuge (Heraeus Fresco 21; Thermo Scientific, Waltham, MA) for 10 min at 4°C to obtain serum. Urine was also collected monthly using metabolic cages. Samples were briefly centrifuged to remove debris and stored at −20°C. Mice were sacrificed 6 or 12 months after treatment. Cardiac perfusion with PBS was performed, and liver, kidney, spleen, heart, and knee joints were collected. Tissue samples were fixed in methacarn solution (30% chloroform, 60% methanol, 10% acetic acid) for 24 hr or were frozen in dry ice (for ARSB activity and GAG quantitative assays).
Immune capture assay for determination of serum ARSB activity
Serum ARSB activity was measured by an immune capture assay based on the use of a specific anti-hARSB polyclonal antibody (Covalab, Villeurbanne, France). Ninety-six-well plates (Nunclon, Roskilde, Denmark) were coated with 5 μg/ml in 0.1 M NaHCO3 (100 μl/well) and incubated overnight (O/N) at 4°C. The following day, plates were washed twice with 0.25 M NaCl/0.02 M Tris, pH 7, and then were blocked with 1% milk in 0.25 M NaCl/0.02 M Tris, pH 7.0 (blocking solution), for 2 hr at room temperature. Plates were washed again as described above and then 50 μl of standard and unknown samples (diluted 1:10) was added to each well. Plates were shaken for 1 hr at room temperature and then incubated at 4°C O/N. The day after, plates were shaken for 1 hr at room temperature and then washed 2× with 0.25 M NaCl/0.02 M Tris, pH 7.0. In total, 100 μl of 5 mM 4-methylumbelliferyl-sulfate potassium salt (4-MUS; Sigma-Aldrich, Milan, Italy) substrate was added to each well and then incubated at 37°C for 4 hr. The reaction was stopped by the addition of 100 μl/well of stop solution (glycine 0.2 M). Plates were shaken for 10 min at room temperature and the fluorescence was read (excitation 365 nm/emission 460 nm) on a multiplate fluorimeter (TECAN Infinite F200, Männedorf, Switzerland). Serum ARSB was determined based on an rhARSB (Naglazyme; BioMarin Europe) standard curve and expressed as pg/mL.
ELISA for detection of anti-ARSB IgG antibodies
The presence of circulating antibodies against the hARSB protein in murine serum was determined in a plate-binding assay, as previously described (Cotugno et al., 2010). Briefly, 96-well plates (Nunclon) were coated with 100 μl rhARSB (Naglazyme; BioMarin Europe) and diluted to 10 μg/mL in PBS O/N at 4°C. Plates were blocked for 2 hr at room temperature with 200 μl of 100% fetal bovine serum (FBS), washed five times with PBS containing 0.05% Tween 20 (washing buffer), and incubated with 100 μl of 1:50 dilutions of serum in PBS–0.05% Tween–10% FBS for 2 hr at room temperature. The washing step was repeated and 100 μl of biotinylated anti-mouse IgG antibody (Vector Laboratories, Burlingam, CA) diluted 1:400 in PBS–0.05% Tween–10% FBS was added and incubated for 1 hr at room temperature. Binding was revealed by the addition of ExtrAvidin–horseradish peroxidase (Sigma-Aldrich, Milan, Italy) followed by o-phenylenediamine dihydrochloride (OPD) substrate (Sigma-Aldrich, Milan, Italy). Absorbance at 450 nm (optical density) was determined on a multiplate reader (TECAN Infinite F200). The optical density, determined in an assay, represented a qualitative measure of antibody affinity.
AAV vector genome distribution
Genomic DNA was extracted from livers using a DNeasy Blood and Tissue Extraction kit (Qiagen). Real-time PCR analysis was performed on 100 ng of genomic DNA using a set of primers/probe specific for the viral genome and TaqMan universal PCR master mix (Applied Biosystems, Foster City, CA). Amplification was run on a 7300 Real-Time PCR system (Applied Biosystems) with standard cycles. All the reactions were performed in triplicate.
Assay for ARSB activity evaluation in tissues
Tissues were homogenized in water and protein concentrations were determined with the bicinchoninic acid (BCA) protein assay reagent (Pierce Protein Research Products; Thermo Fisher Scientific, Rockford, IL). The ARSB assay was performed as previously described (Chang et al., 1981). Briefly, 30 μg of protein was incubated with 40 μl of the fluorogenic 4-methylumbelliferyl sulfate substrate (12.5 mM; Sigma-Aldrich, Saint Louis, MO) for 3 hr at 37°C in the presence of 40 μl silver nitrate (0.75 mM; Carlo Erba, Milan, Italy), which is known to inhibit the activity of other sulfatases. The reaction was stopped by adding 200 μl of carbonate glycine stop buffer and the fluorescence of the 4-methylumbelliferone liberated was measured on a multiplate reader (TECAN Infinite F200) at 365 nm (excitation) and 460 nm (emission). Enzyme activities were calculated with a standard curve of the fluorogenic 4-methylumbelliferone product (12.5 mM; Sigma-Aldrich, Saint Louis, MO). For tissue lysates the activity is expressed as nanomoles per milligram of protein per hour (nmol/mg/hr).
Quantitative analysis of GAG accumulation in tissues and urine
Urine samples were diluted 1:50 in water for measurement of GAG content. One hundred microliters of diluted urine or 250 μg of protein extract from liver, spleen, and kidney was used for the GAG assay, as previously described (De Jong et al., 1989). GAG concentrations were determined on the basis of a dermatan sulfate standard curve (Sigma-Aldrich, Saint Louis, MO). Tissue GAGs are expressed as micrograms of GAG per milligram of protein (μg/mg of protein). Urinary GAGs were normalized to creatinine content. Creatinine was measured with a creatinine assay kit (Quidel, San Diego, CA). Urinary GAGs are reported as percentage of affected (AF) control mice. The urinary GAG levels measured over time were averaged for each mouse and the resulting value was then averaged for each group.
Alcian blue staining
After methacarn fixation, all the tissues (liver, spleen, kidney, heart) were embedded in paraffin and sectioned into 7-μm-thick serial sections on a microtome. Tissue sections were rehydrated and stained with 1% Alcian blue (Sigma-Aldrich, Saint Louis, MO) in hydrochloric acid. Counterstaining was performed for 1 min with nuclear-fast red (Sigma-Aldrich, Saint Louis, MO).
Radiograph analyses
X-rays were performed on 6-month-old treated and untreated control using a 9600 Mobile Imaging System (OEC Medical Systems, Milan, Italy). Skull condylobasal length and the maximal width were measured using the software ImageJ (rsbweb.nih.gov/ij/), and their ratio was calculated. Femur and tibia lengths were also measured.
Behavioral tests
Behavioral analyses were performed 4–5 months postinjection on 5–6-month-old female mice receiving either AAV2/8.TBG.hARSB (n=4) or ERT (n=4), and on age-matched normal (n=13) and affected (n=9) controls (one affected control could not perform the rotarod assay because of technical problems). Mice were housed in Plexiglas cages (18×35×12 cm3) with free access to food and water and kept at a temperature of 20–23°C. All tests were carried out in a behavioral testing room maintained under constant light, temperature, and humidity, during daylight hours (9 a.m.–6.00 p.m.). Before each behavioral task, animals were acclimatized to the testing room for at least 30 min. Mice were subjected to the open-field test on day 1 and to the hanging wire test on day 2 of testing. After 5 days of resting, they were subjected to the rotarod test for 5 consecutive days. In the open-field test, mice were placed in the middle of a Plexiglas arena with a masonite base (43×32×40 cm3) placed on a flat surface 70 cm above the floor. A video camera (Panasonic WV-BP330) hanging over the arena was connected to a video-tracking system (ANY-MAZE-Stoeling, Wheat Lane Wood Dale, IL). Animals were left free to explore the arena for 30 min. The following elements were monitored every 5 min: path length (m), time spent in the fringe area (sec), and time spent in the central area (sec). In the hanging wire test, mice were placed in a wire cage lid that was gently waved, so the mouse gripped the wire, and then turned upside down. Latency to fall off the grid was recorded with a cutoff time of 120 sec; latency was normalized for body weight and reported as sec*log BW. Motor activity was tested by using a rotarod apparatus for mice (Ugo Basile, Comerio, Italy) with five rods, as previously described (Cotugno et al., 2010). Briefly, on the first day each mouse was gently placed on each rod set at a steady slow speed of 4 rpm and trained to remain on the rod for 60 sec. After this habituation trial, each animal was exposed to 4 trials per day for 5 consecutive days, with an intertrial interval of 15 min. The trial started when all mice moved in the correct direction. The rotarod was set at increasing speeds ranging from 5 to 40 rpm over 5 min (Monville et al., 2006) and the animals were left on the rod for an additional 5 min. The latency to fall off the rod within this period was recorded. The observer was blinded to genotype and treatment.
Statistical analyses
All results are expressed as mean±SE. Statistical comparisons were made using either t-test or one-way analysis of variance (ANOVA); the Tukey post hoc test was used when appropriate, except for comparisons related to the behavioral analysis where the Fisher LSD post hoc test was used. Statistical significance was considered if p<0.05.
The exact p-value for each comparison follows:
Table 1. Liver: the ANOVA p-value is 1.29e−7; the p-value of normal (NR) vs. AF is 1.29e−7; the p-value of AAV.hARSB vs. AF is 0.015; the p-value of rhARSB vs. AF is 0.920; the p-value of AAV.hARSB vs. NR is 0.154; the p-value of rhARSB vs. NR is 0.003; Kidney: the ANOVA p-value is 2.13e−14; the p-value of NR vs. AF is <2e−16; Spleen: the ANOVA p-value is <2e−16; the p-value of NR vs. AF is <2e−16.
Table 1.
Serum and Tissue Arylsulfatase B Activity in Mucopolysaccharidosis VI Mice Treated with Either Gene Therapy or Enzyme Replacement Therapy
| Groups of animals | Serum ARSB | Liver ARSB | Spleen ARSB | Kidney ARSB |
|---|---|---|---|---|
| NR** | 21627±576 | 132±12** | 67±6** | 220±18** |
| AF | — | — | — | — |
| AAV.hARSB (n=12)** | 3614±460 | 82±30* | 0.83±0.30 | — |
| rhARSB (n=6) | — | 26±5 | 4.70±0.52 | 0.30±0.16 |
AAV, adeno-associated viral vector; AAV.hARSB, MPS VI affected mice treated with 2×1012 genome copies (gc)/kg of AAV2/8.TBG.hARSB; AF, MPS VI affected controls; ANOVA, analysis of variance; ARSB, arylsulfatase B; MPS, mucopolysaccharidosis; NR, normal controls; rhARSB, MPS VI affected mice treated with 1 mg/kg of rhARSB.
Measurements in tissues were done at the time of sacrifice. Serum ARSB is reported as the mean activity measured over time for each group. ARSB is expressed as pg/mL for sera and nmol/mg protein/hr for tissues. The number of animals within each treated group is indicated inside the table. The number of animals with the NR and AF group varies depending on the assay. Values are represented as mean±SE. Statistical comparisons were made using the one-way ANOVA and the Tukey post hoc test. The p-value vs. AF is *≤0.05 and **≤0.01. The exact p-values obtained are indicated in the Materials and Methods section.
Figure 2. The ANOVA p-value is <2e−16; the p-values of NR, AAV.hARSB, and rhARSB vs. AF are <2e−16; the p-value of AAV.hARSB vs. NR is 0.119; the p-value of rhARSB vs. NR is 0.788.
FIG. 2.

Urinary GAG reduction in MPS VI mice treated with either AAV.TBG.hARSB or rhARSB. Urinary GAGs were measured in MPS VI mice treated with either AAV.TBG.hARSB (AAV.hARSB, light gray bar) or rhARSB (dark gray bar) and in normal (NR, white bar) and affected (AF, black bar) controls. The urinary GAG levels measured over time were averaged for each mouse, and the resulting value was then averaged for each group and is indicated inside each bar. Results are represented as mean±SE. Statistical comparisons were made using the one-way ANOVA and the Tukey post hoc test. The p-value vs. AF is **≤0.01. The exact p-values obtained are indicated in the Materials and Methods section. The number (n) of animals in each group is indicated under each bar. ANOVA, analysis of variance; GAG, glycosaminoglycan.
Figure 3. Liver: the ANOVA p-value is <2e−16; the p-values of NR, AAV.hARSB, and rhARSB vs. AF are <2e−16; the p-value of AAV.hARSB vs. NR is 0.999; the p-value of rhARSB vs. NR is 1.000; Kidney: the ANOVA p-value is <2e−16; the p-values of NR, AAV.hARSB, and rhARSB vs. AF are <2e−16; the p-value of AAV.hARSB vs. NR is 0.795; the p-value of rhARSB vs. NR is 0.997; Spleen: the ANOVA p-value is 4.81e−16; the p-values of NR, AAV.hARSB, and rhARSB vs. AF are <2e−16; the p-value of AAV.hARSB vs. NR is 0.954; the p-value of rhARSB vs. NR is 0.999.
FIG. 3.

GAG reduction in liver, kidney, and spleen of MPS VI mice treated with either AAV.TBG.hARSB or rhARSB. Levels of GAGs in liver, kidney, and spleen were measured in MPS VI mice treated with either AAV.TBG.hARSB (AAV.hARSB, light gray bar) or rhARSB (dark gray bar) and in normal (NR, white bar) and affected (AF, black bar) controls. Results are represented as mean±SE and the corresponding value is indicated inside each bar. Statistical comparisons were made using the one-way ANOVA and the Tukey post hoc test. The p-value vs. AF is **≤0.01. The exact p-values obtained are indicated in the Materials and Methods section. The number (n) of animals in each group is indicated inside the graph.
Figure 5. The ANOVA p-value is 3.52 e−7; the p-value of NR vs. AF is 0.00002.
FIG. 5.

Skull abnormalities in MPS VI mice treated with either AAV.TBG.hARSB or rhARSB. The ratio between the skull condilobasal length and the maximal width was measured in 6-month-old MPS VI mice treated with either AAV.TBG.hARSB (AAV.hARSB, light gray bar) or rhARSB (dark gray bar) and in normal (NR, white bar) and affected (AF, black bar) controls. The skull ratio is reported inside each bar as a percentage of normal controls (% of NR). Results are represented as mean±SE. Statistical comparisons were made using the one-way ANOVA and the Tukey post hoc test. The p-value vs. AF is **≤0.01. The exact p-values obtained are indicated in the Materials and Methods section. The number (n) of animals in each group is indicated under each bar.
Supplementary Table S1. Liver: the p-value is 0.845; Kidney: the p-value is 0.829; Spleen: the p-value is 0.151; Brain: the p-value is 0.110.
Supplementary Table S2. 0.5 month postinjection: the ANOVA p-value is 0.000502; the p-value of NR vs. AF is 0.000586; the p-value of AAV.hARSB vs. AF is 0.025; the p-value of rhARSB vs. AF is 0.645; the p-value of AAV.hARSB vs. NR is 0.607; the p-value of rhARSB vs. NR is 0.012; 1 month postinjection: the ANOVA p-value is 0.062; the p-value of NR vs. AF is 0.177; the p-value of AAV.hARSB vs. AF is 0.070; the p-value of rhARSB vs. AF is 0.971; the p-value of AAV.hARSB vs. NR is 0.899; the p-value of rhARSB vs. NR is 0.665; 2 months postinjection: the ANOVA p-value is 5.33e−6; the p-value of NR vs. AF is 1.06e−5; the p-value of AAV.hARSB vs. AF is 8.42 e−5; the p-value of rhARSB vs. AF is 0.0004; the p-value of AAV.hARSB vs. NR is 0.910; the p-value of rhARSB vs. NR is 0.979; 3 months postinjection: the ANOVA p-value is 0.0001; the p-value of NR vs. AF is 0.0002; the p-value of AAV.hARSB vs. AF is 0.010; the p-value of rhARSB vs. AF is 0.003; the p-value of AAV.hARSB vs. NR is 0.712; the p-value of rhARSB vs. NR is 0.982; 4 months postinjection: the ANOVA p-value is 1.92e−5; the p-value of NR vs. AF is 8.33e−6; the p-value of AAV.hARSB vs. AF is 0.001; the p-value of rhARSB vs. AF is 0.004; the p-value of AAV.hARSB vs. NR is 0.579; the p-value of rhARSB vs. NR is 0.738; 5 months postinjection: the ANOVA p-value is 6.74e−6; the p-value of NR vs. AF is 5.22e−6; the p-value of AAV.hARSB vs. AF is 0.0001; the p-value of rhARSB vs. AF is 0.008; the p-value of AAV.hARSB vs. NR is 0.751; the p-value of rhARSB vs. NR is 0.557; 6 months postinjection: the ANOVA p-value is 8.53e−7; the p-value of NR vs. AF is 2.8e−6; the p-value of AAV.hARSB vs. AF is 0.0007; the p-value of rhARSB vs. AF is 2.2 e−5; the p-value of AAV.hARSB vs. NR is 0.739; the p-value of rhARSB vs. NR is 0.848; 7 months postinjection: the ANOVA p-value is 0.0003; the p-value of NR vs. AF is 0.001; the p-value of AAV.hARSB vs. AF is 0.009; the p-value of rhARSB vs. AF is 0.001; the p-value of AAV.hARSB vs. NR is 0.998; the p-value of rhARSB vs. NR is 0.629; 8 months postinjection: the ANOVA p-value is 0.0006; the p-value of NR vs. AF is 0.0006; the p-value of AAV.hARSB vs. AF is 0.231; the p-value of rhARSB vs. AF is 0.009; the p-value of AAV.hARSB vs. NR is 0.567; the p-value of rhARSB vs. NR is 0.960; 9 months postinjection: the ANOVA p-value is 3.42 e−5; the p-value of NR vs. AF is 5.42e−5; the p-value of AAV.hARSB vs. AF is 0.038; the p-value of rhARSB vs. AF is 0.0008; the p-value of AAV.hARSB vs. NR is 0.901; the p-value of rhARSB vs. NR is 0.652; 10 months postinjection: the ANOVA p-value is 0.0009; the p-value of NR vs. AF is 0.0004; the p-value of AAV.hARSB vs. AF is 0.082; the p-value of rhARSB vs. AF is 0.235; the p-value of AAV.hARSB vs. NR is 0.898; the p-value of rhARSB vs. NR is 0.560; 11 months postinjection: the ANOVA p-value is 0.0019; the p-value of NR vs. AF is 0.0018; the p-value of AAV.hARSB vs. AF is 0.13; the p-value of rhARSB vs. AF is 0.037; the p-value of AAV.hARSB vs. NR is 0.994; the p-value of rhARSB vs. NR is 0.976.
Supplementary Fig. S2. Hanging wire test: the ANOVA p-value is 0.04 (F3/26=3.18); the p-value of NR vs. AF is 0.006; the p-value of AAV.hARSB vs. AF is 0.06; the p-value of rhARSB vs. AF is 0.10; the p-value of AAV.hARSB vs. NR is 0.83; the p-value of rhARSB vs. NR is 0.64. Open field (first 5 min of testing): the ANOVA p-value for the total 30 min testing is (1) groups effect, 0.7 (F3/26=0.49); (2) time interval effect, 0.0001 (F5/130=52.62); (4) groups×time interval effect, 0.01 (F15/130=2.16); the p-value of NR vs. AF is 0.03; the p-value of AAV.hARSB vs. AF is 0.4; the p-value of rhARSB vs. AF is 0.22; the p-value of AAV.hARSB vs. NR is 0.42; the p-value of rhARSB vs. NR is 0.69. Rotarod: the ANOVA p-value is 0.12 (F3/25=2.09); the p-value of NR vs. AF is 0.04; the p-value of AAV.hARSB vs. AF is 0,12; the p-value of rhARSB vs. AF is 0.04; the p-value of AAV.hARSB vs. NR is 0.97; the p-value of rhARSB vs. NR is 0.58. Time spent in the central area: the ANOVA p-value is 0.07 (F3/26=2.638); the p-value of NR vs. AF is 0.037; the p-value of AAV.hARSB vs. AF is 0.42; the p-value of rhARSB vs. AF is 0.52; the p-value of AAV.hARSB vs. NR is 0.42; the p-value of rhARSB vs. NR is 0.026.
N.B. Please note that 2e−16 is the minimum value calculated by the software; p-Values relative to the behavioral analysis are also indicated inside the body text.
Results
Gene therapy results in increased and stable levels of ARSB, while ERT has a typical peak-and-drop serum kinetic
MPS VI mice at postnatal day 30 (p30) received a single intravenous administration of 2×1012 gc/kg of AAV2/8-TBG-hARSB vector, which encodes hARSB under the control of the liver-specific TBG promoter. The dose used is the highest dose administered systemically in clinical trials based on AAVs (Manno et al., 2006; Nathwani et al., 2011b), which, we have previously shown, can provide therapeutic levels of ARSB in the MPS VI feline model (Cotugno et al., 2011; Ferla et al., 2012). A second cohort of MPS VI mice was treated, starting from p30, with weekly intravenous injections of 1 mg/kg rhARSB (Naglazyme; BioMarin Europe), the dose currently used in MPS VI patients. As controls, MPS VI mice received at p30 either a single injection of 2×1012 gc/kg of AAV2/8-TBG-eGFP (which encodes the enhanced green fluorescent protein) or weekly injections of PBS, or were left untreated.
Serum ARSB activity was undetectable in control MPS VI mice. MPS VI mice that received gene therapy had stably increased levels of serum ARSB levels, which were on average 17% of NR (Fig. 1a). To confirm long-term transgene expression we measured ARSB enzyme activity and AAV vector genome copies (gc) in livers from injected and control mice at the end of the study. The ARSB activity in the liver of mice that received AAV2/8.TBG.hARSB was not statistically different from one measured in normal animals (Table 1). In addition, persistence of liver transduction was confirmed by the presence of detectable AAV vector gc (0.9±0.5 gc/molecule of diploid genome) at the end of the study.
FIG. 1.

Serum ARSB levels in MPS VI mice treated with either AAV-mediated gene therapy (a) or enzyme replacement therapy (b). (a) MPS VI mice were injected with 2×1012 gc/kg of AAV2/8.TBG.hARSB at postnatal day 30 (p30). Serum ARSB (pg/ml) was monitored for up to 6 or 12 months. Each curve represents the mean±SE of serum ARSB levels measured over time in normal controls (NR, gray line with triangles) and in MPS VI mice receiving AAV2/8.TBG.hARSB (AAV.hARSB, black line with full symbols). Serum ARSB levels were undetectable in affected controls. The number of normal controls is 21 up to 6 months and 12 up to 12 months. The number of AAV.hARSB-treated mice is 14 up to 6 months and 2 up to 12 months. (b) MPS VI mice were injected with 1 mg/kg of rhARSB and levels of ARSB in the serum were measured after 5 min and 1, 3, 5, 24, and 48 hr. The curve represents the mean±SE of serum ARSB levels measured at the various time points in ERT-treated mice (n=3, except for 24 and 48 hr n=2). AAV, adeno-associated viral vector; ARSB, arylsulfatase B; ERT, enzyme replacement therapy; MPS, mucopolysaccharidosis.
In contrast, as previously reported in cats and humans (Crawley et al., 1996; Harmatz et al., 2005b), rhARSB was rapidly cleared from the circulation after enzyme infusion and dropped to undetectable levels 5 hr after the infusion (Fig. 1b). As predicted, anti-hARSB antibodies were not detected in the serum of mice treated with either gene therapy or ERT in the immune tolerant MPS VI mouse model (data not shown).
Amelioration of biochemical, visceral, cardiac, skeletal, and behavioral abnormalities in MPS VI mice treated with either gene therapy or ERT
Reduction of urinary GAGs is considered to be a sensitive marker of lysosomal storage clearance and therapeutic efficacy in LSDs (Harmatz et al., 2004, 2005a,b, 2006, 2008). GAG levels in the urine were measured monthly in MPS VI mice receiving either gene therapy or ERT as well as in NR and AF controls. The urinary GAG level measured over time were averaged for each mouse and the resulting value was then averaged for each group and reported as a percentage (%) of AF mice, as shown in Fig. 2. Urinary GAGs in NR mice were about 42% of urinary GAGs in affected control mice. Urinary GAGs were reduced to approximately normal levels both in mice treated with gene therapy and those treated with ERT (Fig. 2). Urinary GAGs levels at each time point are reported in the Supplementary Table S2.
We then measured ARSB activity as well as GAG levels in liver, kidney, and spleen of MPS VI treated and control mice (Table 1 and Fig. 3). In MPS VI mice receiving AAV2/8.TBG.hARSB, ARSB activity was detected in spleen but not in kidney. However, GAGs were normalized in both organs, indicating that undetectable levels of enzyme provided by gene therapy clear visceral organs from lysosomal storage (Fig. 3). MPS VI mice treated with ERT were sacrificed 7 days after the last injection of rhARSB to measure the tissue residual enzymatic activity. Although ARSB activity rapidly drops to undetectable levels in the serum of ERT-treated mice, mainly because of enzyme uptake in peripheral tissues, we found ARSB activity in tissues up to 7 days after injection, as previously reported in MPS VI cats (Crawley et al., 1996). In particular, increased but lower than normal ARSB activity was detected in the liver of mice receiving ERT. Nonetheless, ARSB activity was detected in the spleen but not in the kidney similar to the mice treated with gene therapy, and normalization of GAGs occurred in all tissues analyzed (Table 1 and Fig. 3). Alcian blue staining of tissues confirmed normalization of lysosomal GAG storage in liver, kidney, and spleen of MPS VI mice independently of treatment (Supplementary Fig. S1).
Cardiomyopathy and heart valve involvement are serious clinical complications of MPS VI that often negatively affect its prognosis (Braunlin et al., 2011). Peters and associates reported that knockout MPS VI mice (Evers et al., 1996) mimic the human MPS VI cardiac disease (Strauch et al., 2003). We performed Alcian blue staining on heart histological sections from mice receiving either AAV.TBG.hARSB (n=2) or rhARSB (n=2) and found that a dramatic reduction of GAG levels was observed in both heart valves and myocardium after both types of treatment (Fig. 4).
FIG. 4.
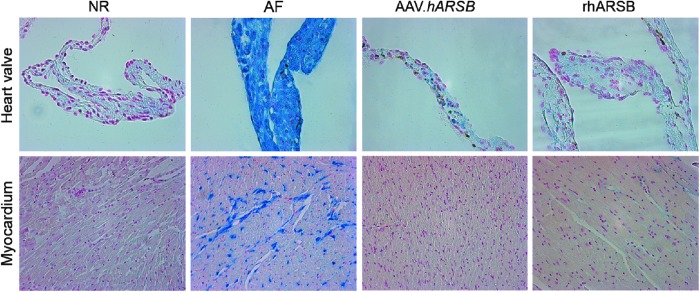
GAG reduction in myocardium and heart valves of MPS VI mice treated with either AAV.TBG.hARSB or rhARSB. Reduction of GAGs storage in myocardium and heart valves was evaluated by Alcian blue staining of heart histological sections obtained from MPS VI mice receiving AAV.TBG.hARSB (AAV.hARSB) or rhARSB and from normal (NR) and affected (AF) mice. Magnification is 40× for heart valves and 20× for myocardium.
One of the major landmarks of MPS VI is severe skeletal dysplasia. Like MPS VI patients and other MPS VI animal models (Yoshida et al., 1993; Evers et al., 1996; Neufeld and Muenzer, 2010; Cotugno et al., 2010, 2011), our MPS VI mice had widened and shortened skulls (Fig. 5) as well as shortened long bones (data not shown). To assess the impact of hARSB liver gene transfer or rhARSB administration on the skeletal phenotype of MPS VI mice, we performed radiographic analysis of both skull and long bones in 6-month-old treated and control animals. Skull abnormalities were assessed by measuring the ratio between the condylobasal length and the maximal zygomatic width and were reported as the percentage of the skull ratio of NR age-matched control. As previously reported in other MPS VI models (Tessitore et al., 2008; Cotugno et al., 2010), the skull length-to-width ratio is significantly reduced in AF mice compared with NR controls (88 vs. 100%). Although neither gene therapy nor ERT significantly improved the skull abnormalities, some mice in the gene therapy-treated group (4 out of 13 animals) had normalized skull ratios (103%, 103%, 102%, and 98% of NR) (Fig. 5). No improvement of long bones' length was observed in treated mice compared with AF controls regardless of the type and age of mice at treatment administration. Indeed, no response in terms of femur growth was observed in mice receiving either AAV.TBG.hARSB at p2, p15 (2×1014 gc/kg), or p30 (2×1012 gc/kg) or weekly injections of rhARSB (1 mg/kg) starting at p15 and p30.
Bone and joint abnormalities are the cause of motor disabilities in MPS VI patients as well as in cat and rat models of the disease (Cotugno et al., 2010, 2011). The behavioral phenotype of MPS VI mice has not been addressed before. We hypothesized that MPS VI mice could have impaired motor activity, as described for other MPS VI animal models (Cotugno et al., 2010, 2011). To test this hypothesis we subjected 5–6-month-old mice to the hanging wire, the open field, and the rotarod tasks. Behavioral performance of MPS VI affected mice (AF, n=9) compared with normal control mice (NR, n=13) was impaired in the hanging wire (ANOVA p-value is 0.01; F1/20=6.81) and in the open field [genotype×time interval (ANOVA p-value is 0.0003; F5/100=5.129]; the difference in the open field was mainly because of the first 5 min testing (ANOVA p-value is 0.039; F1/20=4.85). Consistently, in the rotarod task the difference between AF and NR in the mean latency to fall off the rod almost approached significance (ANOVA p-value is 0.06; F1/19=3.76). Although, the number of treated animals was too low (n=4) to draw definitive conclusions on the behavioral effects of the therapies, the results we obtained are highly consistent among all tasks performed and suggest that both treatments similarly improved motor activity in MPS VI mice evaluated 4–5 months postinjection (see Supplementary Results and Supplementary Fig. S2). A qualitative analysis of the exploratory behavior patterns in the open field of these different groups of animals suggests that AF tend to spend less time in the center of the arena as compared with NR animals. This behavioral pattern is evidenced also in AF animals treated with ERT, but not in AF animals treated with AAV (Supplementary Fig. S2c and e). An altered exploratory pattern/behavior in the open field is commonly reported in the literature as a sign of anxiety or distress (Li et al., 2012). Although further confirmation is needed, these behavioral results suggest that both therapies are comparable in rescuing the motor deficits in MPS VI mice, but gene therapy may be additionally associated with reduced signs of distress compared with ERT.
Discussion
ERT is the current standard of care for several lysosomal storage diseases (Desnick and Schuchman, 2012; Wyatt et al., 2012) and is being developed for additional others (Hemsley et al., 2008; Dickson and Chen, 2011; Sohn et al., 2013) in which evidence of therapeutic efficacy has been shown in preclinical as well as clinical trials. Although ERT has been a major breakthrough in the treatment of patients with LSDs, who until then have only benefited from supportive therapies, ERT efficacy is partial and has several side effects associated with frequent infusions (Desnick and Schuchman, 2012; Wyatt et al., 2012). An additional limitation of ERT is the requirement for multiple administrations, which limits compliance. In addition, ERT utilization is limited in some countries where the health system cannot afford its high costs (Schlander and Beck, 2009).
Studies of in vivo and ex vivo gene therapy in animal models have indeed confirmed the potential of gene delivery to provide sustained therapeutic efficacy after a single administration (Byrne et al., 2012). Since gene therapy is based on a single vector administration and provides stable levels of therapeutic enzyme, it is expected to overcome many of the limitations of ERT.
In this study we compared the efficacy of gene therapy based on AAV-mediated, liver-directed gene transfer with ERT using an MPS VI mouse model, which is the first side-by-side comparison between gene therapy and ERT for an LSD. Here we have shown that a single systemic administration of AAV2/8-TBG-hARSB is as effective as weekly infusions of rhARSB, a result consistent with ARSB uptake by deficient cells with both treatments and comparable enzyme biodistribution among the various tissues between the two treatments.
Clearance of lysosomal storage in visceral organs is one of the main outcomes of ERT in MPS VI, both in animal models (Crawley et al., 1996; Auclair et al., 2003) and in patients (Harmatz et al., 2004, 2005a,b, 2006, 2008). In this study, normalization of lysosomal storage occurred in all tissues in both of the two groups of treated mice, even though very low to undetectable levels of enzyme were found in some organs, such as kidney. These findings support what found in MPS VI rats and cats (Cotugno et al., 2010, 2011), that very low ARSB tissue levels are sufficient to achieve lysosomal storage clearance.
Cardiac valve disease is usually unresponsive or, at best, only stabilized in MPS VI patients undergoing ERT, although very early treatment has been reported to prevent heart valve disease (Braunlin et al., 2011). We previously demonstrated that AAV-mediated gene therapy could reduce heart valve abnormalities in MPS VI cats, independent of ARSB serum levels and age of treatment (Cotugno et al., 2011). Here we show that the stable ARSB levels provided by gene therapy, which are on average 17% of NR, appear to be as effective as the transient supraphysiologic levels provided by weekly infusions of rhARSB in reducing GAG storage in heart muscle and heart valves.
Even if definitive conclusions cannot be drawn because of the low number of treated animals, our data suggest that both therapies similarly improved motor activity and that gene therapy improves the animal performance in the open-field test. This could be because of reduced signs of animal distress or anxiety compared with the multiple injection course of ERT. Indeed, it has been reported that anxiolitic drugs increase the percentage of time spent in the center of the arena in the first minutes of exploration (Silverman et al., 2007; Li et al., 2012). The absence of primary CNS involvement in MPS VI disease suggests that increased anxiety in MPS VI is secondary to motor disturbance (Tessitore et al., 2008). Weekly administrations required by ERT are likely the cause of lack of beneficial effects on distress measures, despite the observed improvement of motor activity. Accordingly, it has been previously shown that mice that had received a single intraperitoneal injection of harmless saline show an increased glucocorticoid (stress) response to a second saline injection (Drude et al., 2007). Although further confirmation is needed, these data suggest that the repeated drug injection procedures might have distressing effects in mice as well as in humans.
Amelioration of LSD skeletal abnormalities is indeed very challenging because of the intrinsically poor vascularization of cartilage and bone (Standring et al., 2004). We observed similar amelioration of skull bone abnormalities in response to both ERT and gene therapy, although the latter tended to perform slightly better. No amelioration of long bones' length and pathology was observed for either treatment. The improved therapeutic outcomes in flat compared with long bones reflect the different vascularization between these two types of bones (Standring et al., 2004). The lack of impact on the skeletal disease in the MPS VI mouse model may be explained by the use of the non-species-specific human transgene, which may be associated with lower biological activity than the species-specific murine enzyme. For instance, it may be a poorer substrate for murine phospho-transferase or sulfatase-modifying factor (SUMF1, Cosma et al., 2003), limiting how much active and phosphorylated enzyme the liver might produce and secrete. Indeed, the advantages of using a species-specific enzyme were clearly demonstrated in MPS VI cats (Bielicki et al., 1999). Alternatively, the product of the inactive transgenic C91S hARSB protein could somehow compete with vector-derived ARSB for crucial posttranslational modifications [i.e., the modification operated by SUMF1 on all sulfatases, including ARSB].
In summary, we used a mouse model of MPS VI to show that a single systemic administration of a gene therapy vector can be as effective long-term as multiple weekly infusions of recombinant enzyme used in ERT. Our study illustrates the potential advantages of AAV-mediated gene therapy for the treatment of LSDs and should encourage further testing of its efficacy in humans.
Supplementary Material
Acknowledgments
We thank Annamaria Carissimo and Luisa Cutillo (Bioinformatics Core, TIGEM) for the statistical analyses; Viola Alba, Monica Doria, and Antonella Ferrara (AAV Vector Core, TIGEM) for AAV vector production; Ellen Abrams and Graciana Diez-Roux (Scientific Office, TIGEM) for the critical reading of the manuscript; and Prof. Christopher Peters (Institute of Molecular Medicine and Cell Research, University of Freidburg) for the kind gift of the MPS VI transgenic mouse.
This work was supported by the European Community's Seventh Framework Programme (FP7/2007–2013)—MeuSIX (304999), the Telethon Foundation (TMAAMT611TT), the Isaac Foundation, and the American MPS society.
Author Disclosure Statement
The authors declare no conflicts of interest.
References
- Auclair D., et al. (2003). Replacement therapy in mucopolysaccharidosis type VI: advantages of early onset of therapy. Mol. Genet. Metab. 78, 163–174 [DOI] [PubMed] [Google Scholar]
- Bielicki J., et al. (1999). Advantages of using same species enzyme for replacement therapy in a feline model of mucopolysaccharidosis type VI. J. Biol. Chem. 274, 36335–36343 [DOI] [PubMed] [Google Scholar]
- Braunlin E.A., et al. (2011). Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J. Inherit. Metab. Dis. 34, 1183–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks D.A., et al. (1995). Two site-directed mutations abrogate enzyme activity but have different effects on the conformation and cellular content of the N-acetylgalactosamine 4-sulphatase protein. Biochem. J. 307 (Pt 2), 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N., and Auricchio A. (2010). Gene therapy of human inherited diseases. In The Online Metabolic and Molecular Bases of Inherited Diseases. Scriver R., ed. (McGraw Hill, New York: ). 5.2, 1–50 [Google Scholar]
- Byrne B.J., et al. (2012). Gene therapy approaches for lysosomal storage disease: next-generation treatment. Hum. Gene Ther. 23, 808–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P.L., et al. (1981). Differential assay of arylsulfatase A and B activities: a sensitive method for cultured human cells. Anal. Biochem. 117, 382–389 [DOI] [PubMed] [Google Scholar]
- Cosma M.P., et al. (2003). The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 113, 445–456 [DOI] [PubMed] [Google Scholar]
- Cotugno G., et al. (2010). Different serum enzyme levels are required to rescue the various systemic features of the mucopolysaccharidoses. Hum. Gene Ther. 21, 555–569 [DOI] [PubMed] [Google Scholar]
- Cotugno G., et al. (2011). Long-term amelioration of feline mucopolysaccharidosis VI after AAV-mediated liver gene transfer. Mol. Ther. 19, 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley A.C., et al. (1996). Enzyme replacement therapy in a feline model of Maroteaux-Lamy syndrome. J. Clin. Invest. 97, 1864–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong J.G., et al. (1989). Dimethylmethylene blue-based spectrophotometry of glycosaminoglycans in untreated urine: a rapid screening procedure for mucopolysaccharidoses. Clin. Chem. 35, 1472–1477 [PubMed] [Google Scholar]
- Desnick R.J., and Schuchman E.H. (2012). Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu. Rev. Genomics Hum. Genet. 13, 307–335 [DOI] [PubMed] [Google Scholar]
- Dickson P.I., and Chen A.H. (2011). Intrathecal enzyme replacement therapy for mucopolysaccharidosis I: translating success in animal models to patients. Curr. Pharm. Biotechnol. 12, 946–955 [DOI] [PubMed] [Google Scholar]
- Drude S., et al. (2007). Side effects of control treatment can conceal experimental data when studying stress responses to injection and psychological stress in mice. Lab. Anim. (NY) 40, 119–128 [DOI] [PubMed] [Google Scholar]
- Evers M., et al. (1996). Targeted disruption of the arylsulfatase B gene results in mice resembling the phenotype of mucopolysaccharidosis VI. Proc. Natl. Acad. Sci. USA 93, 8214–8219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferla R., et al. (2012). Gene therapy for mucopolysaccharidosis type VI is effective in cats without pre-existing immunity to AAV8. Hum. Gene Ther. 24, 163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich G., and Soriano P. (1991). Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 5, 1513–1523 [DOI] [PubMed] [Google Scholar]
- Harmatz P., et al. (2004). Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J. Pediatr. 144, 574–580 [DOI] [PubMed] [Google Scholar]
- Harmatz P., et al. (2005a). Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics 115, e681–e689 [DOI] [PubMed] [Google Scholar]
- Harmatz P., et al. (2005b). Pharmacokinetic profile of recombinant human N-acetylgalactosamine 4-sulphatase enzyme replacement therapy in patients with mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): a phase I/II study. Acta Paediatr. Suppl. 94, 61–68; discussion 57 [DOI] [PubMed] [Google Scholar]
- Harmatz P., et al. (2006). Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J. Pediatr. 148, 533–539 [DOI] [PubMed] [Google Scholar]
- Harmatz P., et al. (2008). Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol. Genet. Metab. 94, 469–475 [DOI] [PubMed] [Google Scholar]
- Hartung S.D., et al. (2004). Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human alpha-L-iduronidase gene. Mol. Ther. 9, 866–875 [DOI] [PubMed] [Google Scholar]
- Hemsley K.M., et al. (2008). Effect of high dose, repeated intra-CSF injection of sulphamidase on neuropathology in MPS IIIA mice. Genes Brain Behav. 7, 740–753 [DOI] [PubMed] [Google Scholar]
- Li Q., Luo T., Jiang X., and Wang J. (2012). Anxiolytic effects of 5-HT1A receptors and anxiogenic effects of 5-HT2C receptors in the amygdala of mice. Neuropharmacology 62, 474–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., et al. (2013). Retro-orbital injection of RAAVs to neonatal mice dramtically enhances gene delivery to the CNS. ASGCT Abstract No. 326 [Google Scholar]
- Manno C.S., et al. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347 [DOI] [PubMed] [Google Scholar]
- Mingozzi F., and High K.A. (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 12, 341–355 [DOI] [PubMed] [Google Scholar]
- Monville C., et al. (2006). Comparison of incremental and accelerating protocols of the rotarod test for the assessment of motor deficits in the 6-OHDA model. J. Neurosci. Methods 158, 219–223 [DOI] [PubMed] [Google Scholar]
- Nathwani A.C., et al. (2011a). Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol. Ther. 19, 876.–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani A.C., et al. (2011b). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 365, 2357.–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld E.F. (1991). Lysosomal storage diseases. Annu. Rev. Biochem. 60, 257–280 [DOI] [PubMed] [Google Scholar]
- Neufeld E., and Muenzer J. (2010). The mucopolysaccharidoses. In The Online Metabolic and Molecular Bases of Inherited Disease. Scriver R., ed. (McGraw Hill, New York: ) 136, 1–73 [Google Scholar]
- Sands M.S., and Davidson B.L. (2006). Gene therapy for lysosomal storage diseases. Mol. Ther. 13, 839–849 [DOI] [PubMed] [Google Scholar]
- Schlander M., and Beck M. (2009). Expensive drugs for rare disorders: to treat or not to treat? The case of enzyme replacement therapy for mucopolysaccharidosis VI. Curr. Med. Res. Opin. 25, 1285–1293 [DOI] [PubMed] [Google Scholar]
- Silverman M.N., et al. (2007). Endogenous glucocorticoids protect against TNF-alpha-induced increases in anxiety-like behavior in virally infected mice. Mol. Psychiatry 12, 408–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn Y.B., et al. (2013). Improvement of CNS defects via continuous intrathecal enzyme replacement by osmotic pump in mucopolysaccharidosis type II mice. Am. J. Med. Genet. A 161A, 1036–1043 [DOI] [PubMed] [Google Scholar]
- Sorrentino N.C., et al. (2013). A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol. Med. 5, 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standring S., et al. (2004). Gray's Anatomy: The Anatomical Basis of Clinical Practice (Churchill Livingstone, New York: ) [Google Scholar]
- Strauch O.F., et al. (2003). Cardiac and ocular pathologies in a mouse model of mucopolysaccharidosis type VI. Pediatr. Res. 54, 701–708 [DOI] [PubMed] [Google Scholar]
- Sun B., et al. (2005). Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol. Ther. 11, 57–65 [DOI] [PubMed] [Google Scholar]
- Tessitore A., et al. (2008). Biochemical, pathological, and skeletal improvement of mucopolysaccharidosis VI after gene transfer to liver but not to muscle. Mol. Ther. 16, 30–37 [DOI] [PubMed] [Google Scholar]
- Thomas C.E., et al. (2004). Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J. Virol. 78, 3110–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D.A., et al. (2011). Direct gene transfer to the CNS prevents emergence of neurologic disease in a murine model of mucopolysaccharidosis type I. Neurobiol. Dis. 43, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt K., et al. (2012). The effectiveness and cost-effectiveness of enzyme and substrate replacement therapies: a longitudinal cohort study of people with lysosomal storage disorders. Health Technol. Assess. 16, 1–543 [DOI] [PubMed] [Google Scholar]
- Yoshida M., et al. (1993). Arylsulfatase B-deficient mucopolysaccharidosis in rats. J. Clin. Invest. 91, 1099–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler R.J., et al. (2007). Correction of the biochemical and functional deficits in fabry mice following AAV8-mediated hepatic expression of alpha-galactosidase A. Mol. Ther. 15, 492–500 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.