

Abstract
In recent years, our understanding of the genetic foundations of incomitant strabismus has grown significantly. Much new understanding has been gleaned since the concept of congenital cranial dysinnervation disorders (CCDDs) was introduced in 2002, and the genetic basis of CCDDs continues to be elucidated. In this review, we aim to provide an update of the genetic and clinical presentation of these disorders. Disorders reviewed include Duane syndrome (DS), HOXA1 and HOXB1 syndromes, Moebius syndrome, congenital fibrosis of the extraocular muscles (CFEOM), and horizontal gaze palsy with progressive scoliosis (HGPPS).
Keywords: congenital cranial dysinnervation disorders, congenital fibrosis of the extraocular muscles, Duane syndrome, Moebius syndrome, horizontal gaze palsy with progressive scoliosis, HOX mutations
Introduction
Congenital cranial dysinnervation disorders (CCDDs) are a group of disorders that result from the abnormal development of cranial motor nuclei and their respective cranial nerves. Because these cranial nerves fail to appropriately innervate their intended target muscles, these muscles often have fibrotic changes, which is now understood to be secondary to either absent or aberrant innervation from other nearby cranial nerves.
Our understanding of the CCDDs has evolved over time. The path to understanding the history of this is best exemplified by congenital fibrosis of the extraocular muscles (CFEOM). Long recognized as a source of ocular pathology, elements of this disorder were first described as early as the late 1800s, but it was not until 1956 that Laughlin first coined the term CFEOM.1 The basis of pathology was initially thought to be the muscles themselves, which showed fibrosis histologically and restrictive strabismus clinically. Through the 1980s and 90s, however, mounting evidence showed that the underlying pathology was the agenesis or hypoplasia of the oculomotor nerve and that the disorder was in fact absent or aberrant innervation of these muscles that caused secondary fibrotic and restrictive changes. Other disorders were also shown to have similar underlying pathogenesis, including Duane syndrome (DS) and Moebius syndrome. This new understanding prompted the reclassification of these disorders under the umbrella term “CCDDs.”
In this review, we discuss the current understanding of the genetic underpinnings of CCDDs. Table 1 summarizes the disorders, their clinical and radiographic findings, and the genes that have been associated with each of them.
Table 1.
Summary of Disorders with Defined Genetic Basis
| Disorder | Associated Genes | Inheritance | Clinical Findings | Orbital and Intracranial MRI Findings |
|---|---|---|---|---|
| Non-Syndromic Duane Syndrome | ||||
| Familial DS | CHN1 | AD | Type 1 or Type 3 DS and/or vertical motility anomalies | Abnormalities in the abducens and oculomotor nerve with or without superior oblique muscle hypoplasia |
| Syndromic Duane Syndrome | ||||
| Duane Radial Ray (Okihiro) Syndrome | SALL4 | AD | DS, radial ray anomaly, with or without deafness | Hypoplastic to absent abducens nerves with normal intracranial and optic nerves. Evidence of aberrant innervation of the lateral rectus muscle |
| Holt-Oram syndrome | SALL4 | AD | Radial ray anomalies with cardiac defects without DS | |
| Acro-renal-ocular syndrome | SALL4 | AD | Radial ray anomalies, kidney defects, DS | |
| Townes-Brock Syndrome | SALL1 | AD | Imperforate anus, hearing impairment, thumb malformations with rare DS association | |
| HOX Mutations | ||||
| Bosley-Salih-Alorainy Syndrome | HOXA1 | AR | Bilateral type 3 DS, sensorineural hearing loss, malformations of the cerebral vasculature, cardiac malformations, autism | Normal extraocular muscles. Hypoplastic abducens nerve Hypoplastic or absent internal carotid arteries. Occasional duplication of the vertebral artery. |
| Athabascan Brain Dysgenesis Syndrome | HOXA1 | AR | Horizontal gaze restriction, intellectual disabilities, sensorineural hearing loss, cardiac malformations, facial weakness, central hypoventilation, cerebral vasculature malformation | |
| HoxB1 | HOXB1 | AR | Esotropia, bilateral facial palsy, deafness | Bilateral absence of the facial nerve |
| Congenital Fibrosis of the Extraocular Muscles | ||||
| CFEOM1 |
KIF21A Rarely TUBB3 |
AD | Bilateral nonprogressive restrictive ophthalmoplegia with blepharoptosis. Eyes are infraducted in resting position with limitation of vertical movements | Hypoplasia of oculomotor nerve > abducens nerve. Hypoplasia of levator palpebrae and superior rectus muscles. Misinnervation of lateral rectus muscle by oculomotor nerve. Reduction of mean optic nerve size compared with normal subjects. |
| CFEOM2 | PHOX2A | AR | Profound ptosis, restrictive ophthalmoplegia with exotropia and poorly reactive pupils | Enlarged lateral rectus muscles with all other extraocular muscles comparatively hypoplastic. Absence of oculomotor and trochlear nerves |
| CFEOM3 |
TUBB3 Rarely KIF21A |
AD | Variable phenotype with unilateral or bilateral blepharoptosis and ophthalmoplegia with some limitation of vertical movements | Variable findings that correlate with clinical features. Hypoplasia of the superior rectus, medial rectus, levator palpebrae, and inferior oblique muscles to varying degrees. Hypoplasia of the oculomotor nerve. Evidence of dysinnervation |
| Horizontal Gaze Palsy with Progressive Scoliosis | ||||
| HGPPS | ROBO3 | AR | Horizontal gaze limitation, scoliosis | Flattened pons, hindbrain midline cleft. Butterfly configuration of brainstem on axial scans |
Key: AD – autosomal dominant, AR – autosomal recessive, DS – Duane Syndrome, CFEOM – congenital fibrosis of extraocular muscles, HGPPS – horizontal gaze palsy with progressive scoliosis
Methods
A literature search of PubMed (covering years 1950-January, 2013) for the following terms was conducted: “congenital cranial dysinnervation disorders,” “Duane syndrome,” “CHN1 and Duane syndrome,” “Duane radial ray syndrome,” “CHN1 and Duane syndrome,” “Goldenhar and Duane syndrome,” “SALL1 and Duane syndrome,” “8q12 duplication and Duane syndrome,” “8q13 and Duane syndrome,” “Wildervanck and Duane syndrome,” “HOXA1 and Bosley-Salih-Alorainy syndrome,” “HOXA1 and Athabascan Brain Dysgenesis syndrome,” “Moebius syndrome,” “HOXB1 and Moebius syndrome,” “congenital fibrosis of the extraocular muscles,” “congenital fibrosis of the extraocular muscles and KIF21A,” “CFEOM and PHOX2A,” “CFEOM and TUBB3,” “horizontal gaze palsy with progressive scoliosis,” and “horizontal gaze palsy with progressive scoliosis and ROBO3.” Reference lists within pertinent articles were reviewed. Only English language papers with relevance to ocular findings were reviewed.
Isolated (Non-Syndromic) Duane Syndrome
The most common CCDD, Duane syndrome (DS), has been reported to account for 1–4% of strabismus cases.2,3 DS is characterized by congenital absence or hypoplasia of the abducens nucleus and subsequent aberrant innervation of the lateral rectus muscle by branches of the oculomotor nerve.2,3 Clinically, this syndrome manifests as decreased horizontal movement of the affected eye with narrowing of the palpebral fissure and globe retraction with attempted adduction.4
Three types of DS have been described. In type 1 DS, the affected eye has limited abduction but preserved adduction, frequently resulting in esotropia in primary gaze. In type 2, the affected eye has limited adduction but preserved abduction, often causing exotropia in primary gaze. In type 3, the affected eye has limitation of both abduction and adduction. In many patients, these types are not distinct once the eye movements have been carefully scrutinized. The primary gaze deviation can be esotropia, exotropia, or orthophoria, depending on the balance between the deficits of the horizontal muscles.
Most individuals with Duane syndrome are the only affected member of their family (simplex, or sporadic, cases), but hereditary forms account for 5–10% of cases.5 Several investigators have described bilateral DS with an autosomal dominant inheritance pattern.6,7 Recent mapping of the phenotype of families with DS inherited as a dominant trait identified the DURS2 locus on chromosome 2 and, subsequently, heterozygous mutations in CHN1.5,8–12 Current evidence suggests that gain of function mutations in CHN1 hyperactivate α2-chimerin and cause disruption in the growth or guidance of cranial axons destined to innervate extraocular muscles during development.11,12
Phenotypic variability has been recognized among individuals harboring heterozygous CHN1 mutations.7,11,12 Many affected individuals have bilateral DS with either manifestations of either type 1 or type 3 DS. Some have one type 1 in one eye and type 3 in the other, but none have type 2.9 In addition, a subset also has vertical deviations associated with DS,9,12–14 and some have vertical deviations in the absence of DS.12 Given these features, it is reasonable for patients with bilateral DS and associated vertical motility anomalies12,14 and patients with familial vertical deviations12 to be screened for CHN1 mutations. Individuals harboring the familial mutation do not always have clinical evidence of disease, indicating that while phenotypic penetrance is very high, it is not complete.14,15
Demer and colleagues performed magnetic resonance imaging (MRI) on 8 family members from two autosomal dominant DS pedigrees who harbored CHN1 mutations and compared the findings with those of 11 control patients who had comitant strabismus. The DS patients showed a variable endophenotype, with most participants showing markedly abnormal lateral rectus muscles and some showing abnormalities in other extraocular muscles as well. The only muscles that were not affected were those supplied by the inferior division of the oculomotor nerve: the inferior rectus, medial rectus, and inferior oblique muscles. This finding suggests that the abnormality in DS may not be limited to just the abducens nerve but may also involve the superior branch of the oculomotor nerve. In addition, there was evidence of superior oblique hypoplasia in half of the individuals tested, suggesting that the trochlear nerve may also be affected in some individuals.13
It is important to note that CHN1 has not been identified as a cause of simplex DS15 nor has it been implicated in Brown syndrome, congenital disorders of the oblique muscles, or vertical retraction syndrome.16 CHN1 hyperactivation has, however, been associated with deficits in supraduction in the absence of DS.12 Abnormalities in chromosomes 10 and 22 have also been implicated in the pathogenesis of DS.17 Therefore, while much information on the genetic basis of DS has been gleaned in recent years, much more remains to be determined.
Syndromic Duane Syndrome
While the majority of DS occurs in isolation, DS is associated with characteristic systemic findings in about 30% of cases. In this section, we describe the clinical features and genetic analysis to date of these syndromes.
A spectrum of overlapping disorders that include DS with radial limb abnormalities, facial asymmetry, hearing deficits, ear abnormalities, anal stenosis, and cardiac and renal abnormalities have been associated with mutation in SALL4.19,20 Individuals with SALL4 mutations have also been found in radial limb anomalies without DS.18 Disorders on this spectrum include Duane radial ray (Okihiro) syndrome, Holt-Oram syndrome, and acro-renal-ocular syndrome, all of which have been shown to have links to SALL4 mutations.19 A SALL4 mutation has also been identified in a man previously thought to have thalidomide embryopathy who then had a daughter with similar limb deformities.19 MRI analysis of a family with a heterozygous SALL4 mutation resulting in Duane radial ray syndrome revealed hypoplastic to absent abducens nerves with normal intracranial oculomotor and optic nerves. In some subjects, a branch of the oculomotor nerve was shown to be proximal to the lateral rectus muscle, implying that it may partially innervate this muscle.13 SALL4 mutations have not been identified in patients with simplex DS.21,22 Table 2a summarizes the clinical differences among SALL4 disorders.
Table 2a.
Quick-Look Differentiation of Syndromes with Similar Clinical Pictures for SALL4 Mutations
| Name of Syndrome | DS | Radial Ray Anomalies | Sensorineural Defects | Cardiac Defects | Kidney Defects |
|---|---|---|---|---|---|
| Duane Radial Ray (Okihiro) Syndrome | Sometimes | X | X | ||
| Holt-Oram syndrome | No | X | X | ||
| Acro-renal-ocular syndrome | Yes | X | X |
Townes-Brocks syndrome is an autosomal dominant condition that is associated with mutations in SALL1.20 This syndrome is characterized by ear, limb, anal, and renal anomalies but has rarely been associated with DS.21
Abnormalities on chromosome 8 have been observed to co-segregate with DS. A reciprocal balanced translocation in chromosome 8q13 implicated CPAH as a possible etiology of simplex DS.22 Three other patients with disruption of this gene locus have exhibited DS with various other systemic associations.23–25 Chromosome 8q12 duplications have also been reported to produce a combination of DS with sensorineural deafness, cardiac defects, hypotonia, and developmental delay.26 Duplications in this chromosome locus can also produce developmental delay and particular facial features, including full cheeks, a specific lip shape, and horizontal and flared eyebrows, with or without DS.27,28 The duplicated region of 8q12 includes CHD7, implying that abnormal dosage of the transcribed protein may be a factor.26,27
Wildervanck syndrome (also known as cervico-oculo-acoustic syndrome) is characterized by Duane syndrome, Klippel-Feil deformity of the spine (congenitally fused cervical vertebrae), and hearing loss. This syndrome has a female predominance, suggesting a possible defect on the X chromosome that is lethal in hemizygous males.29 Genetic evaluation of a simplex case in an affected male revealed a microdeletion on chromosome X involving only a single gene: Fibroblast Growth Factor Homologous Factor 13 (FGFHF13).30
Goldenhar syndrome,31 which occurs in as many as 3% of DS patients,32 has been associated with deletions in chromosome 22q11.2.33,34 While some DS has been linked to chromosome 22, the exact locus responsible for the association of these two disorders is, as yet, unknown. DS has been linked to numerous other defined conditions in isolated case reports (e.g., Marfan syndrome),33 but to date, these do not have an identified genetic basis.
HOXA1 Syndromes
Recessive, homozygous HOXA1 mutations have been reported to cause horizontal gaze palsy, facial weakness, and sensorineural hearing loss sometimes associated with developmental delay and hypoventilation.34 Initially, two syndromes, Bosley-Salih-Alorainy Syndrome (BSAS) and Athabascan Brain Dysgenesis syndrome (ABDS), were described. BSAS was described in the Middle Eastern population and included bilateral type 3 DS, sensorineural hearing loss, malformation of the cerebral vasculature, and autism in select patients.34 MRI findings showed grossly normal orbits and extraocular muscles. The internal carotid artery, however, was hypoplastic or absent in all individuals, and a few patients had duplication of the vertebral artery. The abducens nerve appeared to be absent bilaterally in one patient.35 The second variant, ABDS, was reported in the Native American population. These individuals had horizontal gaze restriction, facial weakness, hearing impairment, cerebral vasculature malformation, cardiac malformations, central hypoventilation, and intellectual disabilities.34,36
Despite some initial differences noted between BSAS and ABDS, overlapping features between the two populations were noted at the outset.34 Subsequently, members of a BSAS family were reported to also have cardiac anomalies (a feature more in line with ABDS), and members of the ABDS family had mild cognitive changes more consistent with BSAS. The study also found that not all patients harboring a homozygous HOXA1 mutation had horizontal gaze limitation or deafness.37 These findings further blurred the distinction between the two entities and widen the HOXA1 human phenotype.
Homozygous HOXA1 mutations result in phenotypes that overlap with type 3 DS and Moebius syndrome (see section on Moebius syndrome below). Evaluation of HOXA1 in DS and Moebius patients, however, did not reveal any mutations, indicating that HOXA1 mutations are not a major cause of simplex DS38 or Moebius syndrome.39 Table 2b summarizes the clinical differences between ABDS and BSAS.
Table 2b.
Quick-Look Differentiation of Syndromes with Similar Clinical Pictures for HOXA1 Mutations
| Name of Syndrome | Horizontal Gaze Limitation | Mental Retardation | Autism | Sensorineural Defects | Cerebral Vascular Malformations | Central Hypoventilation |
|---|---|---|---|---|---|---|
| Bosley-Salih-Alorainy Syndrome | X | X | X | |||
| Athabascan Brain Dysgenesis Syndrome | X | X | X | X | X |
Moebius Syndrome
Moebius syndrome is a nonprogressive, congenital facial paralysis with limited abduction of the eyes; it can be associated with hearing impairment and other cranial nerve dysfunction, as well as other developmental abnormalities.40 The syndrome occurs in simplex cases and is more likely to occur in children exposed to misoprostol in utero.41 While some mutations have been identified in patients with atypical forms of Moebius syndrome, no mutations have been observed in patients with typical findings. The genetic basis of this syndrome, if one exists, remains elusive.
Carta and colleagues45 evaluated a large cohort of patients with this syndrome to identify their clinical characteristics. Three different patterns of eye movements were identified. The most common motility pattern, identified in half of the patients, was a large esotropia with almost complete abduction deficit but relatively conserved adduction. These patients utilized cross-fixation to compensate for their duction deficits. The next most common ocular motility pattern was orthotropia in primary gaze with deficits of abduction and adduction, requiring large head movements to look to either side. Vertical ductions were preserved. Finally, the smallest number of patients (less than 10%) exhibited a large angle exotropia with absence of convergence. These patients also had limited vertical gaze accompanied by torticollis.42
HOXB1
Identified in a conservative German American population, a single homozygous HOXB1 mutation has been associated with comitant strabismus, bilateral facial palsy, and hearing impairment in two families. While affected individuals had facial palsy and esotropia, they did not meet criteria for Moebius syndrome, as they had full abduction of both eyes. The mutation is predicted to result in loss of HOXB1 function.43
Congenital Fibrosis of the Extraocular Muscles
Congenital fibrosis of the extraocular muscles (CFEOM) is a congenital, non-progressive restrictive ophthalmoplegia and ptosis that has been shown to have a neurogenic basis. Three phenotypes have been described: CFEOM1, 2, and 3.
CFEOM1, the most common of the three phenotypes, is defined by the combination of non-progressive, bilateral infraduction of the eyes in resting position, limited vertical eye movements with the inability to elevate either eye over the horizontal midline, and bilateral ptosis with a compensatory “chin up” head posture (Figure 1). Many individuals have been noted to have “A pattern” strabismus.44 Pupil size and response are normal. Autopsy and radiographic studies have shown hypoplasia or agenesis of the oculomotor nerve, in particular its superior division.46,47 These studies support the hypothesis that the primary pathology of CFEOM is dysinnervation, with secondary fibrosis of the target muscles.45
FIGURE 1.

Motility of a patient with a KIF21A mutation. He also has aberrant innervation causing a Marcus Gun jaw wink. Reproduced with permission from Yamada, Hunter, et al., “A novel kif21a mutation in a patient with congenital fibrosis of the extraocular muscles and Marcus Gunn jaw-winking phenomenon,” Arch Ophthalmol. 2005;123(9):1254–1259.
Inheritance of CFEOM1 is autosomal dominant. Mutations in KIF21A on chromosome 12, which encodes a kinesin motor protein, have been identified in most individuals with CFEOM1.46,47 Rare CFEOM1 patients harbor mutations in TUBB3,48 and other genes not yet identified are likely to be responsible for CFEOM1 in a minority of individuals.49 In addition, in rare individuals, mutations in KIF21A result in clinical findings most consistent with CFEOM3 (see description below).50
Individuals with CFEOM2 have profound ptosis, exotropia, and small, poorly reactive pupils.51 The condition was found to result from homozygous mutations in PHOX2A,52 which encodes a homeodomain transcription factor. To date, there has been no evidence of PHOX2A mutations in patients with CFEOM1 or 3,53,54 indicating that this is a genetically distinct entity.
Clinical evaluation of patients with PHOX2A mutations found ptosis often with compensatory chin-up position, pupils that were unreactive to light but that retained response to pharmacologic agents, and large angle exotropia with compensatory head turn away from the fixing eye.55 Convergence and abduction were also almost absent. Neuroimaging revealed large lateral rectus muscles consistent with the exotropic eye position, with all other rectus muscles comparatively small. These studies suggest that the clinical findings in CFEOM2 are the result of isolated but complete dysinnervation of targets of the oculomotor and trochlear nerves, with preservation of all other cranial nerves.
CFEOM3 is a third phenotype that has variable clinical findings that include varying degrees of ptosis and ophthalmoplegia, ranging from mild to complete. In the severest form, these patients have profound ptosis and abducted and infraducted position of the globes with severe restriction of motility (Figure 2). In mildly affected individuals, the only deficit is decreased supraduction with the globes in a normal position in primary gaze. Findings can be unilateral or bilateral. Some of these individuals resemble CFEOM1 or CFEOM2 phenotypes. Familial transmission is autosomal dominant with variable penetrance.
FIGURE 2.

(A–E) and (G, H) Eye manifestations of patients with CFEOM3 and TUBB3 mutations. (F) and (I) Concomitant deformities of the extremities in two patients. (J) MRI of the brainstem at the level of the oculomotor nerve. (K–L) MRI of posterior orbit of a patient with TUBB3 mutation (K) compared with a normal posterior orbit (L). Reproduced with permission from Tischfield MA, Baris HN, Wu C, et al., “Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance,” Cell. Jan 8 2010;140(1):74–87. 153×207mm (300×300 DPI).
CFEOM3 can result from heterozygous missense mutations in TUBB3, which encodes a β-tubulin isotype that is a component of neuronal microtubules.48 While some TUBB3 mutations result in isolated CFEOM3, other specific mutations cause dysfunction of additional cranial and spinal nerves, and social and intellectual disabilities accompanied by maldevelopment of the corpus callosum and morphologic changes in the basal ganglia.48
One specific TUBB3 mutation results in an E410K amino acid substitution and results in what is now referred to as the TUBB3 E410K syndrome.39 Clinical overlap exists between atypical Moebius syndrome and the TUBB3 E410K syndrome. Seven of the eight subjects found to have this mutation previously had a diagnosis of Moebius syndrome until genetic evidence showed de novo heterozygous missense TUBB3 mutations.39 These individuals clinically had ptosis and limited upgaze, more consistent with CFEOM3 than with typical Moebius syndrome.
MRI findings in patients with TUBB3 mutations reveal structural correlations with the clinical phenotypes. In individuals with severe phenotypes that clinically resemble CFEOM1, there is hypoplasia of the superior rectus, medial rectus, and levator palpebrae superioris muscles. In some cases, the inferior oblique muscle is also hypoplastic. Aberrent branches of the oculomotor nerve suggest misinnervation of the lateral rectus muscle. Individuals with milder phenotypes also had fewer abnormalities on MRI imaging, with relatively less hypoplasia of the subarachnoid portion of the oculomotor nerve and its targeted extraocular muscles.56
Recently, a homozygous missense TUBB2B mutation was found to be associated with CFEOM and polymicrogyria. To date this is the only mutation in TUBB2B that has been associated with a CFEOM.57 In addition, rare individuals meeting CFEOM3 criteria harbor KIF21A mutations.53,58,59
While the above genes account for the majority of families with CFEOM, additional mutations causing the disorder are likely yet to be identified and characterized. One mutation-negative pedigree exhibits the phenotype of CFEOM3 and has a translocation that implicates a locus on chromosome 13q12 (FEOM4) as the source of pathogenesis.60 Additional autosomal recessive forms of CFEOM also appear to exist. A consanguineous Turkish family with autosomal recessive transmission of a unilateral, non-progressive ophthalmoplegia and hand abnormalities mapped to chromosome 21;61 however, forced ductions were reportedly normal in this family, suggesting an atypical presentation for a CFEOM. In one Lebanese family, affected members had unilateral ptosis and restrictive strabismus but were found to have no identifiable linkage to the CFEOM1, -2, or -3 locus and had no mutation in KIF21A or PHOX2A genes.62 The full spectrum of mutations that can give rise to CFEOM phenotype, therefore, remains to be elucidated.
Horizontal Gaze Palsy with Progressive Scoliosis
First described in 1975, horizontal gaze palsy with progressive scoliosis (HGPPS) is a rare autosomal recessive disorder that features lateral gaze limitation and scoliosis (Figure 3) as key clinical findings.63 It was not until 2004, however, that the pathogenesis was linked to mutations in ROBO3 in consanguineous families with autosomal recessive inheritance pattern of the disease.64 This gene is necessary for hindbrain axons to appropriately cross the midline. Corresponding defects in MRI imaging of the hindbrain were identified in affected individuals, including a characteristic anterior flattening and midline cleft now referred to as a butterfly configuration on axial cuts (Figure 4).64 HGPPS can also result from compound heterozygous mutations in non-consanguineous families.65 Presenting signs in these patients were plagiocephaly and torticollis, both of which developed before scoliosis. Therefore, infants and children presenting with plagiocephaly or torticollis in addition to lateral gaze palsy would benefit from genetic testing for ROBO3 mutations.65 Because of the ophthalmologic and molecular cues that can help diagnose HGPPS, patients can be diagnosed from an early age and screened for the onset of scoliosis,66 the most debilitating clinical sequela of this disorder.
FIGURE 3.
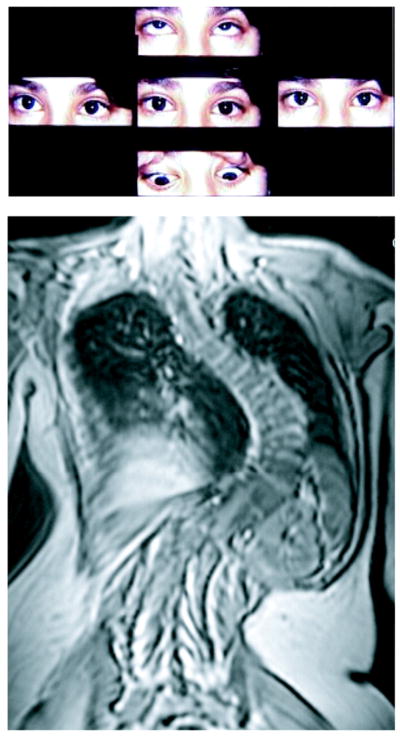
Motility of a patient with horizontal gaze palsy and progressive scoliosis. MRI of spine revealing profound scoliosis below. Reproduced with permission from Jen JC, Chan WM, Bosley TM, et al., “Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis,” Science. Jun 4 2004;304(5676):1509–1513.
FIGURE 4.

MRI studies comparing normal subjects (A–C) with patients with HGPPS (D–F) at similar anatomical levels. CC – corpus callosum, P - pons, M - medulla, * - fourth ventricle. Reproduced with permission from Jen JC, Chan WM, Bosley TM, et al., “Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis,” Science. Jun 4 2004;304(5676):1509–1513. 368×235mm (72×72 DPI).
Discussion
Over the past decade, an explosion of evidence has reinforced the neurogenic etiology of the CCDDs. The identification of specific gene mutations as the pathogenesis for these disorders has increased our knowledge of the importance of the normal protein products of these genes in the normal development of ocular cranial motor neurons and guidance of their axons to the appropriate end muscle targets. The combination of clinical findings, MR imaging, and gene product characterization has led to a greater understanding of the cascade of events that leads ultimately to the clinical presentation of these patients. This cascade starts as a gene mutation coding for an abnormal protein that leads to changes in the normal development of the nervous system that then results in structural abnormalities that culminate in anatomical dysfunction. The effort to gather this knowledge has spanned continents and has involved much collaboration among institutions.
Despite the advances in our understanding, much remains to be learned about these conditions. The variable expression pattern of patients with known mutations reveals the potential importance of additional genetic variation and environmental factors on the end phenotype. The high number of simplex CCDD patients with no identified gene mutation again highlights that environmental factors or somatic mutations may cause disruptions in development that result in a clinical presentation similar to that of underlying germline mutations. In many pedigrees there is little or no genetic data to point to the identity of the mutation. For these reasons, much work is still needed to obtain a better understanding of the environmental and genetic mechanisms that lead to dysinnervation in the CCDDs.
Acknowledgments
Funding: supported by R01 EY12498 and the Children’s Hospital Ophthalmology Foundation, Boston, MA. ECE is an investigator of the Howard Hughes Medical Institute.
References
- 1.Laughlin RC. Congenital fibrosis of the extraocular muscles; a report of six cases. American journal of ophthalmology. 1956 Mar;41(3):432–438. doi: 10.1016/0002-9394(56)91259-4. [DOI] [PubMed] [Google Scholar]
- 2.Hotchkiss MG, Miller NR, Clark AW, Green WR. Bilateral Duane’s retraction syndrome. A clinical-pathologic case report. Archives of ophthalmology. 1980 May;98(5):870–874. doi: 10.1001/archopht.1980.01020030864013. [DOI] [PubMed] [Google Scholar]
- 3.Miller NR, Kiel SM, Green WR, Clark AW. Unilateral Duane’s retraction syndrome (Type 1) Archives of ophthalmology. 1982 Sep;100(9):1468–1472. doi: 10.1001/archopht.1982.01030040446016. [DOI] [PubMed] [Google Scholar]
- 4.Duane A. Congenital Deficiency of Abduction associated with impairment of adduction, retraction movements, contraction of the palpebral fissure and oblique movements of the eye. Archives of ophthalmology. 1905;34:133–150. doi: 10.1001/archopht.1996.01100140455017. [DOI] [PubMed] [Google Scholar]
- 5.Appukuttan B, Gillanders E, Juo SH, et al. Localization of a gene for Duane retraction syndrome to chromosome 2q31. American journal of human genetics. 1999 Dec;65(6):1639–1646. doi: 10.1086/302656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sevel D, Kassar BS. Bilateral Duane syndrome. Occurrence in three successive generations. Archives of ophthalmology. 1974 Jun;91(6):492–494. doi: 10.1001/archopht.1974.03900060506017. [DOI] [PubMed] [Google Scholar]
- 7.Chung M, Stout JT, Borchert MS. Clinical diversity of hereditary Duane’s retraction syndrome. Ophthalmology. 2000 Mar;107(3):500–503. doi: 10.1016/s0161-6420(99)00090-1. [DOI] [PubMed] [Google Scholar]
- 8.Evans JC, Frayling TM, Ellard S, Gutowski NJ. Confirmation of linkage of Duane’s syndrome and refinement of the disease locus to an 8.8-cM interval on chromosome 2q31. Human genetics. 2000 Jun;106(6):636–638. doi: 10.1007/s004390000311. [DOI] [PubMed] [Google Scholar]
- 9.Engle EC, Andrews C, Law K, Demer JL. Two pedigrees segregating Duane’s retraction syndrome as a dominant trait map to the DURS2 genetic locus. Investigative ophthalmology & visual science. 2007 Jan;48(1):189–193. doi: 10.1167/iovs.06-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan WM, Miyake N, Zhu-Tam L, Andrews C, Engle EC. Two novel CHN1 mutations in 2 families with Duane retraction syndrome. Archives of ophthalmology. 2011 May;129(5):649–652. doi: 10.1001/archophthalmol.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyake N, Chilton J, Psatha M, et al. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane’s retraction syndrome. Science. 2008 Aug 8;321(5890):839–843. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyake N, Demer JL, Shaaban S, et al. Expansion of the CHN1 strabismus phenotype. Investigative ophthalmology & visual science. 2011 Aug;52(9):6321–6328. doi: 10.1167/iovs.11-7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demer JL, Clark RA, Lim KH, Engle EC. Magnetic resonance imaging evidence for widespread orbital dysinnervation in dominant Duane’s retraction syndrome linked to the DURS2 locus. Investigative ophthalmology & visual science. 2007 Jan;48(1):194–202. doi: 10.1167/iovs.06-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murillo-Correa CE, Kon-Jara V, Engle EC, Zenteno JC. Clinical features associated with an I126M alpha2-chimaerin mutation in a family with autosomal-dominant Duane retraction syndrome. Journal of AAPOS: the official publication of the American Association for Pediatric Ophthalmology and Strabismus / American Association for Pediatric Ophthalmology and Strabismus. 2009 Jun;13(3):245–248. doi: 10.1016/j.jaapos.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyake N, Andrews C, Fan W, He W, Chan WM, Engle EC. CHN1 mutations are not a common cause of sporadic Duane’s retraction syndrome. American journal of medical genetics. Part A. 2010 Jan;152A(1):215–217. doi: 10.1002/ajmg.a.33168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volk AE, Fricke J, Strobl J, Kolling G, Kubisch C, Neugebauer A. Analysis of the CHN1 gene in patients with various types of congenital ocular motility disorders. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2010 Sep;248(9):1351–1357. doi: 10.1007/s00417-010-1417-7. [DOI] [PubMed] [Google Scholar]
- 17.Smith SB, Traboulsi EI. Duane syndrome in the setting of chromosomal duplications. American journal of ophthalmology. 2010 Dec;150(6):932–938. doi: 10.1016/j.ajo.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 18.Terhal P, Rosler B, Kohlhase J. A family with features overlapping Okihiro syndrome, hemifacial microsomia and isolated Duane anomaly caused by a novel SALL4 mutation. American journal of medical genetics. Part A. 2006 Feb 1;140(3):222–226. doi: 10.1002/ajmg.a.31060. [DOI] [PubMed] [Google Scholar]
- 19.Kohlhase J, Schubert L, Liebers M, et al. Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. Journal of medical genetics. 2003 Jul;40(7):473–478. doi: 10.1136/jmg.40.7.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohlhase J, Wischermann A, Reichenbach H, Froster U, Engel W. Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nature genetics. 1998 Jan;18(1):81–83. doi: 10.1038/ng0198-81. [DOI] [PubMed] [Google Scholar]
- 21.Barry JS, Reddy MA. The association of an epibulbar dermoid and Duane syndrome in a patient with a SALL1 mutation (Townes-Brocks Syndrome) Ophthalmic genetics. 2008 Dec;29(4):177–180. doi: 10.1080/13816810802354224. [DOI] [PubMed] [Google Scholar]
- 22.Pizzuti A, Calabrese G, Bozzali M, et al. A peptidase gene in chromosome 8q is disrupted by a balanced translocation in a duane syndrome patient. Investigative ophthalmology & visual science. 2002 Dec;43(12):3609–3612. [PubMed] [Google Scholar]
- 23.Vincent C, Kalatzis V, Compain S, et al. A proposed new contiguous gene syndrome on 8q consists of Branchio-Oto-Renal (BOR) syndrome, Duane syndrome, a dominant form of hydrocephalus and trapeze aplasia; implications for the mapping of the BOR gene. Human molecular genetics. 1994 Oct;3(10):1859–1866. doi: 10.1093/hmg/3.10.1859. [DOI] [PubMed] [Google Scholar]
- 24.Calabrese G, Stuppia L, Morizio E, et al. Detection of an insertion deletion of region 8q13-q21.2 in a patient with Duane syndrome: implications for mapping and cloning a Duane gene. European journal of human genetics: EJHG. 1998 May-Jun;6(3):187–193. doi: 10.1038/sj.ejhg.5200173. [DOI] [PubMed] [Google Scholar]
- 25.Rickard S, Parker M, van’t Hoff W, et al. Oto-facio-cervical (OFC) syndrome is a contiguous gene deletion syndrome involving EYA1: molecular analysis confirms allelism with BOR syndrome and further narrows the Duane syndrome critical region to 1 cM. Human genetics. 2001 May;108(5):398–403. doi: 10.1007/s004390100495. [DOI] [PubMed] [Google Scholar]
- 26.Lehman AM, Friedman JM, Chai D, et al. A characteristic syndrome associated with microduplication of 8q12, inclusive of CHD7. European journal of medical genetics. 2009 Nov-Dec;52(6):436–439. doi: 10.1016/j.ejmg.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Amouroux C, Vincent M, Blanchet P, et al. Duplication 8q12: confirmation of a novel recognizable phenotype with duane retraction syndrome and developmental delay. European journal of human genetics: EJHG. 2012 May;20(5):580–583. doi: 10.1038/ejhg.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo H, Xie L, Wang SZ, et al. Duplication of 8q12 encompassing CHD7 is associated with a distinct phenotype but without duane anomaly. European journal of medical genetics. 2012 Nov;55(11):646–649. doi: 10.1016/j.ejmg.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Wettke-Schafer R, Kantner G. X-linked dominant inherited diseases with lethality in hemizygous males. Human genetics. 1983;64(1):1–23. doi: 10.1007/BF00289472. [DOI] [PubMed] [Google Scholar]
- 30.Abu-Amero KK, Kondkar AA, Alorainy IA, et al. Xq26.3 Microdeletion in a Male with Wildervanck Syndrome. Ophthalmic genetics. 2013 Feb 1;:1–7. doi: 10.3109/13816810.2013.766218. [DOI] [PubMed] [Google Scholar]
- 31.Miller MT. Association of Duane retraction syndrome with craniofacial malformations. Journal of craniofacial genetics and developmental biology. Supplement. 1985;1:273–282. [PubMed] [Google Scholar]
- 32.Marshman WE, Schalit G, Jones RB, Lee JP, Matthews TD, McCabe S. Congenital anomalies in patients with Duane retraction syndrome and their relatives. Journal of AAPOS: the official publication of the American Association for Pediatric Ophthalmology and Strabismus / American Association for Pediatric Ophthalmology and Strabismus. 2000 Apr;4(2):106–109. doi: 10.1067/mpa.2000.103439. [DOI] [PubMed] [Google Scholar]
- 33.Kothari M, Manurung F, Mithiya B. Simultaneous occurrence of duane retraction syndrome with marfan syndrome. Case reports in ophthalmological medicine. 2011;2011:784259. doi: 10.1155/2011/784259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tischfield MA, Bosley TM, Salih MA, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nature genetics. 2005 Oct;37(10):1035–1037. doi: 10.1038/ng1636. [DOI] [PubMed] [Google Scholar]
- 35.Bosley TM, Salih MA, Alorainy IA, et al. Clinical characterization of the HOXA1 syndrome BSAS variant. Neurology. 2007 Sep 18;69(12):1245–1253. doi: 10.1212/01.wnl.0000276947.59704.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holve S, Friedman B, Hoyme HE, et al. Athabascan brainstem dysgenesis syndrome. American journal of medical genetics. Part A. 2003 Jul 15;120A(2):169–173. doi: 10.1002/ajmg.a.20087. [DOI] [PubMed] [Google Scholar]
- 37.Bosley TM, Alorainy IA, Salih MA, et al. The clinical spectrum of homozygous HOXA1 mutations. American journal of medical genetics. Part A. 2008 May 15;146A(10):1235–1240. doi: 10.1002/ajmg.a.32262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tischfield MA, Chan WM, Grunert JF, Andrews C, Engle EC. HOXA1 mutations are not a common cause of Duane anomaly. American journal of medical genetics. Part A. 2006 Apr 15;140(8):900–902. doi: 10.1002/ajmg.a.31167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chew S, Balasubramanian R, Chan WM, et al. A novel syndrome caused by the E410K amino acid substitution in the neuronal beta-tubulin isotype 3. Brain: a journal of neurology. 2013 Feb;136(Pt 2):522–535. doi: 10.1093/brain/aws345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller G. Neurological disorders. The mystery of the missing smile. Science. 2007 May 11;316(5826):826–827. doi: 10.1126/science.316.5826.826. [DOI] [PubMed] [Google Scholar]
- 41.Pastuszak AL, Schuler L, Speck-Martins CE, et al. Use of misoprostol during pregnancy and Mobius’ syndrome in infants. N Engl J Med. 1998 Jun 25;338(26):1881–1885. doi: 10.1056/NEJM199806253382604. [DOI] [PubMed] [Google Scholar]
- 42.Carta A, Mora P, Neri A, Favilla S, Sadun AA. Ophthalmologic and systemic features in mobius syndrome an italian case series. Ophthalmology. 2011 Aug;118(8):1518–1523. doi: 10.1016/j.ophtha.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 43.Webb BD, Shaaban S, Gaspar H, et al. HOXB1 founder mutation in humans recapitulates the phenotype of Hoxb1−/− mice. American journal of human genetics. 2012 Jul 13;91(1):171–179. doi: 10.1016/j.ajhg.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demer JL, Clark RA, Engle EC. Magnetic resonance imaging evidence for widespread orbital dysinnervation in congenital fibrosis of extraocular muscles due to mutations in KIF21A. Investigative ophthalmology & visual science. 2005 Feb;46(2):530–539. doi: 10.1167/iovs.04-1125. [DOI] [PubMed] [Google Scholar]
- 45.Engle EC, Goumnerov BC, McKeown CA, et al. Oculomotor nerve and muscle abnormalities in congenital fibrosis of the extraocular muscles. Annals of neurology. 1997 Mar;41(3):314–325. doi: 10.1002/ana.410410306. [DOI] [PubMed] [Google Scholar]
- 46.Yamada K, Andrews C, Chan WM, et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1) Nature genetics. 2003 Dec;35(4):318–321. doi: 10.1038/ng1261. [DOI] [PubMed] [Google Scholar]
- 47.Yang X, Yamada K, Katz B, et al. KIF21A mutations in two Chinese families with congenital fibrosis of the extraocular muscles (CFEOM) Molecular vision. 2010;16:2062–2070. [PMC free article] [PubMed] [Google Scholar]
- 48.Tischfield MA, Baris HN, Wu C, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010 Jan 8;140(1):74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan AO, Shinwari J, Omar A, et al. Lack of KIF21A mutations in congenital fibrosis of the extraocular muscles type I patients from consanguineous Saudi Arabian families. Molecular vision. 2011;17:218–224. [PMC free article] [PubMed] [Google Scholar]
- 50.Sener EC, Lee BA, Turgut B, Akarsu AN, Engle EC. A clinically variant fibrosis syndrome in a Turkish family maps to the CFEOM1 locus on chromosome 12. Archives of ophthalmology. 2000 Aug;118(8):1090–1097. doi: 10.1001/archopht.118.8.1090. [DOI] [PubMed] [Google Scholar]
- 51.Wang SM, Zwaan J, Mullaney PB, et al. Congenital fibrosis of the extraocular muscles type 2, an inherited exotropic strabismus fixus, maps to distal 11q13. American journal of human genetics. 1998 Aug;63(2):517–525. doi: 10.1086/301980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakano M, Yamada K, Fain J, et al. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nature genetics. 2001 Nov;29(3):315–320. doi: 10.1038/ng744. [DOI] [PubMed] [Google Scholar]
- 53.Yamada K, Chan WM, Andrews C, et al. Identification of KIF21A mutations as a rare cause of congenital fibrosis of the extraocular muscles type 3 (CFEOM3) Investigative ophthalmology & visual science. 2004 Jul;45(7):2218–2223. doi: 10.1167/iovs.03-1413. [DOI] [PubMed] [Google Scholar]
- 54.Engle EC, McIntosh N, Yamada K, et al. CFEOM1, the classic familial form of congenital fibrosis of the extraocular muscles, is genetically heterogeneous but does not result from mutations in ARIX. BMC genetics. 2002;3:3. doi: 10.1186/1471-2156-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bosley TM, Oystreck DT, Robertson RL, al Awad A, Abu-Amero K, Engle EC. Neurological features of congenital fibrosis of the extraocular muscles type 2 with mutations in PHOX2A. Brain: a journal of neurology. 2006 Sep;129(Pt 9):2363–2374. doi: 10.1093/brain/awl161. [DOI] [PubMed] [Google Scholar]
- 56.Demer JL, Clark RA, Tischfield MA, Engle EC. Evidence of an asymmetrical endophenotype in congenital fibrosis of extraocular muscles type 3 resulting from TUBB3 mutations. Investigative ophthalmology & visual science. 2010 Sep;51(9):4600–4611. doi: 10.1167/iovs.10-5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cederquist GY, Luchniak A, Tischfield MA, et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Human molecular genetics. 2012 Dec 15;21(26):5484–5499. doi: 10.1093/hmg/dds393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu S, Zhao C, Zhao K, Li N, Larsson C. Novel and recurrent KIF21A mutations in congenital fibrosis of the extraocular muscles type 1 and 3. Archives of ophthalmology. 2008 Mar;126(3):388–394. doi: 10.1001/archopht.126.3.388. [DOI] [PubMed] [Google Scholar]
- 59.Lin LK, Chien YH, Wu JY, Wang AH, Chiang SC, Hwu WL. KIF21A gene c.2860C>T mutation in congenital fibrosis of extraocular muscles type 1 and 3. Molecular vision. 2005;11:245–248. [PubMed] [Google Scholar]
- 60.Aubourg P, Krahn M, Bernard R, et al. Assignment of a new congenital fibrosis of extraocular muscles type 3 (CFEOM3) locus, FEOM4, based on a balanced translocation t(2;13) (q37.3; q12.11) and identification of candidate genes. Journal of medical genetics. 2005 Mar;42(3):253–259. doi: 10.1136/jmg.2004.021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tukel T, Uzumcu A, Gezer A, et al. A new syndrome, congenital extraocular muscle fibrosis with ulnar hand anomalies, maps to chromosome 21qter. Journal of medical genetics. 2005 May;42(5):408–415. doi: 10.1136/jmg.2004.026138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Traboulsi EI. Congenital abnormalities of cranial nerve development: overview, molecular mechanisms, and further evidence of heterogeneity and complexity of syndromes with congenital limitation of eye movements. Transactions of the American Ophthalmological Society. 2004;102:373–389. [PMC free article] [PubMed] [Google Scholar]
- 63.Sharpe JA, Silversides JL, Blair RD. Familial paralysis of horizontal gaze. Associated with pendular nystagmus, progressive scoliosis, and facial contraction with myokymia. Neurology. 1975 Nov;25(11):1035–1040. doi: 10.1212/wnl.25.11.1035. [DOI] [PubMed] [Google Scholar]
- 64.Jen JC, Chan WM, Bosley TM, et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004 Jun 4;304(5676):1509–1513. doi: 10.1126/science.1096437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan WM, Traboulsi EI, Arthur B, Friedman N, Andrews C, Engle EC. Horizontal gaze palsy with progressive scoliosis can result from compound heterozygous mutations in ROBO3. Journal of medical genetics. 2006 Mar;43(3):e11. doi: 10.1136/jmg.2005.035436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Volk AE, Carter O, Fricke J, et al. Horizontal gaze palsy with progressive scoliosis: three novel ROBO3 mutations and descriptions of the phenotypes of four patients. Molecular vision. 2011;17:1978–1986. [PMC free article] [PubMed] [Google Scholar]