

Abstract
Background/Aims
Acute phase C-reactive protein (CRP), elevated in obesity and inflammation, is a major binding protein for leptin. It is thought that CRP contributes to leptin resistance by preventing leptin from crossing the blood-brain barrier (BBB). Here we determined how CRP interacts with the BBB and whether it deters leptin from reaching CNS targets.
Methods
BBB permeability, compartmental distribution, tracer stability, and expression of tight junction protein and inflammatory marker were determined.
Results
CRP was stable in blood, but did not permeate the BBB in trace amounts. However, it increased paracellular permeability at a higher dose. Agouti viable (Avy) mice with adult-onset obesity show higher CRP entry into the brain. CRP did not permeate hCMEC/D3 cells nor change zona occludin-1 or cyclooxygenase-2 expression. An intermediate dose of CRP had no effect on leptin transport across the BBB after co-treatment. Thus, acute interactions between CRP and leptin at the BBB level were negligible and did not explain the leptin resistance seen in obesity.
Conclusions
The interactions of CRP and the BBB are a two-phase process, with increased paracellular permeability at a high dose that enables its entry into the CNS and serves to induce reactive gliosis and impair CNS function.
Keywords: CRP, Blood-brain barrier, Permeability, Tight junction, Leptin, Adipokines, Inflammation
Introduction
The primary goal of this study was to determine whether and how CRP interacts with the blood-brain barrier (BBB) and whether it contributes to the phenomenon of leptin resistance at the BBB in obesity. C-reactive protein (CRP) is an acute phase protein in the pentraxin family mainly produced in liver in response to proinflammatory cytokines such as interleukin-6 and interleukin-1β. Obesity is associated with elevation of CRP and leptin in blood [1, 2]. Obesity is also known to induce neuroinflammation in the form of reactive gliosis, particularly in the hypothalamus [3, 4]. In primary culture, CRP induces reactivity of microglia and astrocytes, and exacerbates gliosis after scratch injury [5]. As one of the major proteins bound to leptin as shown by affinity chromatography and verified by mass spectrometry, human CRP inhibits leptin binding to ObRb that is overexpressed in HEK293 cells, and it attenuates pSTAT3 signaling [1]. When infused into ob/ob mice, CRP blocks the effects of leptin in suppressing food intake, weight gain, and liver triglyceride level. Leptin is rendered ineffective in hCRP transgenic mice [1]. This leads to an attractive hypothesis that CRP binding impedes leptin entry into the brain, thus contributing to leptin resistance at the level of BBB transport [1]. It was felt that “it is not yet clear if the inhibitory effects of leptin are due to the interaction of CRP with leptin within the CNS or to the impediment of the transport of leptin in the CNS” [6].
Initially discovered in 1930 [7], CRP is a 21 kD protein structurally similar to serum amyloid P component. It forms a cyclic pentamer of 5 identical nonglycosylated protein subunits. Healthy human adults have a CRP concentration of approximately 800 ng/ml, seldom going above 10 μg/ml. After inflammation or injury, however, CRP concentrations peak 24 – 48 h later and can reach 300 μg/ml [8]. The concentration of CRP correlates with cardiovascular risk and endothelial dysfunction [9]. CRP may also contribute to the loss of brain volume. A correlation between CRP and a loss of brain volume is reported in the Framingham Heart Study [10] and the 3C-Dijon Study [11]. The SEARCH Health Study did not detect an association of CRP level with gray matter, but it did identify a strong negative correlation with cognitive function [12]. High CRP is also associated with a lower performance in visual tasks in subjects from Framingham Heart Study [13], and with lacunar infarcts and white matter ischemia in subjects in the Rotterdam Scan study [14]. All these reports suggest a detrimental role of CRP on CNS function.
Obesity is associated with elevated blood concentrations of prothrombotic - proinflammatory factors and markers of endothelial dysfunction such as CRP, fibrinogen, von Willebrand factor, and homocysteine. There are potential interactions between CRP and the BBB endothelia. In endothelial cells of peripheral origin, CRP binds to FCγRIIB and blocks the effect of HDL, E2, VEGF, and insulin to induce endothelial nitric oxide synthase (eNOS) in caveolae [15]. The inhibition of eNOS by CRP may thus contribute to a higher vascular resistance and therefore be involved in hypertension. In the CNS, eNOS dysfunction is implicated in neurodegenerative diseases. If CRP permeates the endothelial cells composing the BBB to activate glial cells directly, it could serve as a major culprit in the neuroinflammation seen in obesity.
Suppressing feeding behavior, leptin is an essential anorexigenic adipokine that crosses the BBB by receptor-mediated transport [16–18]. It may activate inert transport systems such as that for urocortin [19, 20]. We examined whether a similar situation occurs with CRP. Leptin cellular signaling is subject to potentiation by other anorexigenic signals [21, 22], and its effects can be antagonized by CRP as well as others. Adiponectin is also produced by adipocytes and acts in concert with leptin, but it does not cross the BBB itself [23]. Therefore, we also compared the effects of CRP and fibrinogen on leptin transport with that of adiponectin. The studies of CRP permeation across the BBB and its interactions with leptin at the BBB level are important not only because the concentrations of circulating CRP and leptin are highly upregulated in human obesity and inflammation, but also because both are crucial modulators for disease progression and secondary CNS degeneration.
Materials and Methods
Mice and reagents
Adult C57 from the Jackson Laboratory (Bar Harbor, ME) were used for most BBB permeability assays. The agouti viable yellow (Avy) mouse of C57 background originated from the Jackson Laboratory and has been maintained in our breeding colony. Only male mice were used to avoid confounding effects of the estrous cycle in females. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC). Carrier-free recombinant mouse leptin and CRP were purchased from R & D Systems (Minneapolis, MN). Carrier-free mouse fibrinogen and albumin were obtained from Sigma (St Louis, MO). Mouse adiponectin (Acrp30) was obtained from ProSpec (Rehovot, Israel). 125I (Perkin Elmer, Waltham, MA) was used to radioactively label leptin and CRP, whereas 131I was used to label albumin by the chloramine-T method. 131I-albumin is a marker for the vascular space and serves as a negative control for BBB permeability. The radioactively labeled tracers (radiotracers) were purified by elution on Sephadex G-10 columns. The specific activity of 125I-leptin was 99 Ci/g. The specific activity of 125I-CRP used for transport assays in C57 mice was 33 Ci/g. For the rest of the experiments, specific activity of 125I-CRP from another batch was 48 Ci/g.
Multiple-time regression analysis to determine BBB permeability
Group designs are specified in detail in the Results section. In any study group, 7–10 mice were used, and each mouse represents a time point between 1 and 20 min. The duration was chosen based on CRP stability data shown in subsequent sections of the study, and the known period during which leptin remains stable after intravenous delivery [24]. At time 0, an intravenous bolus of 100 μl of 125I-leptin or 125I-CRP and 131I-albumin (about 1 μCi each) was given through the left jugular vein of an anesthetized mouse. At the designated time point, a mouse was decapitated immediately after collection of arterial blood from the right common carotid artery. Brain was collected to obtain serum. The radioactivity of 125I and 131I in brain and 50 μl of serum were determined with a dual-channel program on a Wallac 1470 Wizzard γ-counter. The disappearance dynamics of 125I-leptin or 125I-CRP were calculated from the serum radioactivity over time. The influx rate (Ki) and volume of distribution (Vi) of the radiotracers were determined from the linear regression of brain/serum ratio of radioactivity over exposure time, a theoretical steady state value [24]. Since the level of 131I-albumin remained constant in blood, its influx rate and volume of distribution were determined by the brain/serum ratio over time.
Capillary depletion
To determine whether the radioactivity present in brain entered parenchymal tissue or remained in the vasculature, cerebral cortex was dissected and homogenized in capillary buffer, mixed thoroughly with 26% dextran (Sigma), and centrifuged at 5400 g for 30 min at 4 °C, as described previously [25, 26]. The pellet represents the capillary fraction whereas the supernatant represents brain parenchyma. The parenchymal uptake over time was calculated after subtraction of the vascular space reflected by 131I-albumin.
Stability assays by reversed phase high performance liquid chromatography (HPLC)
To ensure that the radioactivity in brain remained on the intact material injected, blood and brain were collected 0, 20, and 30 min after intravenous injection of 125I-CRP. Serum was obtained after centrifugation of blood at 4 °C. Brain was homogenized in the presence of complete protease inhibitor cocktail (Halt™ Protease Inhibitor Cocktail Kit, Thermo Scientific, Rockford, IL), and the supernatant was used for HPLC analyses with a Vydac C4 column. The 0 time point represents the processing control, in which 125I-CRP was added ex-vivo before homogenization of the brain tissue. The mobile phase for HPLC was acetonitrile containing 0.1% trifluoroacetic acid. A linear program with acetonitrile increasing from 10% to 90% over 40 min with a flow rate of 1 ml/min was used. The eluant was collected at a rate of 1 ml/min for each fraction.
In-vitro BBB permeability assays
To test the hypothesis that leptin modulates the transport of CRP across the BBB, hCMEC/D3 cells were used as an in-vitro BBB model. The cells at passage 36 were seeded onto Transwell 12-well plates coated with rat tail collagen. The seeding density was about 150%. The cells were cultured in EGM-2 medium containing EBM-2 supplemented with EGM™-2 SingleQuots® (Lonza, Walkersville, MD) and permeability assays with 125I-CRP and 131I-albumin were performed 3 days later. Five groups of cells were studied (n = 3 wells/group): (1) radioactively labeled tracers only, (2) radioactively labeled tracers along with 200-fold molar excess of leptin (3.8 μg/ml), (3) radioactively labeled tracers in cells pretreated with leptin (20 ng/ml) for 8 h, with leptin removed and washed with medium before the transport assays, (4) cells pretreated with leptin at 20 ng/ml for 8 h, followed by co-treatment with 3.8 μg/ml (200-fold excess) of leptin during the transport assays at the 9th h, and (5) additional wells without cells, functioning as a negative control. The upper (donor) chamber of the Transwell system contained 0.5 ml of EGM-2 medium, and the bottom (acceptor) chamber of the Transwell contained 1.5 ml of EGM-2 medium. At time 0, 125I-CRP (2,000,000 cpm/ml, 0.9 μCi/ml) and 131I-Alb (1,000,000 cpm/ml, 0.45 μCi/ml) were placed on the donor chamber of the Transwell maintained at 37 °C with a constant supply of 5 % CO2. At 5, 10, 20, 30, 40, 50, and 60 min, 10 μl of medium from the basolateral side (acceptor chamber) was sampled. The basolateral/apical ratio or radioactivity was determined, and the permeability coefficient was determined as described previously [27].
Western blotting (WB) to determine the effects of hCRP on hCMEC/D3 functions
Because of the human origin of the hCMEC/D3 cell line, human (h) CRP was used to treat the cells for 0, 3, 12, or 24 h after being grown to confluency on 6-well plates (n = 3/group). The dose of 20 μg/ml was chosen because it imitates conditions of mild inflammation [8]. The cells were collected with protein lysis buffer containing complete protease inhibitor cocktail, and subjected to WB for the tight junction protein zona occludin-1 (ZO-1), the inflammatory marker cyclooxygenase (COX)-2, and the reference gene β-actin. Twenty μg of protein was separated on 8.5% SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with Pierce Protein-Free TBS Blocking Buffer (Thermo Scientific, Rockford, IL) with 0.1 % Tween-20 for 1 h. The membranes were incubated overnight with polyclonal rabbit anti ZO-1 (1:700, cat# 40-2300, Invitrogen, Carlsbad, CA), polyclonal goat anti COX-2 (M-19, 1:1000, cat# sc-1747, Santa Cruz Biotechnology, Santa Cruz, CA), or monoclonal mouse anti β-actin (1:8000, cat# A5441, Sigma, St. Louis, MO). After thorough washes, the membranes were incubated with HRP-conjugated secondary antibodies (1:5000 dilution) at room temperature for 1 h. The signals were developed with Pierce ECL Western Blotting Substrate (cat#32209, Thermo Scientific). Densitometry analysis was performed by use of NIH Image J software.
Statistical analyses
Linear regression analysis was performed to determine the influx rate and volume of distribution of radiotracers in the brain and in-vitro BBB permeability assay. The difference between the experimental and control groups and among groups was determined by the least squares method with the Prism GraphPad program. One way ANOVA was used to analyze the permeability coefficient and WB results.
Results
125I-CRP does not cross the BBB in normal mice; leptin does not change this, but excess CRP increases paracellular permeability
Three groups of 3-month old male C57 mice were studied (n = 8–13/group): radiotracer (125I-CRP and 131I-albumin) only; co-administration of unlabeled CRP (5 μg/mouse, about 167-fold excess of the 0.03 μg injected); or co-administration of unlabeled leptin (5 μg/mouse). Within 20 min after intravenous bolus administration, 125I-CRP did not have a significant influx from blood to the brain. The influx rate was not significantly different from that of 131I-albumin. Co-administration of unlabeled leptin did not induce a significant increase of the influx. Co-administration of unlabeled CRP seemed to initially depress the volume of distribution of 125I-CRP in the first 10 min, but later increase it. This suggests that the excess unlabeled CRP initially competed with the 125I-CRP to reach the BBB, but later induced an acute increase of BBB permeability. Unlabeled CRP also seemed to cause an initial reduction of the volume of distribution of 131I-albumin, suggesting that CRP also binds to albumin. At later times, CRP also increased the influx rate of 131I-albumin. This indicates increased paracellular permeability resulting from partial disruption of the BBB (Fig. 1A).
Fig. 1.
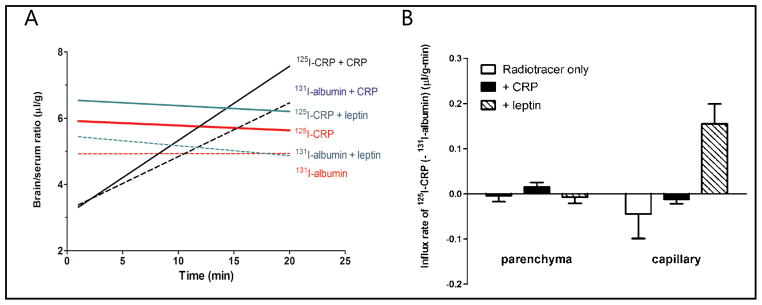
Permeation of 125I-CRP from blood to brain in C57 mice, with 131I-albumin as a reference substance. (A) Multiple-time regression analysis showing the overall influx rate. Trace amounts of CRP did not cross the BBB, shown by a slightly sloping regression line almost parallel to that of albumin. Co-treatment of leptin did not affect the influx rate (slope of the regression line) and the increase of the volume of distribution (y-axis values) was not significant. Co-treatment with CRP initially seemed to depress the volume of distribution of 125I-CRP in the first 10 min, but overall induced an influx of 125I-CRP (Ki = 0.22 ± 0.13 μl/g-min) and 131I-albumin (Ki = 0.16 ± 0.11 μl/g-min). (B) Capillary depletion study showing the distribution of radioactively labeled tracers in cerebral cortical parenchyma and vasculature. The influx rate of 125I-CRP to brain parenchyma, normalized by subtracting the 131I-albumin space, was only positive in the group treated with excess unlabeled CRP. This contrasts with an increased vascular accumulation in the leptin treated group.
Cerebral cortex devoid of circumventricular organs was subjected to capillary depletion to separate microvessel and parenchyma fractions. As shown in Figure 1B, the influx rate of trace amounts of 125I-CRP from blood to brain parenchyma was not significantly different from zero, and was no higher than that of 131I-albumin representing the vascular space. Excess unlabeled CRP increased 125I-CRP entry into brain parenchyma, but statistical significance was not reached in comparison with the controls. The large variability is commonly seen in a situation of minimal permeation. Leptin co-treatment only increased the vascular space without greater parenchymal uptake of 125I-CRP, consistent with a direct binding of leptin with CRP [1] that may further increase physical hindrance.
CRP permeation in the obese agouti viable yellow (Avy) mice
Two groups of 6 month old mice were studied (n = 8 – 12 mice/group): Avy and C57 littermate controls. There was no significant influx of 125I-CRP in C57 mice (Ki = 0.10 ± 0.11 μl/g-min). There was a significant influx of 125I-CRP in the Avy group (Ki = 0.36 ± 0.07 μl/g-min). However, the slopes of these two lines were not significantly different from each other [F(1,14) = 2.81, p = 0.16]. Figure 2A shows that the influx rates of 125I-CRP remained higher than the co-administered albumin, but there was no significantly different change of either substance between the C57 control and obese Avy mice. Similar to that seen in Figure 1, the low permeability at baseline was associated with large standard errors, making it difficult to derive statistically significant changes from multiple-time regression analysis. The higher influx of 125I-CRP in the Avy mice was associated with a higher serum half-life, increased to 6.3 min from the control value of 1.8 min (Fig. 2B). This is consistent with a higher level of endogenous CRP that may delay the degradation of 125I-CRP simply by competition.
Fig. 2.
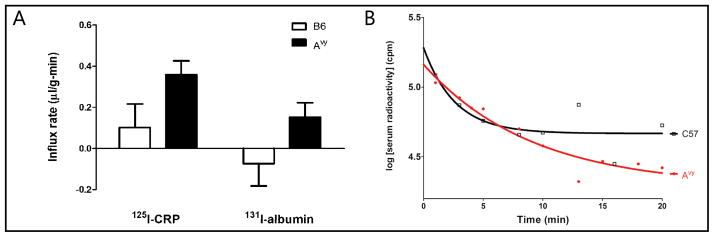
Effects of Avy mutation on the interactions of CRP with the BBB. (A) The obese Avy mice had a positive influx of CRP and albumin from blood to brain, indicating higher paracellular permeability in comparison with the age-matched control C57 mice studied at the same time. (B) The half-life of 125I-CRP in blood was higher in the Avy mice than in the C57 mice.
125I-CRP is stable in the circulation and brain homogenate
In the processing control in which blood and brain were collected into 125I-CRP containing tubes or homogenizers ex-vivo, 125I-CRP mainly eluted at the same retention time as the stock solution, indicating minimal ex-vivo degradation during tissue processing. In sera of C57 and Avy mice, 125I-CRP remained stable at both 20 and 30 min, as shown by HPLC (Fig. 3A). In the brain, there was a small percent degradation in both C57 and Avy mice at 20 min, shown by the emergence of a small peak at retention times of 14–17 min, as well as dissociation of free 125I that eluted at a retention time of 5 min (Fig. 3B). Nonetheless, even the processing control of the brain homogenate sample showed a similar pattern, indicating that such degradation probably derived from ex-vivo metabolism during tissue processing. By 30 min, only about 80% of the radioactivity in the supernatant of the homogenate remained intact 125I-CRP.
Fig. 3.

Stability of 125I-CRP in blood (A) and brain compartment (B). In the 20 and 30 min samples, the elution profile did not differ from that of stock solution or ex-vivo processing control, indicating that 125I-CRP remained intact during the study period.
125I-CRP does not have meaningful transcytosis across the hCMEC/D3 cell monolayer, and leptin has no modulatory effect
To determine whether CRP transcytoses hCMEC/D3 cells of human BBB origin, and whether leptin modulates CRP permeability in-vitro, the following groups of Transwells were studied (n = 3/group): radiotracer only (125I-CRP + 131I-albumin), 200-fold excess leptin (3.8 μg/ml) co-treatment for 1 h, leptin pretreatment (20 ng/ml) for 8 h, and leptin pre- (20 ng/ml) and co-treatment (3.8 μg/ml) for a total of 9 h. The flux rate (Fig. 4A) and permeability coefficient (Fig. 4B) of 125I-CRP were even lower than those of 131I-albumin, indicating a lack of meaningful transcytosis. The lower permeability of CRP is probably because the 21 kD CRP forms a cyclic pentamer that is larger than the globular albumin. Neither co-treatment nor pretreatment of leptin modulated the low permeation of 125I-CRP from apical to basolateral chambers. Thus, although leptin binds to CRP, it did not serve as a carrier (or Trojan horse) to allow CRP to use its transport system. These in-vitro results are consistent with the results in C57 mice (Fig. 1), in which leptin co-treatment did not affect the minimal permeation of 125I-CRP. In all groups with hCMEC/D3 cells, the permeability was 6-fold lower than in the control Transwell without cells.
Fig. 4.

125I-CRP had minimal permeation across the hCMEC/D3 cell monolayer. (A) The influx rate was unchanged by leptin pre- or co-treatment. (B) The permeability coefficient (Papp) of 125I-CRP was lower than that of 131I-albumin.
Effects of CRP on tight junction protein ZO-1 and inflammatory marker COX-2 in hCMEC/D3 cells
hCRP at 20 μg/ml in cell culture medium did not change ZO-1 expression at 3, 12, or 24 h in comparison with the 0 time control (n = 3/group). Similarly, the expression of COX-2 was unchanged at these time intervals (Fig. 5).
Fig. 5.

Lack of significant effect of CRP (20 μg/ml, human origin) on protein expression of ZO-1 (molecular weight: 225 kD) and COX-2 (molecular weight: 70 kD) at 3, 12, and 24 h. The bottom panels show densitometry results after normalization to the reference gene β-actin, and division by the 0 h values.
Effect of CRP co-administration on leptin transport, in comparison with fibrinogen and adiponectin
Leptin had a significant influx from blood to cerebral cortex, 6-fold higher than the co-administered albumin. This was not modulated by co-treatment with CRP (5 μg/mouse), fibrinogen (10 μg/mouse), or adiponectin (5 μg/mouse), these doses being about 300-fold higher than the dose of the injected 125I-leptin (Fig. 6A). Since leptin also plays a significant role in hypothalamic and hippocampal functions, its permeability into these two regions was also measured. The permeability of leptin into either hypothalamus (Fig. 6B) or hippocampus (Fig. 6C) was not modulated by co-injection of CRP, fibrinogen, or adiponectin.
Fig. 6.

125I-leptin influx from blood to the cerebral cortex (A), hypothalamus (B), and hippocampus (C) was not acutely modulated by co-treatment with adiponectin (5 μg/ml), CRP (5 μg/ml), or fibrinogen (10 μg/ml). The shorter lines at the bottom of each graph represent 131I-albumin serving as a reference control for the vascular space and indicating low paracellular permeability.
Discussion
There are compelling indications to study the permeability of the BBB to CRP and its potential modulation of leptin transport. Not only is CRP a biomarker for inflammation and highly elevated in obesity, but it also binds to leptin and was proposed to confer leptin resistance by preventing its transport across the BBB. However, our studies showed a unique situation of a large protein impairing BBB function and increasing its own permeation. We found that CRP does not cross the BBB in trace amounts, but it increases paracellular permeability of the BBB when blood CRP concentrations exceed a threshold to impair BBB function. This threshold dose (about 2.5 μg/ml, as further discussed below), is easily reached in states of systemic inflammation, or even in obesity.
The studies show a dose-dependent effect of CRP. Acute administration of a trace amount of CRP (0.03 μg) by intravenous injection did not cause immediate changes of BBB permeability to albumin. With a plasma volume of 2 ml, the maximal estimated concentration of 125I-CRP would be 15 ng/ml. The concentration in the permeability study on hCMEC/D3 cells was about 19 ng/ml. At this level, there was no barrier disruption, though hCMEC/D3 cells do not form as tight an in-vitro BBB as do primary cerebral microvascular endothelial cells, and the barrier function is much less than occurs in the CNS in-vivo [28]. Nonetheless, tight junction proteins show paracellular distribution in hCMEC/D3 cells, and the Transwell model of hCMEC/D3 has been adequate for the study of transport in many reports [27–29]. The paracellular permeability of 131I-albumin was even higher than that of 125I-CRP, and its permeability coefficient was comparable to that of other studies without a barrier disrupting agent [27]. Thus, the lower permeability of CRP than albumin probably results from the formation of a cyclic CRP pentamer with a molecular weight of 105 kD, higher than that of albumin (about 60 kD). The size differentiation of permeability indicates the presence of a functioning barrier. With a dose of unlabeled CRP (5 μg/mouse, or 2.5 μg/ml of the initial blood concentration), there was an increase of paracellular permeation between 10–20 min for both 125I-CRP and 131I-albumin.
However, the effects of CRP appear to mainly affect tight junction function rather than protein expression. Even at a dose of 20 μg/ml in-vitro, there was no significant downregulation of ZO-1 expression between 3 – 24 h of treatment on hCMEC/D3 cells. Nonetheless, it has been shown that CRP activates surface Fc-γ receptors CD16/32 to facilitate the formation of reactive oxygen species in bovine brain endothelia, acts through myosin light chain kinase, and leads to disruption of tight junction complexes [30]. It is also known that CRP binds to FCγRIIB and protein phosphotase 2A in endothelia from the peripheral vasculature, dephosphorylates and inactivates eNOS, and thus modifies endothelial cell function for monocyte adhesion and endothelial cell migration [15]. Thus, CRP probably induces acute impairment of tight junction function by cytoskeletal rearrangement, rather than by changing the level of protein expression. However, the lack of induction of COX-2, a common indicator of cellular inflammation, suggests that the hCMEC/D3 cells used in our study may not have abundant surface FC-γ receptors, or that COX-2 is not a main pathway downstream to CRP signaling.
Lipopolysaccharide (LPS) can rapidly increase blood leptin concentration [31, 32]. As an acute phase protein, CRP might be a mediator of the effect of LPS on hyperleptinemia. Chronic inflammation in humans is associated with elevation of both CRP and leptin. An increased endogenous leptin concentration could mask upregulation of leptin transport across the BBB reflected by a decreased uptake of 125I-leptin resulting from partial saturation of the leptin transport system even at physiological concentrations [33]. However, the time course of the BBB transport study of 125I-leptin was short, and it is unlikely that co-administration of CRP induced a significant increase of endogenous leptin within this time. Thus, a leptin surge is unlikely to underlie the lack of visible increase of leptin transport after CRP treatment. Nonetheless, nitric oxide isoenzymes have been shown to modulate inflammatory effects and the specific transport system for insulin at the BBB. eNOS and iNOS mediate the LPS-enhanced transport of insulin, whereas nNOS opposes LPS-enhanced insulin transport [34]. Sustained elevation of CRP, though not studied under the current experimental design, may have profound modulatory effects on specific nutrient and hormone transport systems as well as modulating BBB permeability paracellularly.
Once within the CNS, CRP is able to induce reactive gliosis [5]. It might be partially responsible for the neuroinflammation seen in the hypothalamus of obese mice [3, 4] which have high circulating concentrations of both CRP and leptin. Leptin enters the CNS by receptor-mediated transport. However, though leptin binds to CRP, the presence of CRP did not provide acute modulation of leptin transport. Similarly, co-treatment with fibrinogen or adiponectin did not affect leptin influx from blood to its CNS target regions. Despite gender differences in metabolism and response to obesity [35], it is unlikely that the additional use of female mice would change the conclusions from this study.
In summary, although a trace amount CRP did not cross the BBB after intravenous bolus injection, excess CRP impaired BBB function and increased paracellular permeation. Reciprocal interactions between leptin and CRP did not occur at the level of the BBB, but they both can reach the CNS at high concentrations and exert profound neuroinflammatory effects.
Acknowledgments
Grant support was provided by NIH (DK54880 and DK92245 to AJK, and NS62291 to WP).
References
- 1.Chen K, Li F, Li J, Cai H, Strom S, Bisello A, Kelley DE, Friedman-Einat M, Skibinski GA, McCrory MA, Szalai AJ, Zhao AZ. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat Med. 2006;12:425–432. doi: 10.1038/nm1372. [DOI] [PubMed] [Google Scholar]
- 2.Dube MG, Torto R, Kalra SP. Increased leptin expression selectively in the hypothalamus suppresses inflammatory markers CRP and IL-6 in leptin-deficient diabetic obese mice. Peptides. 2008;29:593–598. doi: 10.1016/j.peptides.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan W, Hsuchou H, He Y, Sakharkar A, Cain C, Yu C, Kastin AJ. Astrocyte Leptin Receptor (ObR) and Leptin Transport in Adult-Onset Obese Mice. Endocrinology. 2008;149:2798–2806. doi: 10.1210/en.2007-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsuchou H, He Y, Kastin AJ, Tu H, Markadakis EN, Rogers RC, Fossier PB, Pan W. Obesity induces functional astrocytic leptin receptors in hypothalamus. Brain. 2009;132:889–902. doi: 10.1093/brain/awp029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsuchou H, Kastin AJ, Pan W. Blood-Borne Metabolic Factors in Obesity Exacerbate Injury-Induced Gliosis. J Mol Neurosci. 2012;47:267–277. doi: 10.1007/s12031-012-9734-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Li F, Zhao A. Inflammation and leptin. Drug Discov Today: Disease Mechanisms. 2006;3:387–393. [Google Scholar]
- 7.Tillett WS, Francis T. Serological reactions in pneumonia with a non-protein somatic fraction of pneumococcus. J Exp Med. 1930;52:561–571. doi: 10.1084/jem.52.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pepys MB. C-reactive protein fifty years on. Lancet. 1981;1:653–657. doi: 10.1016/s0140-6736(81)91565-8. [DOI] [PubMed] [Google Scholar]
- 9.Marnell L, Mold C, Du Clos TW. C-reactive protein: ligands, receptors and role in inflammation. Clin Immunol. 2005;117:104–111. doi: 10.1016/j.clim.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Jefferson AL, Massaro JM, Wolf PA, Seshadri S, Au R, Vasan RS, Larson MG, Meigs JB, Keaney JF, Jr, Lipinska I, Kathiresan S, Benjamin EJ, DeCarli C. Inflammatory biomarkers are associated with total brain volume: the Framingham Heart Study. Neurology. 2007;68:1032–1038. doi: 10.1212/01.wnl.0000257815.20548.df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satizabal CL, Zhu YC, Mazoyer B, Dufouil C, Tzourio C. Circulating IL-6 and CRP are associated with MRI findings in the elderly: The 3C-Dijon Study. Neurology. 2012;78:720–727. doi: 10.1212/WNL.0b013e318248e50f. [DOI] [PubMed] [Google Scholar]
- 12.Wersching H, Duning T, Lohmann H, Mohammadi S, Stehling C, Fobker M, Conty M, Minnerup J, Ringelstein EB, Berger K, Deppe M, Knecht S. Serum C-reactive protein is linked to cerebral microstructural integrity and cognitive function. Neurology. 2010;74:1022–1029. doi: 10.1212/WNL.0b013e3181d7b45b. [DOI] [PubMed] [Google Scholar]
- 13.Jefferson AL, Massaro JM, Beiser AS, Seshadri S, Larson MG, Wolf PA, Au R, Benjamin EJ. Inflammatory markers and neuropsychological functioning: the Framingham Heart Study. Neuroepidemiology. 2011;37:21–30. doi: 10.1159/000328864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Dijk EJ, Prins ND, Vermeer SE, Vrooman HA, Hofman A, Koudstaal PJ, Breteler MM. C-reactive protein and cerebral small-vessel disease: the Rotterdam Scan Study. Circulation. 2005;112:900–905. doi: 10.1161/CIRCULATIONAHA.104.506337. [DOI] [PubMed] [Google Scholar]
- 15.Mineo C, Shaul PW. Circulating cardiovascular disease risk factors and signaling in endothelial cell caveolae. Cardiovasc Res. 2006;70:31–41. doi: 10.1016/j.cardiores.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 16.Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides. 1996;17:305–311. doi: 10.1016/0196-9781(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 17.Tu H, Pan W, Feucht L, Kastin AJ. Convergent trafficking pattern of leptin after endocytosis mediated by ObRa - ObRd. J Cell Physiol. 2007;212:215–222. doi: 10.1002/jcp.21020. [DOI] [PubMed] [Google Scholar]
- 18.Tu H, Hsuchou H, Kastin AJ, Wu X, Pan W. Unique leptin trafficking by a tailless receptor. FASEB J. 2010;24:2281–2291. doi: 10.1096/fj.09-143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kastin AJ, Pan W, Akerstrom V, Hackler L, Wang CF, Kotz CM. Novel peptide-peptide cooperation may transform feeding behavior. Peptides. 2002;23:2189–2196. doi: 10.1016/s0196-9781(02)00247-4. [DOI] [PubMed] [Google Scholar]
- 20.Pan W, Akerstrom V, Zhang J, Pejovic V, Kastin AJ. Modulation of feeding-related peptide/protein signals by the blood-brain barrier. J Neurochem. 2004;90:455–461. doi: 10.1111/j.1471-4159.2004.02502.x. [DOI] [PubMed] [Google Scholar]
- 21.Pan W, Tu H, Hsuchou H, Daniel J, Kastin AJ. Unexpected amplification of leptin-induced Stat3 signaling by urocortin: implications for obesity. J Mol Neurosci. 2007;33:232–238. doi: 10.1007/s12031-007-0071-y. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Wu X, He Y, Kastin AJ, Hsuchou H, Rosenblum CI, Pan W. Melanocortin potentiates leptin-induced STAT3 signaling via MAPK pathway. J Neurochem. 2009;110:390–399. doi: 10.1111/j.1471-4159.2009.06144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan W, Tu H, Kastin AJ. Differential BBB interactions of three ingestive peptides: Obestatin, ghrelin, and adiponectin. Peptides. 2006;27:911–916. doi: 10.1016/j.peptides.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 24.Kastin AJ, Akerstrom V, Pan W. Validity of multiple-time regression analysis in measurement of tritiated and iodinated leptin crossing the blood-brain barrier: meaningful controls. Peptides. 2001;22:2127–2136. doi: 10.1016/s0196-9781(01)00569-1. [DOI] [PubMed] [Google Scholar]
- 25.Pan W, Kastin AJ. Interactions of IGF-1 with the blood-brain barrier in vivo and in situ. Neuroendocrinology. 2000;72:171–178. doi: 10.1159/000054584. [DOI] [PubMed] [Google Scholar]
- 26.Yu C, Kastin AJ, Ding Y, Pan W. Gamma glutamyl transpeptidase is a dynamic indicator of endothelial response to stroke. Exp Neurol. 2007;203:116–122. doi: 10.1016/j.expneurol.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 27.Hsuchou H, Kastin AJ, Tu H, Abbott NJ, Couraud PO, Pan W. Role of astrocytic leptin receptor subtypes on leptin permeation across hCMEC/D3 human brain endothelial cells. J Neurochem. 2010;115:1288–1298. doi: 10.1111/j.1471-4159.2010.07028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weksler B, Sublieau E, Perriere N, Charneau P, Holloway K, Leveque M, Tricoire-Leignel H, Nicotra A, Bourdoulous S, Turowski P, Male D, Roux F, Greenwood J, Romero I, Couraud P. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 29.Schrade A, Sade H, Couraud PO, Romero IA, Weksler BB, Niewoehner J. Expression and localization of claudins-3 and -12 in transformed human brain endothelium. Fluids Barriers CNS. 2012;9:6. doi: 10.1186/2045-8118-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuhlmann CR, Librizzi L, Closhen D, Pflanzner T, Lessmann V, Pietrzik CU, de Curtis M, Luhmann HJ. Mechanisms of C-reactive protein-induced blood-brain barrier disruption. Stroke. 2009;40:1458–1466. doi: 10.1161/STROKEAHA.108.535930. [DOI] [PubMed] [Google Scholar]
- 31.Mastronardi CA, Yu WH, Srivastava VK, Dees WL, McCann SM. Lipopolysaccharide-induced leptin release is neurally controlled. Proc Natl Acad Sci U S A. 2001;98:14720–14725. doi: 10.1073/pnas.251543598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mastronardi CA, Srivastava V, Yu WH, Les DW, McCann SM. Lipopolysaccharide-induced leptin synthesis and release are differentially controlled by alpha-melanocyte-stimulating hormone. Neuroimmunomodulation. 2005;12:182–188. doi: 10.1159/000084851. [DOI] [PubMed] [Google Scholar]
- 33.Banks WA, Clever CM, Farrell CL. Partial saturation and regional variation in the blood-to-brain transport of leptin in normal weight mice. Am J Physiol. 2000;278:E1158–E1165. doi: 10.1152/ajpendo.2000.278.6.E1158. [DOI] [PubMed] [Google Scholar]
- 34.Banks WA, Dohgu S, Lynch JL, Fleegal-DeMotta MA, Erickson MA, Nakaoke R, Vo TQ. Nitric oxide isoenzymes regulate lipopolysaccharide-enhanced insulin transport across the blood-brain barrier. Endocrinology. 2008;149:1514–1523. doi: 10.1210/en.2007-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherjee R, Choi JW, Choi DK, Oh TS, Liu H, Yun JW. Gender-dependent protein expression in white adipose tissues of lean and obese rats fed a high fat diet. Cell Physiol Biochem. 2012;29:617–634. doi: 10.1159/000338515. [DOI] [PubMed] [Google Scholar]