

Abstract
Oxidative cleavage of cycloalkene-1-carboxylates, made from the corresponding carboxylic acids, and subsequent oxidation of the resulting ketoaldehyde afforded the important 1-monoesters of 2-ketoalkanedioic acids. Thus ozonolysis of octyl cyclobutene-1-carboxylate followed by sodium chlorite oxidation afforded the 1-monooctyl 2-ketoglutarate. This is a cell-permeable prodrug form of α-ketoglutarate, an important intermediate in the tricarboxylic acid (TCA, Krebs) cycle and a promising therapeutic agent in its own right.
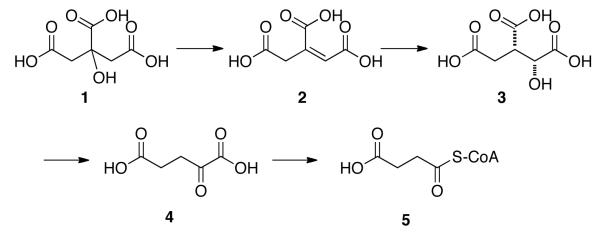
The citric acid cycle, also known as the tricarboxylic acid (TCA) cycle or the Krebs cycle, is the mechanism whereby aerobic organisms generate energy via oxidation of acetate ultimately into carbon dioxide (Fig. 1).1 A key step in this pathway is the conversion of D-isocitrate 3 (formed from citrate 1 via aconitate 2) into α-ketoglutarate 4 via the enzyme isocitrate dehydrogenase in an oxidative decarboxylation process. α-Ketoglutarate 4 is then converted into succinyl CoA 5 and ultimately via oxaloaceate into citrate to complete the cycle. α-Ketoglutarate 4 is also prepared by the deamination of glutamate and is thus an important chemical substance.2 Indeed a recent patent claims its use as a treatment for cancer and other diseases.3 Recently for another project, which was studying the effectiveness of α-ketoglutarate in prolonging the lifespan of C. Elegans,4 we needed to prepare a reasonable amount of a cell-permeable form of α-ketoglutarate and chose the 1-monooctyl ester. Although there are many biological methods for preparing α-ketoglutarate,5 e.g., by carboxylation of succinate using enzymes, we wanted to use a chemical method. Several chemical methods exist for the synthesis of the 1-ester of α-ketoglutarate,6 especially direct esterification of 2-oxo-pentanedioic acid or other methods, but those methods gave very mixed results in our hands. Therefore we developed a new method for the synthesis of the monooctyl ester 6 of α-ketoglutarate 4 via oxidative cleavage of a cyclobutene-1-carboxylate, which guaranteed the formation of the desired 1-monoester. We have further shown that this new method can be applied to the synthesis of a variety of 1-monoesters of α-ketodicarboxylic acids, all of which are analogues of α-ketoglutarate. We believe that the guarantee of obtaining only the desired 1-monoester in generally quite pure form makes up for the additional number of steps that this method employs vs. the direct esterification.
Figure 1.
Part of Citric Acid Cycle
Synthesis of 1-Monooctyl α-ketoglutarate, 6
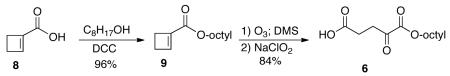
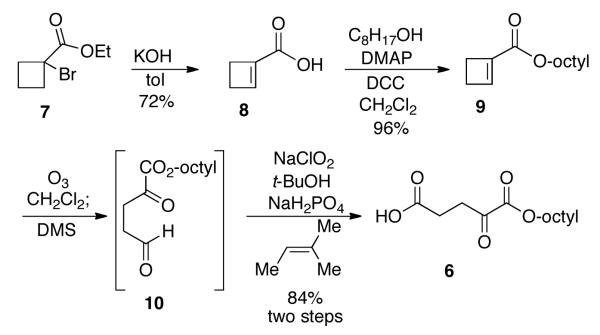
We decided to use the ozonolysis7 of a cycloalkene-1-carboxylate as the penultimate step in preparing the monoesters of the α-ketodicarboxylic acids. This guarantees the formation of only the mono ester and its regiochemistry. The resulting ketoaldehyde would be oxidized to the acid in the last step. Thus for monooctyl α-ketoglutarate 6, one would need the cyclobutene-1-carboxylic acid 8 (Scheme 1). Although this compound is commercially available, it was easily prepared in 72% yield by treatment of the readily available ethyl 1-bromocyclobutane-1-carboxylate 7 with potassium hydroxide in toluene.8 Formation of the ester was carried out using 1-octanol and dicyclohexyl carbodiimide (DCC) with DMAP as base to give the ester 9 in 96% yield. Ozonolysis of the cyclobutene was carried out at −78 °C and dimethyl sulfide (DMS) was used to destroy the ozonide to give the ketoaldehyde 10. This aldehyde was not purified but was immediately oxidized via the Pinnick conditions9 using sodium chlorite to give the desired product, 1-monooctyl α-ketoglutarate 6, in 84% yield for the last two steps. Thus the acid 8 can be converted into the desired monoester of α-ketoglutarate 6 in two operations and an overall yield of 80%.
Scheme 1.
Synthesis of 1-Monooctyl α-Ketoglutarate
Synthesis of Monoesters of α-Ketoalkanedioic Acids, 13a-d
We decided to see how general this new method for the synthesis of monoesters of α-ketoalkanedioic acids was (Scheme 2). For this reason we prepared several cycloalkene-1-carboxylic acids 11a-d by known methods, including the indene-1- and 2-carboxylic acids. Each of these acids were converted into their monoesters using the alcohol and DCC as before to give the esters 12a-d. Ozonolysis and immediate Pinnick oxidation of the resulting ketoaldehyde afforded the desired monoesters of the α-ketodioic acid 13a-d. The overall yields are comparable to those obtained for the monooctyl α-ketoglutarate 6. Thus this method should be generally applicable for the preparation of a variety of monoesters of α-ketoalkanedioic acids.
Scheme 2.

Synthesis of Other Monoesters of α-Ketoalkanedioic Acids
Conclusion
In summary, we have developed a reliable high-yielding method for the synthesis of the 1-mono-esters of α-ketoalkanedioic acids, which involves the ozonolysis and Pinnick oxidation of alkyl cycloalkene carboxylates. The utility of the method has been demonstrated by the facile synthesis of 1-monooctyl α-ketoglutarate 6 in two steps and 80% overall yield from the commercially available acid 8.
Experimental Section
General
All reactions were carried out under an argon atmosphere unless otherwise specified. Tetrahydrofuran (THF) and diethyl ether were distilled from benzoquinone ketyl radical under an argon atmosphere. Dichloromethane, toluene, benzene, pyridine, triethylamine, and diisopropylethylamine (DIPEA) were distilled from calcium hydride under an argon atmosphere. Dimethyl sulfoxide (DMSO) was distilled over calcium hydride and stored over 4 Å molecular sieves. Diisopropylamine was distilled from NaOH and methanol was distilled from magnesium turnings under an argon atmosphere. All other solvents or reagents were purified according to literature procedures. 1H NMR and 13C NMR spectra were obtained at 300 MHz or 500 MHz for proton and 75 MHz or 125 MHz for carbon and are so indicated. The chemical shifts are reported in parts per million (ppm, δ). The coupling constants are reported in Hertz (Hz) and the resonance patterns are reported with notations as the following: br (broad), s (singlet), d (double), t (triplet), q (quartet) and m (multiplet). The peak assignments in the 13C NMR spectra for 6 and 9 were determined by DEPT experiments. High-resolution mass spectra were measured on a time of flight LC-MS. Thin-layer chromatography (TLC) was carried out using precoated silica gel sheets. Visual detection was performed with ultraviolet light, p-anisaldehyde stain, potassium permanganate stain or iodine. Flash chromatography was performed using silica gel P60 (60 α, 40-63 μm) with compressed air.
Ethyl 1-bromo-cyclobutanecarboxylate, 7
N-Bromosuccinimide (0.198 g, 1.1 mmol) and AIBN (8.0 mg, 0.05 mmol) were added to a solution of ethyl cyclobutanecarboxylate (0.12 mL, 0.885 mmol) in dry carbon tetrachloride (4.0 mL) at 21 °C. The mixture was heated to reflux for 4 h. After the mixture was cooled to 21 °C, ethyl acetate (3 × 40 mL) was added to the mixture. The combined organic phases were washed with water and brine, and dried over anhydrous Na2SO4. Flash column chromatography on silica gel eluting with 20/1 hexanes/ethyl acetate gave the known ethyl 1-bromo-cyclobutanecarboxylate 710 (0.169 g, 93%) as a clear oil. 1H NMR (500 MHz, CDCl3): δ 4.20 (q, J = 5.0 Hz, 2H), 2.86 (m, 2H), 2.57 (m, 2H), 2.17 (m, 1H), 1.82 (m, 1H), 1.26 (t, J = 5.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 171.3, 61.8, 54.2, 37.1, 37.0, 16.6, 13.8.
1-Cyclobutene-1-carboxylic acid, 8
Potassium hydroxide (3.024 g, 54 mmol) was dissolved in hot toluene (50 mL), and then the bromoester 7 (2 mL, 0.0124 mol) was added. The mixture was refluxed for 1 h. After the mixture was cooled to 21 °C, water (50 mL) was added, and the mixture was extracted with diethyl ether (30 mL) and ethyl acetate (30 mL). The aqueous phase was acidified with 1.0 M aqueous HCl to pH 1. The acidified aqueous layer was extracted with ethyl acetate (3 × 60 mL) and the combined organic phases were washed with water and brine, and dried over anhydrous Na2SO4. Flash column chromatography on silica gel, eluting with 5/1 hexanes/ ethyl acetate, gave the known 1-cyclobutene-1-carboxylic acid 810 (0.873 g, 72%) which solidified to give a pale solid when stored in fridge. 1H NMR (500 MHz, CDCl3): δ 11.30 (br s, 1H), 6.92 (t, J = 1.8 Hz, 1H), 2.73 (t, J = 5.0 Hz, 2H), 2.48 (td, J = 5.0, 1.8 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ 167.3, 149.9, 138.1, 28.8, 27.2.
Octyl cyclobut-1-enecarboxylate, 9
1-Octanol (0.95 mL, 6.0 mmol), (4-dimethylamino)pyridine (DMAP, 37 mg, 0.3 mmol), and dicyclohexyl carbodiimide (DCC, 0.743 g, 3.6 mmol) were added to a solution of 1-cyclobutene-1-carboxylic acid 8 (0.295 g, 3.0 mmol) in dry dichloromethane (6.0 mL) at 0 °C. After it had stirred for 1 h, the solution was allowed to warm to 21 °C and was stirred for another 8 h. The precipitate was filtered and washed with ethyl acetate (3 × 100 mL). The combined organic phases were washed with water and brine, and dried over anhydrous Na2SO4. Flash column chromatography on silica gel eluting with 80/1 hexanes/ethyl acetate gave octyl cyclobut-1-enecarboxylate 9 as a clear oil (0.604 g, 96%). 1H NMR (500 MHz, CDCl3): δ 6.71 (bs, 1H), 4.07 (t, J = 7.0 Hz, 2H), 2.68 (t, J = 3.5 Hz, 2H), 2.42 (td, J = 3.5, 1.0 Hz, 2H), 1.61 (m, 2H), 1.24 (m, 10H), 0.84 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 162.3, 146.0, 138.8, 64.1 (CH2), 31.7 (CH2), 29.1 (CH2), 29.1 (CH2), 29.0 (CH2), 28.5 (CH2), 26.9 (CH2), 25.8 (CH2), 22.5 (CH2), 14.0 (CH3). HR-MS (ESI) Calcd for [C13H22O2H]+ 211.1698, found 211.1696.
1-Monooctyl α-ketoglutarate, 6
Into a solution of the oil 9 (0.211 g, 1.0 mmol) in dichloromethane (10 mL) cooled to −78 °C was bubbled ozone (in a stream of oxygen) until the solution turned blue. The residual ozone was discharged by bubbling with oxygen and the reaction was warmed to 21 °C and stirred for another 1 h. Dimethyl sulfide (0.11 mL, 1.5 mmol) was added to the mixture and it was stirred for another 2 h. The dichloromethane was removed in vacuo and the crude product 10 was dissolved in a solution of 2-methyl-2-butene (0.8 mL) in tert-butyl alcohol (3.0 mL). To this was added dropwise a solution containing sodium chlorite (0.147 g, 1.3 mmol) and sodium dihydrogen phosphate monohydrate (0.179 g, 1.3 mmol) in water (1.0 mL). The mixture was stirred at 21 °C overnight, and then extracted with ethyl acetate (3 × 50 mL). The combined organic phases were washed with water and brine, and dried over anhydrous Na2SO4. Flash column chromatography on silica gel eluting with 5/1 hexanes/ethyl acetate gave 1-monooctyl α-ketoglutarate 6,6 which became a pale solid when stored in the refrigerator (0.216 g, 84%) but was a clear oil at room temperature. 1H NMR (300 MHz, CDCl3): δ 4.25 (t, J = 7.0 Hz, 2H), 2.95 (m, 2H), 2.72 (t, J = 7.1 Hz, 2H), 1.71 (m, 2H), 1.29 (m, 10H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 192.8, 173.3, 160.2, 65.6 (CH2), 33.9 (CH2), 31.2 (CH2), 28.6 (CH2), 28.5 (CH2), 27.8 (CH2), 27.4 (CH2), 25.2 (CH2), 22.1 (CH2), 13.9 (CH3). HR-MS (ESI) Calcd for [C13H21O5-H]− 257.1389, found 257.1394.
Octyl cyclopent-1-enecarboxylate, 12a
Prepared from 1-octanol and cyclopent-1-ene-carboxylic acid 11a (0.15 g, 1.34 mmol) by the procedure described for the preparation of 9 to give octyl cyclopent-1-enecarboxylate 12a (0.284 g, 95%) as a clear oil.11 1H NMR (500 MHz, CDCl3): δ 6.73 (t, J = 2.4 Hz, 1H), 4.09 (t, J = 6.7 Hz, 2H), 2.53 (m, 2H), 2.46 (m, 2H), 1.92 (pentet, J = 7.6 Hz, 2H), 1.62 (t, J = 7.6 Hz, 2H), 1.38-1.20 (m, 10H), 0.85 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 165.4, 143.3, 136.7, 64.2, 33.2, 31.7, 31.3, 29.2, 29.1, 28.6, 25.9, 23.0, 22. 6, 14.0.
1-Monooctyl α-ketohexanedioic ester, 13a
Prepared from octyl cyclopent-1-enecarboxylate, 12a (0.202 g, 0.9 mmol) by the procedure described for the preparation of 6 to give 1-monooctyl α-ketohexanedioic ester 13a (0.216 g, 89%) as a pale solid. mp 39-40 °C. 1H NMR (500 MHz, CDCl3): δ 4.23 (t, J = 6.8 Hz, 2H), 2.93 (t, J = 7.1 Hz, 2H), 2.43 (t, J = 7.2 Hz, 2H), 1.95 (pentet, J = 7.2 Hz, 2H), 1.71 (pentet, J = 7.2 Hz, 2H), 1.40-1.20 (m, 10H), 0.86 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 193.6, 179.0, 160.9, 66.6, 38.2, 32.5, 31.7, 29.1, 29.0, 28.3, 25.7, 22.6, 17.8, 14.0. HR-MS (ESI) Calcd for [C14H24O5−H]− 271.1545, found 271.1552.
Octyl cyclohex-1-enecarboxylate, 12b
Prepared from 1-octanol and cyclohex-1-ene-carboxylic acid 11b (0.19 g, 1.5 mmol) by the procedure described for the preparation of 9 to give octyl cyclohex-1-enecarboxylate 12b (0.325 g, 91%) as a clear oil.12 1H NMR (500 MHz, CDCl3): δ 6.93 (tt, J = 4.0, 1.5 Hz, 1H), 4.07 (t, J = 6.7 Hz, 2H), 2.22 (m, 2H), 2.15 (m, 2H), 1.65-1.53 (m, 6H), 1.37-1.19 (m, 10H), 0.84 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 167.6, 139.3, 130.5, 64.3, 31.8, 29.22, 29.17, 28.7, 26.0, 25.7, 24.1, 22.6, 22.1, 21.5, 14.0.
1-Monooctyl α-ketoheptanedioic ester, 13b
Prepared from octyl cyclohex-1-enecarboxylate, 12b (0.165 g, 0.7 mmol) by the procedure described in preparation of 6 to give 1-monooctyl α-ketoheptanedioic ester 13b (0.156 g, 78%) as a clear oil. 1H NMR13 (500 MHz, CDCl3): δ 5.38 (m, 1H, major enol), 5.16 (m, 1H, minor enol), 4.23 (t, J = 6.8 Hz, 2H, keto), 4.22 (t, J = 6.8 Hz, minor enol), 4.15 (t, J = 6.8 Hz, major enol), 2.82 (m, 2H), 2.35 (m, 2H), 1.94 (m, 2H), 1.66 (m, 2H), 1.43 (m, 4H), 1.36-1.20 (m, 20H), 0.85 (t, J = 7.1 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ 193.9, 179.2, 167.9 (enol), 161.1, 105.9 (enol), 105.0 (enol), 66.5, 66.0 (enol), 38.8, 33.5, 31.6, 29.03, 29.02, 28.3, 28.2 25.62, 25.55, 22.5, 14.0. HR-MS (ESI) Calcd for [C15H26O5−H]− 285.1702, found 285.1715.
Octyl 1H-indene-3-carboxylate, 12c
Prepared from 1-octanol and 1H-indene-3-carboxylic acid14 11c (0.389 g, 2.4 mmol) by the procedure described for the preparation of 9 to give octyl 1H-indene-3-carboxylate 12c (0.655 g, 99%) as a clear oil. 1H NMR (300 MHz, CDCl3): δ 8.10 (d, J = 7.6 Hz, 1H), 7.48 (d, J = 7.4 Hz, 1H), 7.47 (s, 1H), 7.38 (t, J = 7.4 Hz, 1H), 7.28 (dt, J = 7.4, 1.5 Hz, 1H), 4.34 (t, J = 6.7 Hz, 2H), 3.51 (s, 2H), 1.81 (pentet, J = 6.9 Hz, 2H), 1.55-1.25 (m, 10H), 0.94 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 164.0, 144.1, 143.3, 140.7, 136.4, 126.5, 125.4, 123.6, 122.3, 64.5, 38.2, 31.7, 29.13, 29.09, 28.6, 26.0, 22.5, 14.0. HR-MS (ESI) Calcd for [C18H24O2−H]− 271.1698, found 271.1691.
Octyl 2-(2-(carboxymethyl)phenyl)-2-oxoacetic ester, 13c
Prepared from octyl 1H-indene-3-carboxylate, 12c (0.136 g, 0.5 mmol), by the procedure described for the preparation of 6 to give octyl 2-(2-(carboxymethyl)phenyl)-2-oxoacetic ester 13c (0.135 g, 85%) as pale solid. mp 63-64 °C. 1H NMR (500 MHz, CDCl3): δ 7.76 (d, J = 7.6 Hz, 1H), 7.56 (t, J = 7.4 Hz, 1H), 7.43 (dt, J = 7.7, 0.85 Hz, 1H), 7.32 (d, J = 7.7 Hz, 1H), 4.34 (t, J = 6.9 Hz, 2H), 4.00 (s, 2H), 1.74 (quint, J = 7.2 Hz, 2H), 1.40-1.21 (m, 10H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 188.3, 171.8, 163.8, 137.4, 134.1, 133.2, 132.6, 130.8, 127.4, 66.0, 39.5, 31.2, 28.6, 28.5, 27.9, 25.2, 22.1, 13.9. HR-MS (ESI) Calcd for [C18H24O5−H]− 319.1545, found 319.1557.
Octyl 1H-indene-2-carboxylate, 12d
Prepared from 1-octanol and 1H-indene-2-carboxylic acid15 11d (0.2661 g, 1.66 mmol) by the procedure described for the preparation of 9 to give octyl 1H-indene-2-carboxylate 12d (0.437 g, 97%) as a clear oil. 1H NMR (300 MHz, CDCl3): δ 7.71 (td, J = 1.9, 0.6 Hz, 1H), 7.53-7.47 (m, 2H), 7.35-7.28 (m, 2H), 4.24 (t, J = 6.7 Hz, 2H), 3.67 (d, J = 3.0 Hz, 2H), 1.73 (pentet, J = 6.6 Hz, 2H), 1.36-1.25 (m, 10H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 165.0, 144.7, 142.7, 140.8, 137.4, 127.4, 126.7, 124.1, 123.2, 64.5, 38.2, 31.7, 29.2, 29.1, 28.7, 26.0, 22.6, 14.0. HR-MS (ESI) Calcd for [C18H24O2−H]− 271.1698, found 271.1694.
2-(3-(Octanyloxy)-2,3-dioxopropyl)benzoic acid, 13d
Prepared from octyl 1H-indene-2-carboxylate, 12d (0.137 g, 0.5 mmol) by the procedure described for the preparation of 6 to give 2-(3-(octanyloxy)-2,3-dioxopropyl)benzoic acid 13d (0.125 g, 78%) as a pale solid. mp 67-68 °C. 1H NMR of keto form (500 MHz, CDCl3): δ 8.09 (bd, J = 7.8 Hz, 1H), 7.55 (dt, J = 7.6, 1.2 Hz, 1H), 7.38 (t, J = 7.4 Hz, 1H), 7.26 (d, J = 7.4 Hz, 1H), 4.23 (t, J = 6.7 Hz, 2H), 3.67 (bs, 1H), 3.24 (bs, 1H), 1.63 (pentet, J = 7.0 Hz, 2H), 1.31-1.18 (m, 10H), 0.85 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 190.6, 168.1, 163.3, 136.1, 134.1, 128.9, 128.5, 127.7, 124.2, 66. 7, 35.7, 31.2, 28.5, 28.5, 27.9, 25.1, 22.1, 13.9. HR-MS (ESI) Calcd for [C18H24O5−H]− 319.1545, found 319.1556.
Supplementary Material
Acknowledgements
The LCMS used in this project was supported by Grant Number S10RR025631 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. This NMR spectrometers were supported by the National Science Foundation under equipment grant no. CHE-1048804.
Footnotes
Supporting Information Available: Proton and carbon NMR data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1) (a).Munnich A. Nat. Genet. 2008;40:1148. doi: 10.1038/ng1008-1148. [DOI] [PubMed] [Google Scholar]; (b) Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, Colman RF. Nat. Genet. 2008;40:1230. doi: 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2) (a).Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, Dang L, Fantin VR, Mak TW. J. Exp. Med. 2012;207:339. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wise DR, Ward PS, Shay JES, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Proc. Natl. Acad. Sci. U.S.A. 2011;108:19611. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Townsend CA, Barrabee EB. J. Chem. Soc., Chem. Commun. 1984:1586. [Google Scholar]
- (3).Gottlieb E, Selak MA, Mackenzie ED, Watson DG. PCT Int. Appl. 2006 WO 2006016143 A1. [Google Scholar]
- (4).Chin RM, Pai M, Fu X, Diep S, Jung G, Lomenick B, Deng G, Whelan SA, Monsalve GC, Chang HR, Clarke CF, Frand AR, Jung ME, Huang J. Manuscript submitted.
- (5) (a).Shiio I, Ujigawa-Takeda K. Agric. Biol. Chem. 1980;44:1897. [Google Scholar]; (b) Allison MJ, Robinson IM, Baetz AL. J. Bacteriol. 1979;140:980. doi: 10.1128/jb.140.3.980-986.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Elias BA, Givan CV. Plant Physiol. 1977;59:738. doi: 10.1104/pp.59.4.738. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Buchanan BB, Evans MCW. Proc. Natl. Acad. Sci. U.S.A. 1965;54:1212. doi: 10.1073/pnas.54.4.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Takeuchi Y, Nagao Y, Toma K, Yoshikawa Y, Akiyama T, Nishioka H, Abe H, Harayama T, Yamamoto S. Chem. Pharm. Bull. 1999;47:1284. Natsugari H, Kawano Y, Morimoto A, Yoshioka K, Ochiai M. J. Chem. Soc., Chem. Commun. 1987:62. doi: 10.1248/cpb.35.996. Domagala JM. Tetrahedron Lett. 1980;21:4997. Beyerman HC, van Dijck LA, Levisalles J, Melera A, Veer WLC. Bull. Soc. Chim. France. 1961:1812. (e) for the diester:Hartenstein H, Blitzke T, Sicker D, Wilde H. J. Prakt. Chem. 1993;335:176.
- (7).For reviews and articles on ozonolysis, see: Van Ornum SG, Champeau RM, Pariza R. Chem. Rev. 2006;106:2990. doi: 10.1021/cr040682z. Berglund RA, Kreilein MM. “Ozone” e-EROS Encyclopedia of Reagents for Organic Synthesis. 2001 Schreiber SL, Claus RE, Reagan J. Tetrahedron Lett. 1982;23:3867. Fleming FF, Huang A, Sharief VA, Pu Y. J. Org. Chem. 1999;64:2830. doi: 10.1021/jo9822885. Montgomery J, Savchenko AV, Zhao Y. J. Org. Chem. 1995;60:5699.
- (8) (a).Campbell A, Rydon HN. J. Chem. Soc. 1953:3002. [Google Scholar]; (b) Lee KS, Choi T-L. Org. Lett. 2011;13:3908. doi: 10.1021/ol2014363. [DOI] [PubMed] [Google Scholar]
- (9) (a).Kraus GA, Roth B. J. Org. Chem. 1980;45:4825. [Google Scholar]; (b) Kraus GA, Taschner MJ. J. Org. Chem. 1980;45:1175. [Google Scholar]; (c) Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]
- (10).Witiak DT, Sinha BK, Lee OS, Feller DR. J. Med. Chem. 1972;15:803. doi: 10.1021/jm00278a005. [DOI] [PubMed] [Google Scholar]
- (11).Ovezov A, Charylev M, Sergienko SR, Aidogdyev A, Talalaev EI, Zolotareva Yu. V. Izvest. Akad. Nauk Turkmen. 1996:42. [Google Scholar]
- (12).Gerster M, Mihalic M. PCT Int. Appl. 2009 WO2009034016 A1 20090319. [Google Scholar]
- (13).All of the α-ketoalkanedioic acids exist predominantly as the keto form except for two, namely: 13b, which exists as a mixture of the predominant keto form and both E and Z enol forms (one major and one minor, unassigned); and 13d, which exists as a mixture of all three forms, the keto and both enol forms.
- (14).Pietruszka J, Simon RC, Kruska F, Braun M. Eur. J. Org. Chem. 2009;35:6217. [Google Scholar]
- (15).Varnavas A, Lassiani L, Valenta V, Berti F, Tontini A, Mennuni L, Makovec F. Eur. J. Med. Chem. 2004;39:85. doi: 10.1016/j.ejmech.2003.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.