

Abstract
Na+/H+ exchanger (NHE) proteins are involved in intracellular pH and volume regulation and may indirectly influence neurotransmission. The abundant NHE isoform 1 (NHE1) has also been linked to brain cell damage during metabolic stress. It is not known, however, whether NHE1 or other NHE isoforms play a role in striatal dopamine (DA) neurotransmission under normal or metabolic stress conditions. Our study tested the hypothesis that NHE inhibition with cariporide mesilate (HOE-642) modifies striatal DA overflow and DAergic terminal damage in mice caused by the mitochondrial inhibitor malonate. We also explored the expression of NHE1–5 in the striatum and substantia nigra. Reverse microdialysis of HOE-642 elicited a transient elevation followed by a reduction in DA overflow accompanied by a decline in striatal DA content. HOE-642 pre-treatment diminished the malonate-induced DA overflow without reducing the intensity of the metabolic stress or subsequent DAergic axonal damage. Although NHE isoforms 1–5 are expressed in the striatum and midbrain, NHE1 protein was not co-located on nigrostriatal DAergic neurons. The absence of NHE1 co-location on DAergic neurons suggests that the effects of HOE-642 on striatal DA overflow are either mediated via NHE1 located on other cell types or that HOE-642 is acting through multiple NHE isoforms.
Keywords: cariporide, dopamine, microdialysis, Na+/H+ exchanger, neurotransmission, striatum
The Na+/H+ exchanger (NHE) protein family serves key roles in cellular homeostasis by mediating the electroneutral transport of H+ against an influx of Na+ ions. Ten NHE isoforms (NHE1–10) have been cloned that are variably expressed across mammalian tissues, cell types, and subcellular compartments (Orlowski and Grinstein 2004; Nakamura et al. 2005; Lee et al. 2008). In the brain, NHE1–5 are differentially expressed between regions and cell layers (Ma and Haddad 1997; Attaphitaya et al. 1999; Baird et al. 1999; Xue et al. 2003). Although little is known of the role of most NHE isoforms in the specialized function of brain cells, the abundant NHE1 participates in regulating cytosolic pH and cell volume in neurons and astrocytes (Pizzonia et al. 1996; Yao et al. 1999; Chesler 2003; Pedersen et al. 2006).
The influence of intracellular and extracellular pH changes on neuronal excitability is well established and attributed in part to the H+ sensitivity of neurotransmitter receptors and voltage-gated ion channels (Tang et al. 1990; Takahashi et al. 1993; Pasternack et al. 1996; Makani and Chesler 2007). Although the superfamily of HCO3− transporters and the enzyme carbonic anhydrase are important regulators of brain tissue pH (Chesler 2003), NHE activity mediates H+ extrusion in brain synaptosomes and may also influence neurotransmission (Sauvaigo et al. 1984; Jean et al. 1985; Nachshen and Drapeau 1988). Consistent with this notion NHE inhibition modifies pre-synaptic glutamate and GABA release in dissociated hippocampal neurons (Trudeau et al. 1999; Jang et al. 2006). Given that dopamine (DA) release from synaptosomes is sensitive to pH changes (Drapeau and Nachshen 1988; Cannizzaro et al. 2003), NHE inhibition might also modify DA neurotransmission (Zubieta et al. 1988; Amoroso et al. 1990) yet this possibility has not been previously tested in vivo.
Striatal DA neurotransmission is regulated by vesicular exocytosis and carrier-mediated transmitter re-uptake into pre-synaptic terminals. Activity-dependent exocytosis requires vesicular uptake of cytosolic DA via the vesicular monoamine transporter-2 (VMAT2) (Pothos et al. 2000), while the re-uptake of extracellular DA is mediated by the plasma membrane DA transporter (DAT) (Kilty et al. 1991). Both of these DA uptake processes require the maintenance of transmembrane H+ and Na+ gradients, respectively, which are dependent on energy metabolism and might be influenced by NHE activity. Under conditions of disrupted mitochondrial respiration, accumulation of metabolic acid and failure of the Na+/K+ ATPase lead to dissipated transmembrane H+ and Na+ gradients, which contribute to unregulated exocytosis and DAT-mediated DA efflux (Santos et al. 1996; Buyukuysal and Mete 1999; Moy et al. 2007). Disrupted DA transmission itself is thought to exacerbate striatal tissue damage caused by metabolic stress (Globus et al. 1987; Ferger et al. 1999; Moy et al. 2000; Xia et al. 2001). Similar conditions of metabolic stress may be seen during ischemia/hypoxia and in neurodegenerative disorders such as Parkinson’s disease which have been linked to mitochondrial defects (Parker et al. 1989; Haas et al. 1995).
Interestingly, NHE1 activity during ischemia-reperfusion in the brain and in the heart contributes to intracellular Na+ loading, which favors cell swelling and reversal of the Na+/Ca2+ exchanger causing Ca2+ influx that triggers mitochondria death pathways (Scholz et al. 1995; Luo et al. 2005; Pedersen et al. 2006). In particular, studies have shown that ischemia-induced loss of cortical and hippocampal neurons is attenuated by NHE inhibitors or the genetic knockdown of NHE1 (Vornov et al. 1996; Phillis et al. 1999; Luo et al. 2005). Hence, it is possible that NHE1 may also contribute to disrupted DA neurotransmission and to the ensuing neuronal damage in the striatum under similar conditions of metabolic stress.
The purpose of our study was to test the hypothesis that striatal NHE inhibition modifies DA neurotransmission and DAergic terminal damage caused by metabolic stress in vivo. In addition, we explored the cellular location of NHE1 protein and the expression of other NHE isoforms in the striatum and substantia nigra. We report that the NHE inhibitor cariporide mesilate (HOE-642) modifies striatal DA overflow but not DAergic terminal damage consistent with expression of NHE1–5 in the nigrostriatal regions and the surprising absence of NHE1 co-location on DAergic neurons.
Materials and methods
Animals
Male Swiss-Webster mice aged 3–4 months from Taconic Farms (Germantown, NY, USA) were used in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and as approved by the Institutional Animal Care and Use Committee.
Drugs and reagents
Malonate (MAL) disodium salt, ethylisopropylamiloride (EIPA), mazindol, lactate dehydrogenase, NAD+, and common reagents were from Sigma-Aldrich (St Louis, MO, USA). HOE-642 was generously provided by Sanofi-Aventis (Frankfurt, Germany).
Primary antibodies
Affinity-purified rabbit polyclonal antibody XB-17 raised against amino acids 639–746 of the cytoplasmic region of human NHE1 was a generous gift by Dr M. Musch (University of Chicago). The specificity of the XB-17 antibody for NHE1 has been demonstrated in numerous cell types using western blot, immunoprecipitation, and immunocytochemistry (McSwine et al. 1994; Coupaye-Gerard et al. 1996; Pedersen et al. 2003). Mouse monoclonal antibody raised against tyrosine hydroxylase (TH) was from Calbiochem (San Diego, CA, USA).
Intracerebral cannula implantation and in vivo microdialysis
CMA/7 guide cannulae and microdialysis probes were from CMA Microdialysis (North Chelmsford, MA, USA). Mice were anesthetized with isofluorane, guide cannulae implanted with stereotaxic surgery at AP +0.6 mm, L +2.2 mm, and DV −1.8 mm relative to bregma. After 3–5 days of recovery, the microdialysis probe was inserted through the implanted guide cannula into the striatum and perfused at a flow rate of 1 μL/min with artificial CSF (aCSF: 143 mM NaCl, 2.7 mM KCl, 1.2 mM CaCl2, 0.85 mM MgCl2, and 2.0 mM Na2HPO4, pH ~7.3). After approximately 2 h of probe equilibration, dialysate fractions were collected every 20 min. Drugs were prepared in aCSF and delivered via reverse microdialysis into the striatum of freely moving mice. Dialysate levels of DA and its metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) were determined for 1 h prior to the beginning of drug delivery and served as baseline controls. Animals were killed 4–6 days later for histological evaluation of probe placement or quantification of striatal DAergic terminal damage.
Measurement of DA and metabolites in microdialysis and tissue samples
Levels of DA, DOPAC, and homovanillic acid (HVA) in microdialysis and tissue samples were measured with HPLC and electrochemical detection (Antec Leyden Zoeterwoude, The Netherlands) as previously described (Moy et al. 2000).
Determination of TH content in striatal tissue
Tyrosine hydroxylase was measured with ELISA (Alfinito et al. 2003). Briefly, samples were co-incubated with rabbit polyclonal anti-TH (1 : 500; Calbiochem) and polyclonal anti-rabbit horseradish peroxidase (HRP) (1 : 3000; Amersham Biosciences, Piscataway, NJ, USA) in 96-well microplates (Thermo Labsystems, Franklin, MA, USA), which had been coated with monoclonal antiTH (1 : 500) and blocked with 5% non-fat dry milk. Reaction products from co-exposure to Amplex Red cocktail (Molecular Probes, Eugene, OR, USA) were measured fluorometrically (excitation/emission ratio of 530/580 nm).
Lactate measurement in microdialysis samples
Lactate was quantified by following the enzymatic reduction of NAD+ as previously described (Nicklas and Browning 1978) on a microplate spectrophotometer. Briefly, 6.25 μL sample aliquots were mixed with 3.5 mM NAD+ in 200 μL 0.1 M Tris buffer containing 0.4 M hydrazine hydrate, 10 mM MgCl2, and 5 mM EDTA, pH 8.75. The reaction was started by adding 2.5 μL of 5 mg/mL lactate dehydrogenase (6.25 U in final volume) at 30°C and delta optical density measured at 340 nm.
Synaptosomal 3H-DA uptake assay
3H-DA uptake into striatal synaptosomes in the presence or absence of drugs was performed as previously described (Hogan et al. 2000) on both rat and mouse preparations. No species differences in basal 3H-DA uptake or drug effects were observed and the data were combined.
Monoamine oxidase activity assay
Enzymatic activity of monoamine oxidase-A and B (MAO-A or -B) in mouse striatal homogenates was determined with the Amplex Red MAO Assay kit (Molecular Probes). Briefly, tissue samples in the presence or absence of drugs were pre-incubated with isoform-specific MAO inhibitors (deprenyl or clorgyline) to isolate MAO-A or -B activities, respectively. The reaction was started by adding MAO substrates (tyramine or benzylamine), HRP, and Amplex Red. Reaction products were measured fluorometrically (excitation/emission ratio of 530/580 nm).
Histological evaluation of probe placement
Brains were snap frozen in methylbutane at −30°C and coronal sections (40 μm) were stained with cresyl violet for the verification of microdialysis probe sites. In experiments where the striata had to be freshly dissected for neurochemistry, representative mice from each treatment group were used for histological verification of dialysis sites.
Western blot analysis of subcellular fractions from mouse brain
Sample preparation
Crude synaptosomes were prepared with differential centrifugation as published (Sandoval et al. 2002) with minor modifications. Dissected brain regions were homogenized in 0.32 M sucrose containing a protease inhibitor cocktail (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and spun down at 900 g (10 min). The supernatant was spun at 22 000 g (20 min) to pellet down the crude synaptosomal fraction (P2) from the cytosolic contents (S2). Sample protein concentration was determined using the Bio-Rad Protein assay (Hercules, CA, USA).
Protein electrophoresis and western blot
Samples (10–20 μg of protein) were separated with sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis and transferred electrophoretically to polyvinylidene fluoride membranes (Invitrogen, Carlsbad, CA, USA) that were blocked for 1 h with 5% non-fat dry milk in Tris-buffered saline with 0.05% Tween 20. Membranes were incubated with the anti-NHE1 antibody (1 : 1000) in blocking solution for 1 h at room temperature (20–23°C), washed, exposed to HRP-conjugated goat anti-rabbit IgG antibody (Biosource International, Camarillo, CA, USA), and washed again. Band visualization was achieved with enhanced chemiluminescence (Amersham Biosciences) and exposure to Kodak Biomax film (Rochester, NY, USA).
Double-label immunofluorescence for TH and NHE1
Mice (n = 3) were deeply anesthetized and transcardially perfused with phosphate-buffered saline (PBS), followed by 4% p-formaldehyde in 0.1 M phosphate buffer (PB). Brains were post-fixed in 4% p-formaldehyde/0.1 M PB for 2 h and stored in 30% sucrose/0.1 M PB at 4°C. Cryostat coronal sections (20 μm) were mounted onto glass slides, heated at 60°C in 0.01 M citrate buffer for 45 min, and then blocked for 30 min in PBS containing 0.3% Triton X-100 (PBS-T) with 2% normal goat serum (NGS) and 0.25% bovine serum albumin. Slides were incubated with anti-NHE1 (1 : 100) or anti-TH (1 : 5000) antibodies in 1% NGS–PBS-T overnight at room temperature (20–23°C). After washing with 0.05 M PB, sections were incubated with biotinylated goat anti-rabbit IgG or Cy3-conjugated goat anti-mouse IgG in 1% NGS–PBS-T for 1 h (Jackson ImmunoResearch Labs, West Grove, PA, USA) and washed in 0.05 M PB. Secondary biotinylated antibody labeling was achieved with Cy2-streptavidin (Jackson ImmunoResearch Labs). Procedures were repeated for the second primary/secondary antibody combination. Controls included omission of one or both primary antibodies. Immunofluorescence was visualized with a Zeiss LSM510 Meta confocal laser scanning microscope (Thornwood, NY, USA) using a 63×, 1.2 NA objective, and z-stack images were similarly acquired for all conditions examined.
NHE primer design, isolation of RNA, and RT-PCR
GenBank accession numbers for mouse NHE1–5 were retrieved using the National Center for Biotechnology Information resource Nucleotide: NHE1, NM_016981; NHE2, NM_001033289; NHE3, NM_001081060; NHE4, NM_177084; and NHE5, NM_001081332. Primer pairs specific for NHE1–5 were designed using PrimerQuest software from and synthesized by Integrated DNA Technologies (Coralville, IA, USA): NHE1 (forward 5′-CCTTTTTGCAGTTTG-GACTTTGA and reverse 5′-TTAGCGAAGTGCGTGTAAGACCT, bases 590–741); NHE2 (forward 5′-TGTTCATCACGGCTGCT-ATTG and reverse 5′-TGATCAAAAAACCGACAGTGGAT, bases 1541–1691); NHE3 (forward 5′-TTGGATGCTGGATACTTCATGC and reverse 5′-TCAGTTCACCCATCAAGCCA, bases 358–508); NHE4 (forward 5′-ACCAGGCCCTTCTTTGAGATAT and reverse 5′-GCAGATTGTGTAGCAGTTGATGT, bases 731–881); and NHE5 (forward 5′-CGTCTCAGAACTGGGAATCCA and reverse 5′-GAGTGACACTTGTTCATCTGGCTC, bases 4902–5052).
Total RNA was isolated from mouse striatum and ventral midbrain using the RNEasy Mini Kit (Qiagen, Valencia, CA, USA). RNA concentration was determined by standard spectrophotometric analysis and 1 μg of total RNA was used for cDNA synthesis with the Invitrogen Superscript II kit (Bedford, MA, USA) according to the manufacturer’s protocol. PCR was performed in a total volume of 25 μL with a master mix (Invitrogen) containing 0.2 mM dNTP, 1.5 mM MgCl2, 0.5 μM of each primer, 1 μL template cDNA, and 1 U Taq polymerase. Thermal cycling conditions included 3 min at 95°C, followed by 35 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s. The last cycle included 5 min at 72°C. The samples were cooled to 4°C, analyzed by agarose gel electrophoresis, and visualized by ethidium bromide staining.
Data analysis
For microdialysis data, within-group effects were analyzed with repeated measures one-way ANOVA and a post hoc test for a linear trend to examine dose–effect relationships. Between-group effects on total DA overflow during specific time intervals were analyzed by comparison of area under the curve with Student’s t-test. Tissue DA and metabolites measured 40 min after HOE-642 delivery were compared with levels in the control untreated side using Student’s t-test. Striatal DA or TH content between groups 1 week after microdialysis was compared with one-way ANOVA with post hoc Tukey’s test. All values at p < 0.05 were considered statistically significant.
Results
Striatal delivery of HOE-642 causes a bi-phasic effect on DA overflow
To examine the local effects of NHE inhibition on striatal DA overflow in vivo, the specific NHE inhibitor HOE-642 was delivered via reverse microdialysis in mice with determination of DA in the collected dialysate fluid. HOE-642 was dialyzed in successive 20-min periods at different dosing rates (0.3, 1, and 3 nmol/min) that were separated by drug washout intervals (aCSF perfusion only) of at least 1 h. Striatal DA overflow peaked in time points corresponding to HOE-642 tissue delivery and decreased to or below baseline during each drug washout interval (Fig. 1a). The amplitude of peaks in DA overflow (approximately 250%, 380%, and 500% of baseline) was dependent on the dose of HOE-642 (Fig. 1b). In contrast, DA overflow was progressively reduced starting in the second drug washout interval and reached approximately 50% of pre-drug baseline levels at the end of the experiment (Fig. 1c). The data indicate that striatal delivery of HOE-642 elicits a bi-phasic effect on DA overflow that consists of an increase followed by a decrease in extracellular DA levels.
Fig. 1.

Effects of microdialysis delivery of HOE-642 on striatal DA overflow. (a) Striatal DA overflow in 20-min intervals throughout reverse microdialysis of HOE-642 in one representative experiment. DA levels are expressed as percentage of pre-drug baseline levels (y-axis). The microdialysis probe was perfused with 0.3, 1, or 3 nmol HOE-642/min in successive 20-min periods as shown in the hatched boxes (a, b, and c) followed by drug washout intervals (aCSF perfusion only) of at least 1 h (d, e, and f). (b and c) Bar graphs showing striatal DA overflow (mean ± SEM, n = 3) during each of the 20-min delivery periods of HOE-642 or at 1 h washout time-points after HOE-642 delivery (points a–c or d–f, respectively, illustrated in a). ***p < 0.001 and **p < 0.01 denote overall treatment effects (repeated measures one-way ANOVA and test for a linear trend).
Prolonged delivery of HOE-642 modifies the overflow of DA and its metabolites followed by reduced striatal tissue DA content
To further characterize the effects of NHE inhibition on striatal DA neurotransmission, reverse microdialysis of HOE-642 (3 nmol/min) was extended from 20 to 120 min. Perfusion of HOE-642 caused a rapid elevation in striatal DA overflow that peaked within 20 min (approximately 800% of baseline) but progressively decreased below baseline in spite of the extended drug delivery (Fig. 2a). There were no temporal changes in DA overflow in the aCSF control group.
Fig. 2.

Effects of prolonged local delivery of HOE-642 on striatal overflow of DA, DOPAC, and HVA. (a) Time course of striatal DA overflow (y-axis: logarithmic scale) throughout reverse microdialysis of HOE-642 (■, 3 nmol/min) for 120 min (hatched box) or continuous perfusion with aCSF (□). Inset: Total DA overflow during the periods of HOE-642 treatment (area under the curve, 100–220 min) or washout (area under the curve, 220–320 min). (b) Striatal overflow of DOPAC (●) and HVA (▼) throughout reverse microdialysis of HOE-642 for 120 min (y-axis: linear scale). All values are mean ± SEM (n = 3–5). There was a statistically significant temporal effect on levels of DA, DOPAC, and HVA associated with HOE-642 treatment (■, ●, ▼, p < 0.001 with repeated measures one-way ANOVA). *p < 0.05 relative to control group (t-test).
The onset of HOE-642 delivery also modified the overflow of DOPAC and HVA, which increased to approximately 200% of baseline 40 and 80 min later, respectively (Fig. 2b). Although DOPAC levels returned to baseline over time, HVA remained elevated. These findings are indicative of an increase in DA turnover.
To determine whether the observed decline in DA overflow was due to a reduction in striatal DA stores, we measured striatal tissue DA content 40 min after the end of HOE-642 delivery, at which time DA overflow was below baseline. Reverse microdialysis of HOE-642 reduced the whole striatal DA content to 65% of the control untreated side (Table 1). DOPAC/DA ratios in the infused side were significantly greater than in the control untreated side. There was also a trend for increased HVA tissue levels although it did not reach statistical significance. Together these data indicate that prolonged delivery of HOE-642 modifies DA neurotransmission with an increase in DA turnover and a decline in striatal DA stores.
Table 1.
Striatal content of DA and metabolites after microdialysis delivery of HOE-642
| Treated side | Untreated side | |
|---|---|---|
| DA (% of control) | 65 ± 3* | 100 ± 4 |
| DOPAC (% of control) | 127 ± 17 | 100 ± 11 |
| HVA (% of control) | 140 ± 11 | 100 ± 9 |
| DOPAC/DA ratio | 0.16 ± 0.04* | 0.08 ± 0.02 |
HOE-642 was reverse dialyzed (3 nmol/min) for 2 h. Forty minutes after discontinuation of drug delivery, the mice were killed. Striata were dissected and tissue content of DA, DOPAC, and HVA were measured. Data are expressed as percent of control (untreated side) and are the mean ± SEM (n = 3). Control values (μg/g of tissue) were 14.3 ± 0.5 for DA, 1.1 ± 0.1 for DOPAC, and 0.9 ± 0.1 for HVA. The DOPAC to DA ratio was calculated for both striata in each animal.
Significantly different (p < 0.05) from control side. HOE-642, cariporide mesilate; DA, dopamine; DOPAC, dihydroxyphenylacetic acid.
HOE-642 lacks potent inhibitory effects on synaptosomal DA uptake and MAO activity
The NHE inhibitors of the amiloride family non-selectively inhibit striatal DA re-uptake and the enzyme MAO (Zubieta et al. 1988; Callahan et al. 2001), which might contribute to increased DA overflow in vivo. We tested the ability of HOE-642 to block 3H-DA uptake in striatal synaptosomes (see Table 2). 3H-DA uptake was not significantly inhibited by HOE-642 at concentrations up to 100 μM. In comparison, the IC50 for inhibition of 3H-DA uptake by the amiloride analog EIPA was approximately 3 μM. Nearly complete blockade of 3H-DA uptake was observed with the specific DAT inhibitor mazindol at 1 μM.
Table 2.
Effects of HOE-642 and EIPA on 3H-DA uptake in striatal synaptosomes
| Concentration (μM) |
3H-DA uptake, % of control
|
||
|---|---|---|---|
| HOE-642 | EIPA | Mazindol | |
| 1 | 101 ± 10 | 75 ± 10 | 3 ± 0 |
| 3 | 108 ± 14 | 49 ± 10 | – |
| 10 | 105 ± 11 | 15 ± 6 | 0 |
| 30 | 98 ± 8 | 1 ± 1 | |
| 100 | 92 ± 3 | – | |
Synaptosomal preparations were incubated with varying concentrations of HOE-642 or EIPA and 3H-DA. Data are the mean ± SD (n = 2–4) expressed as the percentage of DA uptake in untreated controls. Mazindol is a potent DAT inhibitor. HOE-642, cariporide mesilate; EIPA, ethylisopropylamiloride; DA, dopamine; DAT, dopamine transporter.
Cariporide mesilate (30–300 μM) was also tested for effects on the activity of MAO-A and -B in striatal tissue homogenates. MAO-A and -B activities were unaffected by HOE-642 at concentrations from 30 to 100 μM. Slight reductions to 80 ± 7% (MAO-A) and 86 ± 1% (MAO-B) of control levels were observed with 300 μM of HOE-642 (mean ± SEM, n = 3–4). Together these data indicate that HOE-642 is not a potent inhibitor of striatal DA uptake or MAO activity.
Striatal delivery of HOE-642 attenuates DA overflow during metabolic stress but does not modify damage to DAergic axon terminals
It has been previously shown that striatal infusion of the mitochondrial inhibitor MAL causes a large DA efflux, which has been correlated with the extent of damage to DAergic axon terminals (Zeevalk et al. 1997; Ferger et al. 1999; Moy et al. 2000). To determine the effects of NHE inhibition on disruption of DA transmission caused by metabolic stress, local delivery of HOE-642 was followed by that of MAL. Intrastriatal dialysis of MAL (1 μmol/min) for 20 min caused a maximal ~270-fold increase in striatal DA overflow relative to baseline levels (Fig. 3a). However, the maximal DA overflow induced by MAL was only ~10-fold above pre-drug baseline levels in animals that were pre-treated with dialysis of HOE-642 (3 nmol/min) for 120 min. Moreover, HOE-642 pre-treatment also attenuated the total DA overflow elicited by MAL in comparison to the control group (Fig. 3a, inset).
Fig. 3.

Effects of striatal HOE-642 pre-treatment on malonate-induced DA and lactate overflow. (a) Striatal DA overflow in 20 min intervals expressed as a percentage of pre-drug baseline levels (y-axis: logarithmic scale) during the reverse microdialysis of HOE-642 (3 nmol/min, hatched box) or malonate (1 μmol/min, filled box). The mito-chondrial inhibitor malonate was applied for 20 min after a 120 min exposure to HOE-642 (■) or aCSF perfusion alone (□). Inset: Total striatal DA overflow elicited by malonate in animals pre-treated with HOE-642 (■) or aCSF (□) (area under the curve, 240–320 min). (b) Striatal levels of lactate (nmol) in 40 min intervals from the same experiment as in (a). All values are mean ± SEM (n = 3–6). There was a statistically significant temporal effect on levels of DA and lactate in both treatment groups (repeated measures one-way ANOVA, p < 0.01 or 0.001). *p < 0.05 relative to the control group (t-test).
To further evaluate whether HOE-642 attenuated the MAL-induced DA efflux because of a reduction in the associated metabolic stress, lactate levels were measured in the same experiments. Previous studies have demonstrated that lactate overflow in the striatum is markedly increased during inhibition of mitochondrial respiration (Rollema et al. 1988). We found that MAL caused a significant increase in striatal lactate overflow that was not modified by HOE-642 pre-treatment (Fig. 3b).
Next we posed the question of whether the observed effects of HOE-642 on MAL-induced DA overflow might modify subsequent damage to DAergic terminals. To that end, the tissue content of DA and TH in the striatum of mice was determined 5–7 days after the microdialysis experiments (see Fig. 4), a length of time sufficient to observe loss of DAergic terminals. Intrastriatal dialysis of HOE-642 alone had no effect on markers of striatal DAergic terminals at this later time point. MAL reduced both DA and TH levels on the dialyzed striatal side by more than 50% of the control untreated side, an effect that was not modified with HOE-642 pre-treatment.
Fig. 4.

Assessment of DAergic terminal damage in the striatum after microdialysis studies. Mice were killed 5–7 days after microdialysis experiments, their striata dissected and homogenized for quantification of DA (a) and TH (b) to assess loss of DAergic terminals. Striatal DA and TH levels are shown for the dialyzed and contralateral untreated sides. All values are mean ± SEM (n = 3–4). ***p < 0.001 and **p < 0.01 relative to the dialyzed side of the HOE/aCSF group (Tukey’s comparison test).
NHE1 protein is located in striatal synaptosomal fractions
Earlier evidence suggests that NHE proteins are important regulators of intracellular pH in axon terminals and might play a role in modulating neurotransmitter release. Western blot was performed on subcellular fractions containing synaptosomes to explore whether the brain abundant NHE1 protein is found at the synaptic level in the mouse striatum (see Fig. 5). The XB-17 anti-NHE1 antibody labeled a single protein band in striatal homogenates that is consistent with the predicted molecular weight of 87–91 kDa for the NHE1 protein (Counillon et al. 1994). The NHE1 signal was further detected in the fraction containing synaptosomes but not in the cytosolic fraction. A similar band distribution pattern was also observed in synaptosomal fractions prepared from mouse cortex and hippocampus (Fig. 5, cortex and hippocampus) which were used as positive controls for NHE1 protein detection in the brain.
Fig. 5.

Western blot characterization of NHE1 protein in the mouse striatum. Subcellular fractions with cytosolic content (S2) and synaptosomes (P2) were prepared from mouse striatum, hippocampus (HPC), and cortex (CTX). Proteins were resolved by 10% SDS–PAGE and detected by western blotting with the anti-NHE1 antibody XB-17 (1 : 1000). The blot shown for striatal samples is representative of three separate experiments (n = 3). A similar band pattern was found in HPC and CTX samples from two separate control experiments. Molecular weight markers are shown on the right.
NHE1 protein is found in the neuropil adjacent to TH-positive neuronal structures
The findings that striatal HOE-642 delivery modifies DA overflow but does not prevent MAL-induced DAergic terminal damage raised the question of whether NHE1 was located on DAergic fibers. Double-label immunofluorescence was performed to explore the association between NHE1 protein and the DAergic neuronal marker TH in coronal sections from adult mouse brain. Confocal microscopy revealed dense NHE1 immunoreactivity in fibrillar and punctate structures adjacent to TH-positive neuronal fibers throughout the striatum [Fig. 6II(A1–2)]. However, examination of serial optical planes showed that NHE1-positive structures were for the most part not superimposed with TH-positive neuron processes in the striatum [Fig. 6II(A3)]. We also examined NHE1 immunoreactivity in relation to DAergic cell bodies in the substantia nigra pars compacta at the level of the ventral midbrain. Consistent with findings in the striatum, NHE1-positive structures were observed in the neuropil with no apparent co-localization with TH-positive neuronal cell bodies or processes [Fig. 6II(B1–3)].
Fig. 6.

Immunofluorescence of NHE1 and TH in the striatum and substantia nigra. Immunofluorescence images of mouse coronal brain sections were obtained with (I) 25×, 0.8 NA or (II) 63×, 1.2 NA objectives on a LSM Zeiss confocal system. (I) Brain sections at the level of the striatum (*) were incubated in (a) the presence or (b) absence of anti-NHE1 antibody. Some non-specific edge effect in (b) was observed in the lining of the lateral ventricle (LV). (II) NHE-1 labeling in the striatum (A) or substantia nigra (B) is shown in green (A1 and B1) and TH labeling in red (A2 and B2). NHE1 labeling appeared to be present in the neuropil (A1 and B1) while TH labeling was found on axonal fibers (A2) and neuronal cell bodies (B2). Overlay of NHE1 and TH labeling (A3 and B3) revealed that most structures expressing NHE1 (arrow) were closely associated but not directly co-localized with those expressing TH (arrowhead).
NHE isoforms 1–5 are expressed in the mouse striatum and ventral midbrain
The apparent absence of NHE1 protein on nigrostriatal DAergic neurons suggests that the observed effects of HOE-642 on DA neurotransmission might be mediated via alternative NHE isoforms. To begin exploring this possibility, RT-PCR was employed to detect mRNA expression of NHE1–5 in the striatum and ventral midbrain (Fig. 7). Gel electrophoresis revealed PCR products of the expected length (~150 bp) for all five NHE isoforms in both regions. These data indicate that multiple NHE isoforms are expressed in the striatum and ventral midbrain of adult mice.
Fig. 7.
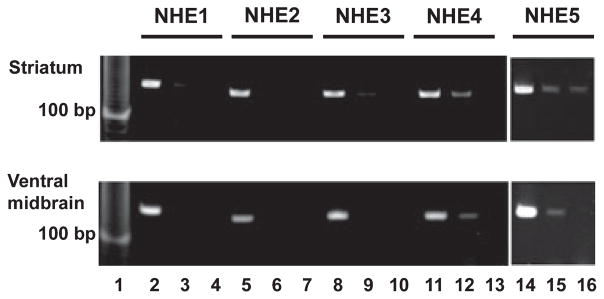
Detection of mRNA expression of NHE isoforms 1–5 in the mouse striatum and ventral midbrain. Striatum (upper panel) and ventral midbrain (lower panel) from mice were dissected for mRNA extraction and RT-PCR detection with specific primer pairs for NHE isoforms 1–5. Lanes 2, 5, 8, 11, and 14 contained reverse transcribed mRNA from each brain region with the respective specific primer sets. Lanes 3, 6, 9, 12, and 15 represent RT-PCR products with the mRNA template and primers in the absence of reverse transcriptase. Lanes 4, 7, 10, 13, and 16 represent primers only. Molecular weight marker is shown in lane 1. Data are representative of two independent experiments.
Discussion
The role of NHE proteins in DA neurotransmission is essentially unknown. We have explored the effects of the NHE inhibitor HOE-642 on striatal DA neurotransmission using in vivo microdialysis. In addition, we have examined mRNA expression of NHE1–5 as well as the location of NHE1 protein in the striatum and midbrain. Intrastriatal delivery of HOE-642 caused an increase followed by a decrease in DA overflow with a concomitant decline in striatal DA content. HOE-642 delivery also attenuated DA overflow induced by the mitochondrial inhibitor MAL without modifying the subsequent DAergic terminal damage. Expression of all five NHE isoforms was detected in the striatum and midbrain. Moreover, NHE1 protein was found in striatal synaptosomes but was not directly co-located with nigrostriatal DAergic neurons. The apparent absence of NHE1 protein on DAergic neurons suggests that the effects of HOE-642 on striatal DA overflow are either mediated via NHE1 located on other cell types or that HOE-642 is acting through NHE isoforms other than NHE1. The latter is a likely possibility because the HOE-642 related effects were seen at concentrations that might inhibit multiple NHE isoforms.
Effects of HOE-642 on striatal DA neurotransmission under normal conditions
The NHE inhibitor HOE-642 belongs to the family of benzoylguanidines that exhibit higher affinities for NHE1, NHE2, and NHE5 (IC50 values of 0.05, 3, and 9.1 μM, respectively), and lower affinities for NHE4 and NHE3 (IC50 values of 250 μM and 1 mM, respectively) (Scholz et al. 1995; Szabo et al. 2000; Luo et al. 2005). Expression of the five NHE isoforms was found in the striatum and ventral midbrain (present study). Based on diffusion characteristics in brain tissue and drug recovery rates in vitro (Nicholson and Rice 1986; Lindefors et al. 1989; Westerink and De Vries 2001), we estimate that reverse microdialysis of HOE-642 (0.3–3 nmol/min) would achieve concentrations of 15–150 μM in the striatum that fall well within the range to block multiple NHE isoforms.
Prolonged HOE-642 delivery caused an early increase followed by a progressive reduction in DA overflow with a concomitant increase in DA turnover and reduced striatal DA content. These findings, which are similar to those seen with the administration of VMAT2 inhibitors such as tetra-benazine (TBZ) or Ro4-1284, indicate a disruption in vesicular DA storage. The inhibition of vesicular DA uptake causes transient accumulation of DA in the cytosol which leads to DAT-mediated DA release, increased intraneuronal DA metabolism and a decrease in pre-synaptic DA stores (Colzi et al. 1993; Moy et al. 2000; Staal and Sonsalla 2000; Andersson et al. 2006). While TBZ and Ro4-1284 directly inhibit the VMAT2 transporter, we propose that HOE-642 disrupts transvesicular ion gradients and indirectly affects vesicular DA storage. This might occur via two non-exclusive mechanisms. First, inhibition of plasma membrane NHE activity on DAergic terminals would cause cytosolic acidification that could reduce the transvesicular pH gradient necessary for adequate DA transport into vesicles (Johnson et al. 1982; Drapeau and Nachshen 1988; Camacho et al. 2006). Secondly, inhibition of any NHE located on synaptic vesicles (Szaszi et al. 2002) could also play a role in the regulation of vesicular volume (via Na+ or K+ influx) as is seen with the organelle NHE isoforms 6–9 (Orlowski and Grinstein 2004; Nakamura et al. 2005). Reduction of vesicular volume is thought to reduce vesicular DA storage (Pothos 2002). While both mechanisms may be in effect, the slower reduction in DA overflow after onset of reverse microdialysis of HOE-642 (> 2 h) compared with the onset of reverse microdialysis of TBZ (30 min, see Andersson et al. 2006) may reflect the need for a sufficient build-up of cytosolic H+ to disrupt vesicular DA uptake.
Although an effect of HOE-642 on vesicular disruption might contribute to the observed changes in DA neurotrans-mission, additional factors are likely to account for the initial large increase in DA overflow. This notion is underscored by the findings that delivery of HOE-642 causes an eightfold increase in DA overflow compared with the maximal effect of TBZ, which elicits only a twofold increase in DA overflow (Andersson et al. 2006). Several potential mechanisms might account for this observation. First, as DA overflow elicited by Ro4 administration is much greater during MAO-A inhibition because of reduced DA degradation (Colzi et al. 1993), inhibition of MAO after HOE-642 delivery could similarly contribute to the increase in DA overflow. However, while amiloride and its analogs inhibit MAO activity (Zubieta et al. 1988), HOE-642 appeared to be a poor inhibitor of either MAO-A or -B in vitro (present findings). Moreover, the significant increases in the overflow of DOPAC and HVA, and DA turnover elicited by HOE-642 indicate a lack of MAO inhibition in vivo.
Another possible mechanism by which HOE-642 could produce elevations in striatal DA overflow is by blockade of DAT and consequent inhibition of extracellular DA re-uptake. This is an important consideration in light of an earlier report that amiloride analogs block striatal synapto-somal 3H-DA uptake (Callahan et al. 2001). However, the present data do not support this hypothesis. HOE-642 did not inhibit 3H-DA uptake into synaptosomes up to 100 μM in contrast to the amiloride analog EIPA that completely blocked 3H-DA uptake at 30 μM (Table 2). In addition, prolonged reverse microdialysis of DAT inhibitors, such as cocaine or nomifensine, causes sustained increases in striatal DA overflow (Tolliver et al. 1999; Westerink and De Vries 2001) in contrast to the HOE-642 mediated bi-phasic effect. Moreover, DAT inhibition protects against MAL-induced damage (Moy et al. 2007), whereas HOE-642 did not provide protection. Hence it is unlikely that HOE-642 modified striatal DA overflow via blockade of extracellular DA re-uptake.
A third possibility is that HOE-642 induces exocytotic DA release in addition to the DAT-mediated DA release, which is typically associated with disruption of vesicular DA stores. This is in accordance with brain synaptosome studies suggesting that NHE inhibition causes exocytotic DA release. Amiloride analogs stimulate synaptosomal 3H-DA efflux that is abolished in Ca2+-free media and is unchanged by DAT inhibition (Cannizzaro et al. 2003), and cytosolic acidification enhances basal DA release in the presence of a DAT inhibitor (Drapeau and Nachshen 1988). Thus, striatal NHE inhibition might trigger exocytotic DA release accompanied by alterations in vesicular DA storage. Such effects invoke the presence of one or more NHE isoforms on the DAergic neurons. Although NHE1 protein was not co-localized on DAergic neurons (Fig. 6), we found that NHE2–5 were also expressed in the striatum and therefore could mediate the effects of HOE-642 on DA release (Fig. 7).
Finally, it is possible that the changes in DA neurotrans-mission seen with HOE-642 also occur via inhibition of NHE activity in non-DAergic cell types in the striatum. For example, NHE activity contributes to H+ extrusion in cultured astrocytes (Kimelberg et al. 1979; Shrode and Putnam 1994; Pizzonia et al. 1996), which actively modulate neurotransmission (Rothstein et al. 1996; Kang et al. 1998). In particular, astrocytes have been implicated in regulating interstitial pH in the brain by secreting acid equivalents (Chesler and Kraig 1989; Chesler 2003), which down-regulate neuronal activity in part via blockade of pre-synaptic voltage-dependent Ca2+ channels (Prod’hom et al. 1989). Although an acid secretory function of glia has been mostly attributed to the Na+-HCO3− cotransporter in other brain regions (Grichtchenko and Chesler 1994; Newman 1996), it is tempting to speculate that NHE inhibition might reduce the ability of astrocytes to secrete H+ surrounding DAergic varicosities, thereby enhancing activity-dependent DA release. The impact of NHE inhibition on corticostriatal projections and striatal interneurons might also affect local release of other transmitters such as glutamate and GABA (Trudeau et al. 1999; Jang et al. 2006) which could indirectly modify nigrostriatal DA activity. While future studies are needed to characterize the cellular mechanisms involved in the HOE-642 induced DA release, our data is consistent with the notion that striatal NHE inhibition triggers a combination of events that contribute to modify DA neurotransmission in vivo.
Effects of HOE-642 on malonate-induced DA overflow and DAergic terminal damage
Earlier studies demonstrated that depletion of striatal DA content (Globus et al. 1987; Ferger et al. 1999; Moy et al. 2000) or administration of DAT inhibitors (Xia et al. 2001; Moy et al. 2007) prior to a metabolic stress attenuate the associated increase in extracellular DA levels and striatal tissue damage. Conversely, disruption of vesicular DA stores during a metabolic stress enhances MAL-induced damage (Albers et al. 1996; Burrows et al. 2000; Nixdorf et al. 2001). Such evidence implies a direct correlation between the extent of DA release during metabolic stress and striatal tissue damage. In the present study, we tested the hypothesis that HOE-642 might protect DAergic terminals against metabolic stress. To eliminate the confounding of HOE-642-induced DA release with MAL-induced toxicity, the mitochondrial inhibitor was infused only after DA overflow had stabilized at or below pre-drug levels. This approach revealed that the MAL-induced DA overflow was attenuated by HOE-642 pre-treatment likely because of the reduced striatal DA pools (Table 1). However, HOE-642 pre-treatment did not modify the MAL-induced DAergic terminal damage, nor did it alter striatal lactate overflow, suggesting that the changes in DA overflow were not because of an attenuation of the metabolic stress per se. While it is not clear how DA itself might contribute to neuronal damage caused by mitochondrial inhibition, one view is that mishandling of striatal DA exacerbates oxidative stress (Hastings et al. 1996; Ferger et al. 1999; Xia et al. 2001). However, the lack of striatal DA terminal protection in mice pre-treated with HOE-642 suggests that the magnitude of DA release during metabolic stress is not predictive of neuronal damage.
The finding that striatal HOE-642 delivery did not modify MAL-induced DA terminal damage is in accordance with previously reported data that systemic EIPA administration does not prevent loss of DAergic terminals caused by the neurotoxin MPTP in mice (Callahan et al. 2001). Together these data are in contrast to studies of animal models of brain ischemia reporting neuroprotection with systemic pre-treatment of HOE-642 or EIPA (Phillis et al. 1999; Castella et al. 2005; Luo et al. 2005). Although such different outcomes might be because of the variation between the experimental models utilized, another possibility is that NHE inhibition does not attenuate DAergic terminal damage because these neurons do not express NHE1.
NHE1 localization and expression of other NHE isoforms in the striatum and midbrain
Na+/H+ exchanger isoform 1 is the most abundant NHE isoform in the brain yet it is differentially expressed across brain regions and neuronal subpopulations (Ma and Haddad 1997; Douglas et al. 2001). The present findings are the first to demonstrate NHE1 protein in the striatum and substantia nigra as well as its localization to striatal synaptosomes. These data indicate that NHE1 is located at the synaptic level, which is in line with earlier demonstrations of amiloride-sensitive NHE activity in whole brain synaptosomes (Sauvaigo et al. 1984; Jean et al. 1985). Together these observations bring further support to the notion that NHE inhibition modifies neurotransmission in general (Trudeau et al. 1999; Jang et al. 2006).
Consistent with our western blot results, immunofluorescence confocal imaging revealed widespread NHE1 labeling in the striatal neuropil. Surprisingly, double-label immunofluorescence revealed that NHE1 signal was predominantly found in TH-negative cell processes. These data indicate that nigrostriatal DAergic neurons express low levels of NHE1 protein, if any, in relation to neighboring subpopulations of cells. These findings do not exclude the possibility that NHE1 protein is found in nigrostriatal DAergic fibers at levels below the limits of our detection method. However, the apparent lack of co-location of NHE1 on nigrostriatal DAergic neurons is consistent with the observation that striatal HOE-642 delivery does not protect DAergic terminals against metabolic stress as it does against ischemic damage to other neuronal populations (Luo et al. 2005).
The differential localization of NHE1 between DAergic and non-DAergic cells in the adult mouse brain is in agreement with a comprehensive demonstration of uneven NHE1 expression in other rat brain regions such as the cerebellum, where NHE1 mRNA signal was found concentrated in the granule and Purkinje cells compared with very weak signals in the cells of the molecular layer (Ma and Haddad 1997). Absence of NHE1 immunoreactivity in epithelial cells of the human choroid plexus has also been reported (Praetorius and Nielsen, 2006). Hence, evidence from our study and others underscores the differential expression of NHE1 protein across subpopulations of cells throughout the brain. Interestingly, both NHE1 activity and gene expression are, respectively, reduced by reactive oxygen species and H2O2 (Mulkey et al. 2004; Kumar et al. 2007). Given the high level of oxidative stress in nigrostriatal DAergic neurons, it is possible that NHE1 expression or activity might be reduced in these cells.
The presence of NHE activity in many other neuronal subtypes (Chesler 2003) in contrast to the apparent absence of NHE1 location on DAergic neurons in our studies would suggest that DAergic neurons express alternative NHE isoforms. In addition to NHE1, we have documented mRNA expression of NHE2–5 in the striatum and ventral midbrain. Future studies are needed to characterize which NHE isoforms are found on nigrostriatal DAergic neurons. A likely candidate seems to be the brain specific NHE5 as it has been localized in hippocampal neuronal processes and speculated to be involved in modulating neurotransmission (Baird et al. 1999; Szaszi et al. 2002). Localization studies of NHE isoforms in non-DAergic cells in the striatum and substantia nigra will also be important to elucidate the role of NHE proteins in DA neurotransmission under physiological and pathological conditions.
Acknowledgments
This work was supported by the National Institutes of Health Grants (NS052733 and ES0050221) and also made possible thanks to the generous gifts of the XB-17 antibody from Dr Mark Mush (University of Chicago) and the compound HOE-642 (Sanofi-Aventis). The authors thank Mr Lawrence Manzino and Ms Lindsay Leveille for their technical support. We also thank Dr Gail Zeevalk for helpful discussions as well as Dr Noriko Kane-Goldsmith (Keck Center for Collaborative Neuroscience, Rutgers University) for assistance with laser scanning confocal microscopy.
Abbreviations used
- aCSF
artificial CSF
- DA
dopamine
- DAT
dopamine transporter
- DOPAC
dihydroxyphenylacetic acid
- EIPA
ethylisopropylamiloride
- HOE-642
cariporide mesilate
- HRP
horseradish peroxidase
- HVA
homovanillic acid
- MAL
malonate
- MAO
monoamine oxidase
- NGS
normal goat serum
- NHE
Na+/H+ exchanger
- PB
phosphate buffer
- PBS-T
PBS containing 0.3% Triton X-100
- TBZ
tetrabenazine
- TH
tyrosine hydroxylase
- VMAT2
vesicular monoamine transporter-2
References
- Albers DS, Zeevalk GD, Sonsalla PK. Damage to dopaminergic nerve terminals in mice by combined treatment of intrastriatal malonate with systemic methamphetamine or MPTP. Brain Res. 1996;718:217–220. doi: 10.1016/0006-8993(96)00135-7. [DOI] [PubMed] [Google Scholar]
- Alfinito PD, Wang SP, Manzino L, Rijhsinghani S, Zeevalk GD, Sonsalla PK. Adenosinergic protection of dopaminergic and GABAergic neurons against mitochondrial inhibition through receptors located in the substantia nigra and striatum, respectively. J Neurosci. 2003;23:10982–10987. doi: 10.1523/JNEUROSCI.23-34-10982.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoroso S, Taglialatela M, Canzoniero LM, Cragoe EJ, Jr, di Renzo G, Annunziato L. Possible involvement of Ca++ ions, protein kinase C and Na(+)-H+ antiporter in insulin-induced endogenous dopamine release from tuberoinfundibular neurons. Life Sci. 1990;46:885–894. doi: 10.1016/0024-3205(90)90119-c. [DOI] [PubMed] [Google Scholar]
- Andersson DR, Nissbrandt H, Bergquist F. Partial depletion of dopamine in substantia nigra impairs motor performance without altering striatal dopamine neurotransmission. Eur J Neurosci. 2006;24:617–624. doi: 10.1111/j.1460-9568.2006.04953.x. [DOI] [PubMed] [Google Scholar]
- Attaphitaya S, Park K, Melvin JE. Molecular cloning and functional expression of a rat Na+/H+ exchanger (NHE5) highly expressed in brain. J Biol Chem. 1999;274:4383–4388. doi: 10.1074/jbc.274.7.4383. [DOI] [PubMed] [Google Scholar]
- Baird NR, Orlowski J, Szabo EZ, Zaun HC, Schultheis PJ, Menon AG, Shull GE. Molecular cloning, genomic organization, and functional expression of Na+/H+ exchanger isoform 5 (NHE5) from human brain. J Biol Chem. 1999;274:4377–4382. doi: 10.1074/jbc.274.7.4377. [DOI] [PubMed] [Google Scholar]
- Burrows KB, Nixdorf WL, Yamamoto BK. Central administration of methamphetamine synergizes with metabolic inhibition to deplete striatal monoamines. J Pharmacol Exp Ther. 2000;292:853–860. [PubMed] [Google Scholar]
- Buyukuysal RL, Mete B. Anoxia-induced dopamine release from rat striatal slices: involvement of reverse transport mechanism. J Neurochem. 1999;72:1507–1515. doi: 10.1046/j.1471-4159.1999.721507.x. [DOI] [PubMed] [Google Scholar]
- Callahan BT, Cord BJ, Yuan J, McCann UD, Ricaurte GA. Inhibitors of Na(+)/H(+) and Na(+)/Ca(2+) exchange potentiate methamphetamine-induced dopamine neurotoxicity: possible role of ionic dysregulation in methamphetamine neurotoxicity. J Neurochem. 2001;77:1348–1362. doi: 10.1046/j.1471-4159.2001.00341.x. [DOI] [PubMed] [Google Scholar]
- Camacho M, Machado JD, Montesinos MS, Criado M, Borges R. Intragranular pH rapidly modulates exocytosis in adrenal chromaffin cells. J Neurochem. 2006;96:324–334. doi: 10.1111/j.1471-4159.2005.03526.x. [DOI] [PubMed] [Google Scholar]
- Cannizzaro C, Monastero R, Vacca M, Martire M. [3H]-DA release evoked by low pH medium and internal H+ accumulation in rat hypothalamic synaptosomes: involvement of calcium ions. Neurochem Int. 2003;43:9–17. doi: 10.1016/s0197-0186(02)00211-5. [DOI] [PubMed] [Google Scholar]
- Castella M, Buckberg GD, Tan Z. Neurologic preservation by Na+-H+ exchange inhibition prior to 90 minutes of hypothermic circulatory arrest. Ann Thorac Surg. 2005;79:646–654. doi: 10.1016/j.athoracsur.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kraig RP. Intracellular pH transients of mammalian astrocytes. J Neurosci. 1989;9:2011–2019. doi: 10.1523/JNEUROSCI.09-06-02011.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colzi A, D’Agostini F, Cesura A, Borroni E, Da Prada M. Monoamine oxidase-A inhibitors and dopamine metabolism in rat caudatus: evidence that an increased cytosolic level of dopamine displaces reversible monoamine oxidase-A inhibitors in vivo. J Pharmacol Exp Ther. 1993;265:103–111. [PubMed] [Google Scholar]
- Counillon L, Pouyssegur J, Reithmeier RA. The Na+/H+ exchanger NHE-1 possesses N- and O-linked glycosylation restricted to the first N-terminal extracellular domain. Biochemistry. 1994;33:10463–10469. doi: 10.1021/bi00200a030. [DOI] [PubMed] [Google Scholar]
- Coupaye-Gerard B, Bookstein C, Duncan P, Chen XY, Smith PR, Musch M, Ernst SA, Chang EB, Kleyman TR. Biosynthesis and cell surface delivery of the NHE1 isoform of Na+/H+ exchanger in A6 cells. Am J Physiol. 1996;271:C1639–C1645. doi: 10.1152/ajpcell.1996.271.5.C1639. [DOI] [PubMed] [Google Scholar]
- Douglas RM, Schmitt BM, Xia Y, Bevensee MO, Biemesderfer D, Boron WF, Haddad GG. Sodium-hydrogen exchangers and sodium-bicarbonate co-transporters: ontogeny of protein expression in the rat brain. Neuroscience. 2001;102:217–228. doi: 10.1016/s0306-4522(00)00473-5. [DOI] [PubMed] [Google Scholar]
- Drapeau P, Nachshen DA. Effects of lowering extracellular and cytosolic pH on calcium fluxes, cytosolic calcium levels, and transmitter release in presynaptic nerve terminals isolated from rat brain. J Gen Physiol. 1988;91:305–315. doi: 10.1085/jgp.91.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferger B, Eberhardt O, Teismann P, de Groote C, Schulz JB. Malonate-induced generation of reactive oxygen species in rat striatum depends on dopamine release but not on NMDA receptor activation. J Neurochem. 1999;73:1329–1332. doi: 10.1046/j.1471-4159.1999.0731329.x. [DOI] [PubMed] [Google Scholar]
- Globus MY, Ginsberg MD, Dietrich WD, Busto R, Scheinberg P. Substantia nigra lesion protects against ischemic damage in the striatum. Neurosci Lett. 1987;80:251–256. doi: 10.1016/0304-3940(87)90463-0. [DOI] [PubMed] [Google Scholar]
- Grichtchenko II, Chesler M. Depolarization-induced acid secretion in gliotic hippocampal slices. Neuroscience. 1994;62:1057–1070. doi: 10.1016/0306-4522(94)90343-3. [DOI] [PubMed] [Google Scholar]
- Haas RH, Nasirian F, Nakano K, Ward D, Pay M, Hill R, Shults CW. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann Neurol. 1995;37:714–722. doi: 10.1002/ana.410370604. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan KA, Staal RG, Sonsalla PK. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: disparity between homogenates and vesicle preparations. J Neurochem. 2000;74:2217–2220. doi: 10.1046/j.1471-4159.2000.0742217.x. [DOI] [PubMed] [Google Scholar]
- Jang IS, Brodwick MS, Wang ZM, Jeong HJ, Choi BJ, Akaike N. The Na+/H+ exchanger is a major pH regulator in GABAergic presynaptic nerve terminals synapsing onto rat CA3 pyramidal neurons. J Neurochem. 2006;99:1224–1236. doi: 10.1111/j.1471-4159.2006.04168.x. [DOI] [PubMed] [Google Scholar]
- Jean T, Frelin C, Vigne P, Barbry P, Lazdunski M. Biochemical properties of the Na+/H+ exchange system in rat brain synaptosomes. Interdependence of internal and external pH control of the exchange activity. J Biol Chem. 1985;260:9678–9684. [PubMed] [Google Scholar]
- Johnson RG, Carty S, Scarpa A. A model of biogenic amine accumulation into chromaffin granules and ghosts based on coupling to the electrochemical proton gradient. Fed Proc. 1982;41:2746–2754. [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kilty JE, Lorang D, Amara SG. Cloning and expression of a cocaine-sensitive rat dopamine transporter. Science. 1991;254:578–579. doi: 10.1126/science.1948035. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Biddlecome S, Bourke RS. SITS-inhibitable Cl− transport and Na+-dependent H+ production in primary astroglial cultures. Brain Res. 1979;173:111–124. [PubMed] [Google Scholar]
- Kumar AP, Chang MK, Fliegel L, Pervaiz S, Clement MV. Oxidative repression of NHE1 gene expression involves iron-mediated caspase activity. Cell Death Differ. 2007;14:1733–1746. doi: 10.1038/sj.cdd.4402176. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kim T, Park ES, Yang S, Jeong D, Choi Y, Rho J. NHE10, a novel osteoclast-specific member of the Na+/H+ exchanger family, regulates osteoclast differentiation and survival. Biochem Biophys Res Commun. 2008;369:320–326. doi: 10.1016/j.bbrc.2008.01.168. [DOI] [PubMed] [Google Scholar]
- Lindefors N, Amberg G, Ungerstedt U. Intracerebral microdialysis: I. Experimental studies of diffusion kinetics. J Pharmacol Methods. 1989;22:141–156. doi: 10.1016/0160-5402(89)90011-9. [DOI] [PubMed] [Google Scholar]
- Luo J, Chen H, Kintner DB, Shull GE, Sun D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J Neurosci. 2005;25:11256–11268. doi: 10.1523/JNEUROSCI.3271-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma E, Haddad GG. Expression and localization of Na+/H+ exchangers in rat central nervous system. Neuroscience. 1997;79:591–603. doi: 10.1016/s0306-4522(96)00674-4. [DOI] [PubMed] [Google Scholar]
- Makani S, Chesler M. Endogenous alkaline transients boost postsynaptic NMDA receptor responses in hippocampal CA1 pyramidal neurons. J Neurosci. 2007;27:7438–7446. doi: 10.1523/JNEUROSCI.2304-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwine RL, Babnigg G, Musch MW, Chang EB, Villereal ML. Expression and phosphorylation of NHE1 in wild-type and transformed human and rodent fibroblasts. J Cell Physiol. 1994;161:351–357. doi: 10.1002/jcp.1041610220. [DOI] [PubMed] [Google Scholar]
- Moy LY, Zeevalk GD, Sonsalla PK. Role for dopamine in malonate-induced damage in vivo in striatum and in vitro in mesencephalic cultures. J Neurochem. 2000;74:1656–1665. doi: 10.1046/j.1471-4159.2000.0741656.x. [DOI] [PubMed] [Google Scholar]
- Moy LY, Wang SP, Sonsalla PK. Mitochondrial stress-induced dopamine efflux and neuronal damage by malonate involves the dopamine transporter. J Pharmacol Exp Ther. 2007;320:747–756. doi: 10.1124/jpet.106.110791. [DOI] [PubMed] [Google Scholar]
- Mulkey DK, Henderson RA, III, Ritucci NA, Putnam RW, Dean JB. Oxidative stress decreases pHi and Na+/H+ exchange and increases excitability of solitary complex neurons from rat brain slices. Am J Physiol Cell Physiol. 2004;286:C940–C951. doi: 10.1152/ajpcell.00323.2003. [DOI] [PubMed] [Google Scholar]
- Nachshen DA, Drapeau P. The regulation of cytosolic pH in isolated presynaptic nerve terminals from rat brain. J Gen Physiol. 1988;91:289–303. doi: 10.1085/jgp.91.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Tanaka S, Teko Y, Mitsui K, Kanazawa H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem. 2005;280:1561–1572. doi: 10.1074/jbc.M410041200. [DOI] [PubMed] [Google Scholar]
- Newman E. Acid efflux from retinal glial cells generated by sodium bicarbonate cotransport. J Neurosci. 1996;16:159–168. doi: 10.1523/JNEUROSCI.16-01-00159.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Rice ME. The migration of substances in the neuronal microenvironment. Ann NY Acad Sci. 1986;481:55–71. doi: 10.1111/j.1749-6632.1986.tb27139.x. [DOI] [PubMed] [Google Scholar]
- Nicklas WJ, Browning ET. Amino acid metabolism in glial cells: homeostatic regulation of intra- and extracellular milieu by C-6 glioma cells. J Neurochem. 1978;30:955–963. doi: 10.1111/j.1471-4159.1978.tb12387.x. [DOI] [PubMed] [Google Scholar]
- Nixdorf WL, Burrows KB, Gudelsky GA, Yamamoto BK. Enhancement of 3,4-methylenedioxymethamphetamine neurotoxicity by the energy inhibitor malonate. J Neurochem. 2001;77:647–654. doi: 10.1046/j.1471-4159.2001.00262.x. [DOI] [PubMed] [Google Scholar]
- Orlowski J, Grinstein S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 2004;447:549–565. doi: 10.1007/s00424-003-1110-3. [DOI] [PubMed] [Google Scholar]
- Parker WD, Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- Pasternack M, Smirnov S, Kaila K. Proton modulation of functionally distinct GABAA receptors in acutely isolated pyramidal neurons of rat hippocampus. Neuropharmacology. 1996;35:1279–1288. doi: 10.1016/s0028-3908(96)00075-5. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, King SA, Rigor RR, Zhuang Z, Warren JM, Cala PM. Molecular cloning of NHE1 from winter flounder RBCs: activation by osmotic shrinkage, cAMP, and calyculin A. Am J Physiol Cell Physiol. 2003;284:C1561–C1576. doi: 10.1152/ajpcell.00562.2002. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, O’Donnell ME, Anderson SE, Cala PM. Physiology and pathophysiology of Na+/H+ exchange and Na+-K+-2Cl− cotransport in the heart, brain, and blood. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1–R25. doi: 10.1152/ajpregu.00782.2005. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Estevez AY, Guyot LL, O’Regan MH. 5-(N-Ethyl-N-isopropyl)-amiloride, an Na(+)-H(+) exchange inhibitor, protects gerbil hippocampal neurons from ischemic injury. Brain Res. 1999;839:199–202. doi: 10.1016/s0006-8993(99)01705-9. [DOI] [PubMed] [Google Scholar]
- Pizzonia JH, Ransom BR, Pappas CA. Characterization of Na+/H+ exchange activity in cultured rat hippocampal astrocytes. J Neurosci Res. 1996;44:191–198. doi: 10.1002/(SICI)1097-4547(19960415)44:2<191::AID-JNR12>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Pothos EN. Regulation of dopamine quantal size in midbrain and hippocampal neurons. Behav Brain Res. 2002;130:203–207. doi: 10.1016/s0166-4328(01)00419-3. [DOI] [PubMed] [Google Scholar]
- Pothos EN, Larsen KE, Krantz DE, Liu Y, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius J, Nielsen S. Distribution of sodium transporters and aquaporin-1 in the human choroid plexus. Am J Physiol Cell Physiol. 2006;291:59–67. doi: 10.1152/ajpcell.00433.2005. [DOI] [PubMed] [Google Scholar]
- Prod’hom B, Pietrobon D, Hess P. Interactions of protons with single open L-type calcium channels. Location of protonation site and dependence of proton-induced current fluctuations on concentration and species of permeant ion. J Gen Physiol. 1989;94:23–42. doi: 10.1085/jgp.94.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollema H, Kuhr WG, Kranenborg G, De Vries J, Van den Berg C. MPP+-induced efflux of dopamine and lactate from rat striatum have similar time courses as shown by in vivo brain dialysis. J Pharmacol Exp Ther. 1988;245:858–866. [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Sandoval V, Riddle EL, Hanson GR, Fleckenstein AE. Methylphenidate redistributes vesicular monoamine transporter-2: role of dopamine receptors. J Neurosci. 2002;22:8705–8710. doi: 10.1523/JNEUROSCI.22-19-08705.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MS, Moreno AJ, Carvalho AP. Relationships between ATP depletion, membrane potential, and the release of neurotransmitters in rat nerve terminals. An in vitro study under conditions that mimic anoxia, hypoglycemia, and ischemia. Stroke. 1996;27:941–950. doi: 10.1161/01.str.27.5.941. [DOI] [PubMed] [Google Scholar]
- Sauvaigo S, Vigne P, Frelin C, Lazdunski M. Identification of an amiloride sensitive Na+/H+ exchange system in brain synaptosomes. Brain Res. 1984;301:371–374. doi: 10.1016/0006-8993(84)91106-5. [DOI] [PubMed] [Google Scholar]
- Scholz W, Albus U, Counillon L, Gogelein H, Lang HJ, Linz W, Weichert A, Scholkens BA. Protective effects of HOE642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res. 1995;29:260–268. [PubMed] [Google Scholar]
- Shrode LD, Putnam RW. Intracellular pH regulation in primary rat astrocytes and C6 glioma cells. Glia. 1994;12:196–210. doi: 10.1002/glia.440120305. [DOI] [PubMed] [Google Scholar]
- Staal RG, Sonsalla PK. Inhibition of brain vesicular monoamine transporter (VMAT2) enhances 1-methyl-4-phenyl-pyridinium neurotoxicity in vivo in rat striata. J Pharmacol Exp Ther. 2000;293:336–342. [PubMed] [Google Scholar]
- Szabo EZ, Numata M, Shull GE, Orlowski J. Kinetic and pharmacological properties of human brain Na+/H+ exchanger isoform 5 stably expressed in Chinese hamster ovary cells. J Biol Chem. 2000;275:6302–6307. doi: 10.1074/jbc.275.9.6302. [DOI] [PubMed] [Google Scholar]
- Szaszi K, Paulsen A, Szabo EZ, Numata M, Grinstein S, Orlowski J. Clathrin-mediated endocytosis and recycling of the neuron-specific Na+/H+ exchanger NHE5 isoform: regulation by phosphatidylinositol 3′-kinase and the actin cytoskeleton. J Biol Chem. 2002;277:42623–42632. doi: 10.1074/jbc.M206629200. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Dixon DB, Copenhagen DR. Modulation of a sustained calcium current by intracellular pH in horizontal cells of fish retina. J Gen Physiol. 1993;101:695–714. doi: 10.1085/jgp.101.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CM, Dichter M, Morad M. Modulation of the N-methyl-D-aspartate channel by extracellular H+ Proc Natl Acad Sci USA. 1990;87:6445–6449. doi: 10.1073/pnas.87.16.6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolliver BK, Newman AH, Katz JL, Ho LB, Fox LM, Hsu K, Jr, Berger SP. Behavioral and neurochemical effects of the dopamine transporter ligand 4-chlorobenztropine alone and in combination with cocaine in vivo. J Pharmacol Exp Ther. 1999;289:110–122. [PubMed] [Google Scholar]
- Trudeau LE, Parpura V, Haydon PG. Activation of neurotransmitter release in hippocampal nerve terminals during recovery from intracellular acidification. J Neurophysiol. 1999;81:2627–2635. doi: 10.1152/jn.1999.81.6.2627. [DOI] [PubMed] [Google Scholar]
- Vornov JJ, Thomas AG, Jo D. Protective effects of extracellular acidosis and blockade of sodium/hydrogen ion exchange during recovery from metabolic inhibition in neuronal tissue culture. J Neurochem. 1996;67:2379–2389. doi: 10.1046/j.1471-4159.1996.67062379.x. [DOI] [PubMed] [Google Scholar]
- Westerink BH, De Vries JB. A method to evaluate the diffusion rate of drugs from a microdialysis probe through brain tissue. J Neurosci Methods. 2001;109:53–58. doi: 10.1016/s0165-0270(01)00401-0. [DOI] [PubMed] [Google Scholar]
- Xia XG, Schmidt N, Teismann P, Ferger B, Schulz JB. Dopamine mediates striatal malonate toxicity via dopamine transporter-dependent generation of reactive oxygen species and D2 but not D1 receptor activation. J Neurochem. 2001;79:63–70. doi: 10.1046/j.1471-4159.2001.00525.x. [DOI] [PubMed] [Google Scholar]
- Xue J, Douglas RM, Zhou D, Lim JY, Boron WF, Haddad GG. Expression of Na+/H+ and HCO3−-dependent transporters in Na+/H+ exchanger isoform 1 null mutant mouse brain. Neuroscience. 2003;122:37–46. doi: 10.1016/s0306-4522(03)00598-0. [DOI] [PubMed] [Google Scholar]
- Yao H, Ma E, Gu XQ, Haddad GG. Intracellular pH regulation of CA1 neurons in Na(+)/H(+) isoform 1 mutant mice. J Clin Invest. 1999;104:637–645. doi: 10.1172/JCI6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Hoppe J, Sonsalla P. In vivo vulnerability of dopamine neurons to inhibition of energy metabolism. Eur J Pharmacol. 1997;320:111–119. doi: 10.1016/s0014-2999(96)00892-8. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Cragoe EJ, Jr, Cubeddu LX. Effects of amiloride and 5-(N, N-hexamethylene)amiloride on dopaminergic and cholinergic neurotransmission. J Pharmacol Exp Ther. 1988;247:88–95. [PubMed] [Google Scholar]