

Abstract
Helicases are motor proteins that use the chemical energy of NTP hydrolysis to drive mechanical processes such as translocation and nucleic acid strand separation. Bacteriophage T7 helicase functions as a hexameric ring to drive the replication complex by separating the DNA strands during genome replication. Our studies show that T7 helicase unwinds DNA with a low processivity, and the results indicate that the low processivity is due to ring opening and helicase dissociating from the DNA during unwinding. We have measured the single-turnover kinetics of DNA unwinding and globally fit the data to a modified stepping model to obtain the unwinding parameters. The comparison of the unwinding properties of T7 helicase with its translocation properties on single-stranded (ss)DNA has provided insights into the mechanism of strand separation that is likely to be general for ring helicases. T7 helicase unwinds DNA with a rate of 15 bp/s, which is 9-fold slower than the translocation speed along ssDNA. T7 helicase is therefore primarily an ssDNA translocase that does not directly destabilize duplex DNA. We propose that T7 helicase achieves DNA unwinding by its ability to bind ssDNA because it translocates unidirectionally, excluding the complementary strand from its central channel. The results also imply that T7 helicase by itself is not an efficient helicase and most likely becomes proficient at unwinding when it is engaged in a replication complex.
Helicases are ubiquitous proteins that are involved in various DNA and RNA metabolic processes that require the separation of double-stranded (ds)DNA into single strands, the removal of secondary structures in RNA, or the dissociation of proteins from nucleic acids (1-4). To perform these functions, helicases use the chemical energy from NTP hydrolysis to drive the mechanical processes of translocation and nucleic acid strand separation. In this paper, we study the mechanism of DNA unwinding by bacteriophage T7 helicase that is involved in DNA replication.
During replication, the helicase has to unwind a long stretch of DNA, and that requires the helicase to couple strand-separation activity to translocation. The mechanisms of these critical processes of the helicase reaction are largely unknown. It is becoming evident that helicases can move unidirectionally along nucleic acid and displace bonded moieties along their path without specifically interacting with these moieties (5-8). Thus, unidirectional translocation is a basic activity that helicases can perform without requiring interactions with the duplex DNA. Nucleic acid strand separation is a thermodynamically unfavorable process and it is made feasible by the binding of the helicase to the newly unwound strands. Numerous mechanisms of unwinding have been proposed (2-4, 9-13), but additional experimental data are needed to distinguish between these mechanisms. For the monomeric or dimeric helicases such as the Escherichia coli PcrA and Rep helicases (14-16), it has been proposed that unwinding occurs by an active mechanism where the helicase binds and destabilizes the duplex region, although the exact mechanism of DNA destabilization is not understood. If in addition to binding the single-stranded (ss)DNA, the helicase lowers the transition state energy for base pair melting, the mechanism is defined as active (2). If the helicase does not lower the energy barrier of base pair separation, the mechanism is classified as passive. The rate of spontaneous base pair opening is fast (17) and it has been shown that a 5-bp hairpin loop can open at rates >3,000 s-1 (18), which exceeds the rate of translocation and unwinding by any helicase (19-24). Thus, it would be difficult to distinguish between active and passive helicase mechanisms simply from the kinetics of unwinding. One can, however, compare the kinetics of unwinding with the kinetics of translocation along ssDNA, because the latter process occurs unimpeded by the duplex DNA, to probe the mechanism of helicases.
Structurally, helicases can be divided into two classes, depending on whether they form a hexamer ring (3). Viruses, bacteria, and eukaryotes encode hexameric ring helicases that are involved in cellular processes such as DNA replication, recombination, and transcription termination. Bacteriophage T7 encodes a 63-kDa primase-helicase protein (gp4A′), which is a paradigm for studying the mechanisms of ring helicases. Gp4A′, referred to as T7 helicase, assembles into a ring-shaped hexamer in the presence of nucleotides such as dTTP, deoxythymidine 5′-diphosphate, and deoxythymidine 5′-β,γ-methylenetriphosphate (25, 26). Crystal structures show that the nucleotide binds at the hexamer subunit interface (27, 28). T7 helicase binds preferentially to ssDNA (29) within the central channel of the ring (26), a DNA-binding mode that appears to be general to ring helicases. T7 helicase translocates unidirectionally along ssDNA with a rate of 130 nt/s (at 18°C) (30). The unidirectional movement of the helicase is fueled by dTTP hydrolysis, and T7 helicase hydrolyzes on an average one dTTP per 3-nt movement along ssDNA. To unwind a duplex DNA, T7 helicase, like other ring helicases, requires two noncomplementary tails at one end of the DNA. During DNA unwinding, T7 helicase ring surrounds the 5′ strand and excludes the 3′ strand from its central channel (31, 32). No DNA unwinding is observed when the ring helicase surrounds both strands of the DNA (6, 33, 34).
In this paper, we study the DNA-unwinding reaction catalyzed by T7 helicase at different dTTP concentrations. We measure the single-turnover kinetics of DNA unwinding and propose a modified stepping model that allows global fitting of the unwinding kinetics to obtain the helicase's kinetic step size, stepping rate, and processivity. We show that T7 helicase has a low processivity of DNA unwinding and we discuss its origin. We measure the translocation rate of T7 helicase while unwinding a duplex DNA. Comparison of the kinetics of translocation along ssDNA and dsDNA provides important insights into the mechanism by which ring helicases catalyze DNA unwinding. Rather than interacting with and destabilizing the dsDNA like monomeric or dimeric helicases, we propose that T7 helicase unwinds DNA by its ability to bind ssDNA and to translocate unidirectionally along ssDNA.
Materials and Methods
Protein and Buffers. The gp4A′ (T7 helicase) is an M64L mutant of T7 helicase-primase protein that was overexpressed in E. coli and purified as described (35, 36). T7 helicase concentration was determined from its absorbance at 280 nm in 8 M urea by using an extinction coefficient of 76,100 M-1·cm-1. Buffer A [50 mM Tris·HCl (pH 7.6)/40 mM NaCl/10%(vol/vol) glycerol] was used in all of the experiments unless specified otherwise. Quenching solution consisted of 100 mM EDTA, 0.4% (vol/vol) SDS, and 20% glycerol.
Oligodeoxynucleotides. The unmodified and carboxytetramethylrhodamine (TAMRA)-modified DNAs were purchased from Integrated DNA Technologies (Coralville, IA). The Cy5-modified DNA was purchased from Synthegen (Houston). The DNAs were purified on a 15% polyacrylamide gel/8 M urea, the major DNA band was excised, and the DNA was electroeluted by using an Elutrap apparatus (Schleicher & Schuell). The DNA concentration was determined from its absorbance at 260 nm in buffer containing 8 M urea. The helicase substrate was prepared by mixing the radiolabeled 5′ strand with a 1.5-fold excess of the complementary nonradiolabeled 3′ strand. The DNA mixture was heated at 95°C for 5 min and allowed to slowly cool down to room temperature to form the dsDNA.
Single-Turnover Kinetics of DNA Unwinding. A solution containing T7 helicase (20 nM hexamer), EDTA (5 mM), dTTP (2 mM), and 5′ 32P-labeled forked DNA (5 nM) was rapidly mixed with an equal volume of MgCl2 (13 mM), dTTP (2 mM), and trap DNA (3 μM of unlabeled 5′ strand) in a rapid chemical quench flow instrument at 18°C (RQF3, KinTek, State College, PA). After predetermined times, the reactions were stopped with the quenching solution. The concentration of free MgCl2 in the reactions was 2 mM. Therefore, when experiments were carried out at 200 μM dTTP, the second syringe contained 9.4 mM MgCl2. The dsDNA and ssDNA were resolved by electrophoresis on a 4-20% gradient nondenaturing polyacrylamide gel. The radioactivity was quantified by using a Molecular Dynamics PhosphorImager. The fraction of ssDNA was calculated as described (31).
DNA Unwinding by Fluorescence Resonance Energy Transfer (FRET). T7 helicase (50 nM hexamer), EDTA (5 mM), dTTP, and the TAMRA-Cy5 labeled ds60 DNA (20 nM) were loaded into one syringe of the stopped-flow instrument (KinTek) at 18°C. The reactions were started by rapidly mixing with an equal volume of a solution containing MgCl2 and dT100 trap DNA (2.5 μM). The given concentrations are the final ones after mixing, and the free Mg(II) concentration was 2 mM. The samples were excited at 555 nm, and donor and acceptor fluorescence were measured with a 580 ± 10 band-pass filter and a long-pass filter (LP 665 filter, Oriel), respectively, with two photomultiplier detectors. The experiment was carried out at dTTP from 10 to 2,000 μM, and the rate versus [dTTP] was fit to a hyperbola to obtain the dTTP Km.
Data Analysis. The incomplete γ-function equation (Eq. 1) was used to fit the unwinding kinetics obtained by the radiometric assay.
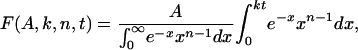 |
1 |
where F is a fraction of unwound DNA substrate molecules, A is the amplitude of unwinding, k is the stepping rate, and t is the reaction time. The number of steps, n, taken by the helicase to unwind the substrate was calculated from Eq. 2.
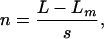 |
2 |
where L is the number of base pairs in the DNA substrate, Lm is the length of the shortest DNA duplex that can stay together under the experimental conditions, and s is the step size. matlab software with the Optimization toolbox (Mathworks, Natick, MA) was used for all of the calculations. Processivity per step (P) was determined from the plot of amplitude (A) versus dsDNA length (l = L - Lm) using Eq. 3 as described (22).
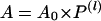 |
3 |
A0 is the maximum amplitude that provides a measure of the fraction of helicase-DNA complex initially present in the reaction.
Results and Discussion
Single-Turnover Kinetics of DNA Unwinding. T7 helicase requires two ssDNA tails at one end of the DNA (fork DNA) to unwind the duplex region (31, 37). A series of fork DNAs with 18- to 90-bp duplex regions (ds18 to ds90) were prepared and each contained a dT35 5′ tail and a dT15 3′ tail (Table 1). The unwinding kinetics were measured under single-turnover conditions using the radiometric assay (22) at 2 mM and 200 μM dTTP. The helicase was preincubated with the fork DNA in the presence of dTTP without Mg(II) to preform the helicase-DNA complex. The reaction was initiated by the addition of Mg(II) and a trap. The 5′ strand of the fork DNA was used as a trap for binding up free protein and to prevent the reannealing of the unwound radiolabeled strand. Other DNAs such as dT80, dT100, or ssM13 were efficient protein traps as well. We verified that the trap did not perturb the unwinding kinetics. The initial time course of unwinding was the same with or without the trap, although without the trap almost all DNAs were unwound, whereas in the presence of the trap, only a fraction were unwound (data not shown).
Table 1.
Helicase DNA substrates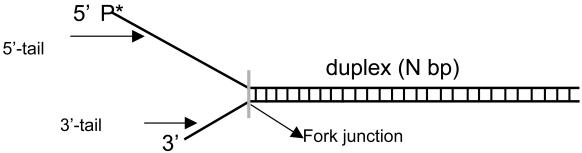
| Substrate | Structure | 5′ Strand sequence |
|---|---|---|
| ds18 |  |
5′T35GAGCGGATTACTATACTA |
| ds30 |  |
5′T35GAGCGGATTACTATACTACATTAGAATTCA |
| ds40 |  |
5′T35GAGCGGATTACTATACTACATTAGAATTCAGAGTGTAGAG |
| ds60 |  |
5′T35GAGCGGATTACTATACTACATTAGAATTCAGAGTGTAGAGATTCGGTAAGTAGGATCATG |
| ds90 |  |
5′T35GAGCGGATTACTATACTACATTAGAATTCAGAGTGTAGAGATTCGGTAAGTAGGATCATGTAGACCAGAGATGTAGTATGTAGCCGAAGA |
| ds60-TAMRA-Cy5 |  |
Same as ds60 |
| ds60-BS |  |
Same as ds60 |
| ds30-BS |  |
Same as ds30 |
B, Biotin linked by C6-dT; S, streptavidin; X, TAMRA linked by C6-dT; Y, Cy5.
Fig. 1 A and B show the single-turnover kinetics of ds18 to ds90 DNA unwinding. The radiometric assay is an all-or-none assay and measures only the completely separated dsDNA (22). Therefore, the unwinding kinetics shows a pre-steady-state kinetic lag. The fact that the pre-steady-state kinetic lag becomes longer as the duplex length increases implies that the helicase unwinds DNA by a multistep process. The amplitude of unwinding decreases with increasing duplex length. Overall, the amplitudes are higher at 2 mM dTTP compared with 200 μM dTTP, because the concentration of the initial helicase-DNA complex is greater at the higher dTTP concentration (38).
Fig. 1.

Single-turnover kinetics of unwinding different lengths of DNA at 2 mM and 200 μM dTTP. The unwinding of ds18 (•), ds30 (▾), ds40 (▪), ds60 (♦), and ds90 (▴) DNAs (Table 1) was measured at 18°C in the presence of a trap at 2 mM (A) or 200 μM (B) dTTP. (A) The data represent an average of three measurements and were globally fit to Eqs. 1 and 2 to obtain s = 9.0 (± 0.7) bp per step, k = 1.7 (± 0.14) steps per s, and Lm = 11.8 (± 0.34) bp. (B) The global fit of the kinetics at 200 μM dTTP provided s = 6.0 (± 0.8) bp per step, k = 1.9 (±0.3) steps per s, and Lm = 11.5 (± 0.5) bp.
Global Fitting to the Stepping Model. To determine the unwinding rate, the kinetic data were analyzed by using the stepping model shown in Fig. 2. This model is similar to the one used to analyze the UvrD helicase kinetics (22, 39), with some modifications. The stepping model assumes that the helicase separates a particular length L of dsDNA in n steps; thus, the step size s is L/n. In this model, it is also assumed that all of the strand-separating species have identical stepping properties, that is there is a homogeneous population of the helicase molecules, and that each step occurs with the same stepping rate (k) and step size (s). The incomplete γ-function (Eq. 1) describes the stepping model in Fig. 2. It is continuous with respect to n the number of steps required to unwind a particular length of DNA and this result makes the incomplete γ-function a more convenient function for globally fitting the unwinding kinetics.
Fig. 2.

Stepping model of DNA unwinding. The model assumes that T7 helicase separates the dsDNA in kinetically discrete steps. The number of steps (n) that a helicase needs to separate a DNA depends on the duplex length (L) and the step size (s) of the helicase. It is assumed that each step is of the same size and occurs at the same rate (k) until the helicase reaches the minimal duplex length (Lm) when the DNA separates spontaneously. The stepping model is described by Eqs. 1 and 2.
Initially, we assumed that the entire length, L, of the DNA is separated by the helicase action. With this assumption, however, we were unable to globally fit the unwinding kinetics. When we examined the individual fits, we observed that the overall unwinding rate (k × s) was decreasing with increasing duplex length. Because all of the DNAs contain a fork junction of a similar structure and DNA sequence, it is highly unlikely that the helicase adjusts its unwinding rate according to the duplex length it is going to unwind. We therefore modified the stepping model as shown in Fig. 2 and included a parameter Lm (minimal duplex length), which is the shortest DNA duplex that can stay together under the experimental conditions. With the modified stepping model, we were able to globally fit the kinetics to Eqs. 1 and 2 as shown by the solid lines in Fig. 1 A and B. This finding indicates that the helicase does not have to separate the entire length of the duplex DNA, and when the helicase reaches the length L - Lm, the DNA separates spontaneously.
The global fitting of the unwinding data at 2 mM dTTP to the modified stepping model provided a step size of 9 ± 0.7 bp per step, a stepping rate of 1.7 ± 0.14 step per s, and a minimal duplex length of 11.8 ± 0.3 bp (Fig. 1 A). At 200 μM dTTP (Fig. 1B), the step size was determined as 6.0 ± 0.8 bp per step with a stepping rate of 1.9 ± 0.3 steps per s and a minimal duplex length of 11.5 ± 0.5 bp. Therefore, T7 helicase unwinds DNA at an average rate of 15 bp/s at 2 mM dTTP and 11 bp/s at 200 μM dTTP. The value of Lm depends on the GC content, reaction temperature, and any strain placed by the helicase on the duplex ahead. The nearest-neighbor analysis (40) indicated that the average melting temperature for the terminal 12 bp of the fork DNA is 12°C. Thus, Lm of 12 bp obtained from our global fitting is quite reasonable because the experiments were carried out at 18°C.
The reason for the decrease in the step size of T7 helicase from ≈9 to ≈6 bp when the dTTP concentration is decreased from 2 mM to 200 μM is not obvious and requires an understanding of the meaning of the kinetic step size. The kinetic step size is defined as the average number of base pairs unwound between two successive rate-limiting steps in the helicase reaction (22, 39). If the rate-limiting step of T7 helicase at low and high dTTP is different, then this might be the reason for the dependence of the kinetic step size on dTTP. Alternatively, the step size may depend on the number of dTTPs bound per hexamer, which also depends on the concentration of dTTP (35, 38). Another possibility that will be investigated in more detail in future studies is the existence of multiple populations of the helicase. If helicase populations exist that have different stepping rates, then the unwinding kinetics will appear to have fewer steps and the estimation of the step size will be exaggerated and unreliable. For these reasons, it is difficult to interpret the dependence of step size on [dTTP] and to relate it to any physical model of unwinding.
The Unwinding Amplitude Decreases with Increasing Duplex Length. The unwinding amplitude is a measure of the fraction of DNA molecules that are completely unwound before the helicase dissociates from the DNA. As shown in Fig. 1 A and B, the amplitude decreases as the length of the duplex length increases from 18 to 90 bp, which indicates low processivity. However, the decrease in amplitude with increasing duplex could be due to any of the following reasons: (i) dTTP depletion during unwinding, (ii) dissociation of the helicase during initiation, (iii) helicase stalling and remaining bound to the DNA, (iv) reannealing of the newly separated DNA strands behind the helicase, or (v) dissociation of the helicase during unwinding. Although the concentration of the helicase substrate is 2.5 nM, the ssDNA trap is present in micromolar amount, which raises the possibility of dTTP depletion during the reaction. We eliminate this possibility, based on the fact that the steady-state dTTP hydrolysis rate ranges from 3 to 10 s-1, depending on the oligo length (30). Therefore, the 10-nM helicase would hydrolyze only a fraction of dTTP during the unwinding reaction (3-10 μM in 100 s) and this result was verified experimentally (data not shown). Dissociation of the helicase during initiation should lower the amplitudes of all of the duplexes equally, which is not the case. The experiment that eliminates the possibility that the helicase stalls and remains bound during unwinding is the one with and without trap mentioned above. Even though a fraction of the DNA is unwound in the presence of the trap, almost all DNAs were unwound in the absence of the trap, albeit at a slower rate. If the helicase stalls but remains bound to DNA then the unwinding amplitudes should be the same with or without trap. Thus, the amplitude decrease with increasing duplex length is either due to DNA strands reannealing behind the helicase or the helicase dissociating from the DNA during unwinding or both.
Does the DNA Reanneal Behind the Helicase? The large helicase ring prevents the unwound DNA strands from reannealing behind the helicase; however, it is possible that after the helicase has unwound 42 ± 5 bp of DNA, which is Lm plus 30 ± 5 nt that is occluded by the helicase (29), the complementary strands come together. To investigate this possibility, we prepared a 60-bp fork DNA containing a FRET pair (TAMRA and Cy5) at the fork junction (Table 1, ds60-TAMRA-Cy5). As the helicase unwinds the DNA, the distance between the FRET dye pair increases resulting in a time-dependent enhancement of donor fluorescence and a corresponding quenching of the acceptor fluorescence (41). If the DNA strands reanneal behind the helicase, the initially separated FRET dye pair will come back together, and thus the decrease in the FRET signal will be transient rather than a stable one. As shown in Fig. 3A, a stable increase in the donor fluorescence and a similar decrease in the acceptor fluorescence was observed upon ds60 unwinding. This result indicates that the lower amplitude of ds60 unwinding is not due to the reannealing of the unwound strands behind the helicase. The unwinding rate increased with [dTTP] and provided a dTTP Km of 82 ± 9 μM (Fig. 3B). A small rise in the acceptor and a dip in the donor fluorescence were observed at the beginning of the reaction. By using singly dye-labeled DNAs, we have determined that the fast initial phase is due to the interaction of T7 helicase with the TAMRA dye on the 5′ strand as the helicase moves along that strand. We conclude from these experiments that the DNA does not reanneal behind the T7 helicase on short duplexes up to 60 bp and the protein's large size makes it unlikely that the 90-bp duplex will reanneal behind the helicase during unwinding. Therefore, the decrease in amplitude with increasing duplex length is due to the dissociation of the helicase from the DNA during unwinding.
Fig. 3.

DNA unwinding at various [dTTP] by FRET. Unwinding of the ds60-TAMRA-Cy5 DNA (Table 1) was measured in a stopped-flow instrument at 18°C. (A) The representative real-time fluorescence changes of the donor TAMRA and the acceptor Cy5 as the DNA unwinds. The experiment was carried out at various dTTP. (B) The observed rate versus dTTP dependency that was fit to a hyperbola with a dTTP Km of 82 (± 9) μM.
How Does the Helicase Dissociate from the DNA? The helicase ring can dissociate from the fork DNA in two ways. First, the hexamer ring can open and the open ring can dissociate from the DNA during unwinding. Second, the hexamer ring remains intact, but the ring slides backward to dissociate from the end of the 5′ ssDNA tail. Although it is unlikely that the helicase moves backward, we introduced a biotin-streptavidin block at the end of the 5′ tail; with the rationale being that the bulky adduct will prevent the dissociation of the helicase from the end of the 5′ tail, and therefore the unwinding amplitude will be greater for the DNA complexed to streptavidin and lower without streptavidin. We found that the amplitude and the unwinding rate of ds30-BS and ds60-BS (Table 1) were the same as the uncomplexed DNAs (data not shown). In addition, streptavidin dissociation from the DNA was not observed in our reactions. We therefore conclude that the unwinding amplitude decreases with increasing duplex length because the helicase dissociates from the DNA during unwinding. Because this event occurs from internal regions of the DNA, it requires the helicase ring to open.
Processivity of DNA Unwinding. The amplitude versus duplex length (Fig. 4) was fit to Eq. 3 to obtain the unwinding processivity P = 0.9835 (± 0.0014) at 2 mM dTTP and 0.9703 (± 0.0029) at 200 μM dTTP. The processivity of DNA unwinding is defined as the probability of the helicase unwinding a base pair versus helicase dissociating from that position on the DNA. The measured processivity shows that the helicase unwinds, on an average, 60 bp (at 2 mM dTTP) before it dissociates from the DNA [1/(1 - P)] and 33 bp at 200 μM dTTP. The helicase processivity therefore depends on the [dTTP] to some extent. Such a dependency was observed for the NPH II helicase (42) and the RecBCD helicase (23). By using the P value of 0.9835 and the relationship P = k/(kd + k) where k is the single-base pair unwinding rate and kd is the dissociation rate, we calculate a rate of 0.25 s-1 for the dissociation of T7 helicase from the DNA during unwinding. This value is ≈100× faster than the dissociation rate of T7 helicase from ssDNA during translocation (30).
Fig. 4.

Unwinding processivity of the T7 helicase at 2 mM (A) and 200 μM(B) dTTP. Amplitudes from the global fitting (Fig. 1 A and B) are plotted against corrected duplex length (L - Lm) and fit to Eq. 3 to obtain the processivity of 0.9835 (± 0.0014) at 2 mM dTTP and 0.9703 (± 0.0029) at 200 μM dTTP. The initial fraction of productive helicase-DNA complex was A0 = 0.65 (± 0.02) at 2 mM dTTP and 0.31 (± 0.018) at 200 μM dTTP.
Contrary to expectations for a ring helicase, the DNA-unwinding studies in this paper reveal a surprisingly low processivity of unwinding for the T7 helicase. We had expected that because the T7 helicase ring binds DNA in its central channel (43), it would have a high processivity. This finding is true when T7 helicase is translocating at 130 nt/s along ssDNA where it travels kilobases before dissociating (30). However, when T7 helicase is translocating more slowly at 15 bp/s to separate the strands of a duplex DNA, it has a low processivity. We have eliminated the alternatives that the low amplitude of unwinding is due to the reannealing of the DNA strands behind the helicase or due to the helicase ring dissociating from the end of the 5′ tail of the fork DNA, or helicase stalling during unwinding. Our studies indicate that the helicase ring must open during DNA unwinding. Ring opening may occur normally by fluctuations in the subunit-subunit interactions of the hexamer or the open ring might be an intermediate in the reaction. In either case, it appears that when the helicase is traveling at a fast rate on ssDNA, it dissociates less often, but when it is traveling more slowly through duplex DNA it dissociates more frequently. It is known that T7 helicase interacts with T7 DNA polymerase (a complex of T7 gp5 and E. coli thioredoxin) and T7 gp2.5 protein during DNA replication (44, 45). The interactions with these proteins at the fork junction can increase the efficiency of unwinding by T7 helicase. It is possible that the complex of T7 helicase and T7 DNA polymerase is a better motor to catalyze the unwinding reaction during DNA replication. Future studies will address these issues.
Conclusions
To unwind a long stretch of DNA, a helicase has to move unidirectionally along the DNA and couple translocation to local base pair separation. As the helicase moves along the DNA, it successively makes and breaks interactions with the DNA. This process is facilitated by NTP hydrolysis, which likely dictates the velocity of the helicase movement supported by the fact that fast helicases have fast NTPase rates (19-24). One of the important parameters to determine for understanding the helicase's mechanism is its translocation rate. We have previously determined (30) that T7 helicase translocates unidirectionally along ssDNA with a rate of 130 nt/sat18°C. In this paper, we have determined that under similar reaction conditions, T7 helicase unwinds DNA with a 9-fold slower rate of 15 bp/s. These results show that T7 helicase does not move at its maximum speed through the duplex region at the fork junction.
Separating the strands of a nucleic acid is a thermodynamically unfavorable process at physiological temperatures. T7 helicase and most helicases make the unwinding process thermodynamically feasible by binding to the ssDNA (4, 13). In addition to ssDNA binding, it has been proposed that helicases such as Rep and PcrA bind directly to the duplex DNA to destabilize it (14-16). It is known that T7 helicase cannot separate an ss/ds junction lacking a noncomplementary 3′ tail (31, 32), and this is a common property of many ring helicases (33, 34, 46). In addition, it was shown that the duplex DNA region of the ss/dsDNA substrate does not contribute to the free energy of E. coli DnaB binding to the helicase substrate (47). The results indicate that these ring helicases do not bind and destabilize duplex DNA as proposed above for Rep and PcrA helicases.
Many helicases have been shown to move unidirectionally along nucleic acid (5, 30, 48) as well as to displace bonded moieties along their path without specifically interacting with these moieties (5-8). Thus, unidirectional translocation is a basic activity of helicases and base pair separation might occur as a consequence of the movement. It is known that the base pairs at the fork junction open and close at very fast rates (17). A 5-bp hairpin DNA at 300 K was shown to open at a rate of 3,000 s-1 (18) that exceeds the translocation speeds of helicases. Thus, helicases do not need to increase the rate of base pair opening but simply need to stabilize the open ssDNA regions. Because base pair opening and closing rates are faster than the rate of helicase movement, the base pair opening reaction (bpclosed ⇌ bpopen) can be considered a rapid equilibrium step or a fluctuating force directed against the movement of the helicase. Our results show that the duplex slows down the movement of the helicase roughly by a factor predicted from the relationship, kunwinding = Kfraying × kssDNA translocation, where the value of Kfraying is determined by the free energy of base pair formation and equal to the ratio of the rate of base pair opening and closing (kopen/kclosed). The 9-fold difference in ssDNA translocation and unwinding rates in terms of a single base pair unwound or a single base translocated (kunwinding/kssDNA transloation) corresponds to a free-energy difference of 1.2 kcal/mol, a value close to the average free energy of opening a single base pair of DNA (2, 13). These results imply that T7 helicase is primarily a ssDNA translocase and the duplex DNA poses a barrier to its movement along ssDNA, although we cannot rule out the possibility that the physical presence of the large helicase hexamer at the fork junction might perturb the duplex region at the junction. We argue that if the helicase were to destabilize the duplex DNA by interacting with it or by increasing the lifetime of the open base pair, the unwinding rate of the helicase would be closer to its rate of translocation along ssDNA. We therefore propose that T7 helicase unwinds DNA by its ability to bind ssDNA as it moves unidirectionally along the DNA. We propose that any protein capable of unidirectional translocation along one strand of the duplex DNA while excluding the other strand can unwind DNA. Our results also imply that the free energy of nucleotide hydrolysis is used mainly to drive unidirectional movement of the helicase along ssDNA and to offset the energetically unfavorable portions of the reaction of translocation and unwinding.
Acknowledgments
This work was supported by National Institutes of Health Grant GM55310 (to S.S.P.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ds, double-stranded; ss, single-stranded; FRET, fluorescence resonance energy transfer; TAMRA, carboxytetramethylrhodamine.
References
- 1.Matson, S. W., Bean, D. W. & George, J. W. (1994) BioEssays 16, 13-22. [DOI] [PubMed] [Google Scholar]
- 2.Lohman, T. M. & Bjornson, K. P. (1996) Annu. Rev. Biochem. 65, 169-214. [DOI] [PubMed] [Google Scholar]
- 3.Patel, S. S. & Picha, K. M. (2000) Annu. Rev. Biochem. 69, 651-697. [DOI] [PubMed] [Google Scholar]
- 4.Levin, M. K. & Patel, S. S. (2003) in Molecular Motors, ed. Schliwa, M. (Wiley-VCH, Verlag GmbH, Weinheim, Germany), pp. 179-198.
- 5.Morris, P. D. & Raney, K. D. (1999) Biochemistry 38, 5164-5171. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan, D. L. & O'Donnell, M. (2002) Mol. Cell 10, 647-657. [DOI] [PubMed] [Google Scholar]
- 7.Jankowsky, E., Gross, C. H., Shuman, S. & Pyle, A. M. (2001) Science 291, 121-125. [DOI] [PubMed] [Google Scholar]
- 8.Morris, P. D., Byrd, A. K., Tackett, A. J., Cameron, C. E., Tanega, P., Ott, R., Fanning, E. & Raney, K. D. (2002) Biochemistry 41, 2372-2378. [DOI] [PubMed] [Google Scholar]
- 9.Betterton, M. D. & Julicher, F. (2003) Phys. Rev. Lett. 91, 258103. [DOI] [PubMed] [Google Scholar]
- 10.Doering, C., Ermentrout, B. & Oster, G. (1995) Biophys. J. 69, 2256-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li, D., Zhao, R., Lilyestrom, W., Gai, D., Zhang, R., DeCaprio, J. A., Fanning, E., Jochimiak, A., Szakonyi, G. & Chen, X. S. (2003) Nature 423, 512-518. [DOI] [PubMed] [Google Scholar]
- 12.Chen, Y. Z., Zhuang, W. & Prohofsky, E. W. (1992) J. Biomol. Struct. Dyn. 10, 415-427. [DOI] [PubMed] [Google Scholar]
- 13.von Hippel, P. H. & Delagoutte, E. (2001) Cell 104, 177-190. [DOI] [PubMed] [Google Scholar]
- 14.Amaratunga, M. & Lohman, T. M. (1993) Biochemistry 32, 6815-6820. [DOI] [PubMed] [Google Scholar]
- 15.Soultanas, P., Dillingham, M. S., Wiley, P., Webb, M. R. & Wigley, D. B. (2000) EMBO J. 19, 3799-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Velankar, S. S., Soultanas, P., Dillingham, M. S., Subramanya, H. S. & Wigley, D. B. (1999) Cell 97, 75-84. [DOI] [PubMed] [Google Scholar]
- 17.Gueron, M. & Leroy, J. L. (1995) Methods Enzymol. 261, 383-413. [DOI] [PubMed] [Google Scholar]
- 18.Bonnet, G., Krichevsky, O. & Libchaber, A. (1998) Proc. Natl. Acad. Sci. USA 95, 8602-8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim, S., Dallmann, H. G., McHenry, C. S. & Marians, K. J. (1996) Cell 84, 643-650. [DOI] [PubMed] [Google Scholar]
- 20.Raney, K. D., Carver, T. E. & Benkovic, S. J. (1996) J. Biol. Chem. 271, 14074-14081. [DOI] [PubMed] [Google Scholar]
- 21.Cheng, W., Brendza, K. M., Gauss, G. H., Korolev, S., Waksman, G. & Lohman, T. M. (2002) Proc. Natl. Acad. Sci. USA 99, 16006-16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali, J. A. & Lohman, T. M. (1997) Science 275, 377-380. [DOI] [PubMed] [Google Scholar]
- 23.Bianco, P. R., Brewer, L. R., Corzett, M., Balhorn, R., Yeh, Y., Kowalczykowski, S. C. & Baskin, R. J. (2001) Nature 409, 374-378. [DOI] [PubMed] [Google Scholar]
- 24.Nanduri, B., Byrd, A. K., Eoff, R. L., Tackett, A. J. & Raney, K. D. (2002) Proc. Natl. Acad. Sci. USA 99, 14722-14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel, S. S. & Hingorani, M. M. (1993) J. Biol. Chem. 268, 10668-10675. [PubMed] [Google Scholar]
- 26.Egelman, E. H., Yu, X., Wild, R., Hingorani, M. M. & Patel, S. S. (1995) Proc. Natl. Acad. Sci. USA 92, 3869-3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sawaya, M. R., Guo, S., Tabor, S., Richardson, C. C. & Ellenberger, T. (1999) Cell 99, 167-177. [DOI] [PubMed] [Google Scholar]
- 28.Singleton, M. R., Sawaya, M. R., Ellenberger, T. & Wigley, D. B. (2000) Cell 101, 589-600. [DOI] [PubMed] [Google Scholar]
- 29.Hingorani, M. M. & Patel, S. S. (1993) Biochemistry 32, 12478-12487. [DOI] [PubMed] [Google Scholar]
- 30.Kim, D. E., Narayan, M. & Patel, S. S. (2002) J. Mol. Biol. 321, 807-819. [DOI] [PubMed] [Google Scholar]
- 31.Ahnert, P. & Patel, S. S. (1997) J. Biol. Chem. 272, 32267-32273. [DOI] [PubMed] [Google Scholar]
- 32.Hacker, K. J. & Johnson, K. A. (1997) Biochemistry 36, 14080-14087. [DOI] [PubMed] [Google Scholar]
- 33.Kaplan, D. L., Davey, M. J. & O'Donnell, M. (2003) J. Biol. Chem. 278, 49171-49182. [DOI] [PubMed] [Google Scholar]
- 34.Shin, J. H., Jiang, Y., Grabowski, B., Hurwitz, J. & Kelman, Z. (2003) J. Biol. Chem. 278, 49053-49062. [DOI] [PubMed] [Google Scholar]
- 35.Hingorani, M. M. & Patel, S. S. (1996) Biochemistry 35, 2218-2228. [DOI] [PubMed] [Google Scholar]
- 36.Patel, S. S., Rosenberg, A. H., Studier, F. W. & Johnson, K. A. (1992) J. Biol. Chem. 267, 15013-15021. [PubMed] [Google Scholar]
- 37.Matson, S. W., Tabor, S. & Richardson, C. C. (1983) J. Biol. Chem. 258, 14017-14024. [PubMed] [Google Scholar]
- 38.Picha, K. M. & Patel, S. S. (1998) J. Biol. Chem. 273, 27315-27319. [DOI] [PubMed] [Google Scholar]
- 39.Lucius, A. L., Maluf, N. K., Fischer, C. J. & Lohman, T. M. (2003) Biophys. J. 85, 2224-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vesnaver, G. & Breslauer, K. J. (1991) Proc. Natl. Acad. Sci. USA 88, 3569-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng, W., Hsieh, J., Brendza, K. M. & Lohman, T. M. (2001) J. Mol. Biol. 310, 327-350. [DOI] [PubMed] [Google Scholar]
- 42.Jankowsky, E., Gross, C. H., Shuman, S. & Pyle, A. M. (2000) Nature 403, 447-451. [DOI] [PubMed] [Google Scholar]
- 43.Yu, X., Hingorani, M. M., Patel, S. S. & Egelman, E. H. (1996) Nat. Struct. Biol. 3, 740-743. [DOI] [PubMed] [Google Scholar]
- 44.Lee, J., Chastain, P. D., Kusakabe, T., Griffith, J. D. & Richardson, C. C. (1998) Mol. Cell 1, 1001-1010. [DOI] [PubMed] [Google Scholar]
- 45.Nakai, H. & Richardson, C. C. (1986) J. Biol. Chem. 261, 15208-15216. [PubMed] [Google Scholar]
- 46.Kaplan, D. L. (2000) J. Mol. Biol. 301, 285-299. [DOI] [PubMed] [Google Scholar]
- 47.Jezewska, M. J., Rajendran, S. & Bujalowski, W. (1998) Biochemistry 37, 3116-3136. [DOI] [PubMed] [Google Scholar]
- 48.Dillingham, M. S., Wigley, D. B. & Webb, M. R. (2000) Biochemistry 39, 205-212. [DOI] [PubMed] [Google Scholar]