

Abstract
Malondialdehyde, a genotoxic byproduct of lipid peroxidation, reacts with guanine in DNA to form pyrimido[1,2-α]purin-10(3H)one (M1dG), the first endogenous DNA lesion found to be a target of nucleotide excision repair enzymes. A subpathway of nucleotide excision repair, transcription-coupled repair, is thought to occur when RNA polymerase (RNAP) is arrested at damage in transcribed DNA strands and might function for efficient removal of M1dG in active genes. Results presented here show that M1dG and its stable, exocyclic analog 1,N2-propanodeoxyguanine (PdG), arrest translocation of T7 RNAP and mammalian RNAPII when located in the transcribed strand of a DNA template. M1dG paired with thymine is exocyclic and poses a stronger block to transcription than the acyclic N2-(3-oxo-1-propenyl)-dG, formed upon cytosine-catalyzed opening of M1dG in duplex DNA. PdG is a complete block to RNAPII regardless of base pairing. The elongation factor TFIIS (SII) induces reversal and RNA transcript cleavage by RNAPII arrested at PdG. Thus, arrested RNAPII complexes may be stable at M1dG in cells and may resume transcription once the offending adduct is removed. The conclusion from this work is that malondialdehyde adducts in the transcribed strand of expressed genes are strong blocks to RNAPs and are targets for cellular transcription-coupled repair. If so, then M1dG, already known to be highly mutagenic in human cells, also may contribute to apoptosis in the developing tissues of individuals with Cockayne's syndrome, a hereditary disorder characterized by transcription-coupled repair deficiency.
Transcription-coupled repair (TCR) specifically eliminates DNA damage from the transcribed strand of active genes, and the arrest of elongating RNA polymerase (RNAP) complexes seems to initiate this process (1). RNAP of Escherichia coli, RNAP I and II of Saccharomyces cerevisiae, and mammalian RNAPII mediate TCR of UV-induced cyclobutane pyrimidine dimers (CPDs) (2-5). Because most lesions that arrest RNAPs are also targets for global nucleotide excision repair (NER), TCR is viewed as a specialized recognition mechanism for removal of DNA-distorting damage from transcriptionally active genes by NER.
A positive correlation exists between in vitro transcriptional arrest by a form of DNA damage and the TCR of that damage in cells. CPDs and cisplatin-intrastrand DNA crosslinks, which cause significant structural changes in DNA and are repaired by TCR, are strong blocks to transcript elongation by both T7 RNAP and mammalian RNAPII (6-8). Oxidized and alkylated bases, produced during normal cellular metabolism, are generally cleared from DNA by base excision repair (9, 10). Whereas TCR of oxidative damage in eukaryotic cells has been suggested (11), neither 7,8-dihydro-8-oxoguanine nor thymine glycol significantly affects translocation of RNAPII in vitro (12, 13). Therefore, a role for TCR in the repair of endogenous oxidative DNA damage remains to be clearly established.
Lipid peroxidation and prostaglandin biosynthesis in cells generate the genotoxic byproduct malondialdehyde (MDA) (14). MDA and its tautomer β-hydroxyacrolein react with guanine in DNA to form the pyrimidopurinone adduct, pyrimido[1,2-α]purin-10(3H)-one (M1dG) (Fig. 1). M1dG is also formed by base propenals (15), which are produced under conditions of oxidative stress and subsequent to DNA nicking by bleomycin, an antitumor antibiotic used in treating human cancers (16). In healthy human tissues, M1dG adducts occur as frequently as 1 in 106 nucleotides and are present in pancreas at levels comparable to those of 7,8 dihydro-8-oxoguanine (17).
Fig. 1.

Formation of M1dG and the structures of M1dG and PdG.
M1dG formation results in two chemically distinct DNA adducts (Fig. 1). When correctly paired with cytosine in duplex DNA, M1dG undergoes a base-catalyzed conversion to an acyclic structure, N2-(3-oxo-1-propenyl)-dG (N2-OPdG) (18). In single-stranded DNA or when mispaired with thymine, M1dG remains exocyclic. A synthetic, saturated analog of M1dG, N2-propanodeoxyguanine (PdG), which remains exocyclic regardless of base pairing, has been used to study the effects of M1dG on purified DNA polymerases and on mutation frequency in cells.
M1dG and PdG are highly mutagenic in bacteria and mammalian cells, inducing both frameshifts and base substitutions (19-22). Therefore, the efficient repair of MDA adducts is essential for genomic stability. Although base excision repair removes exocyclic guanine adducts from DNA, the PdG-induced mutation frequency in E. coli is not affected by the absence of AlkA, the primary glycosylase for ethenopurine and ethenocytosine adducts (23, 24). On the other hand, uvrA- mutants, lacking NER, exhibit a 4-fold increase in base substitutions due to PdG, suggesting that NER has a significant role in the repair of M1dG (23, 25). Indeed, purified UvrABC exinuclease and extracts from Chinese hamster ovary cells excise PdG adducts from DNA in fragments of sizes consistent with NER activity (23). Mismatch repair, which removes misincorporated bases and small loops from DNA, also may contribute to the repair of MDA adducts (26). The bacterial mismatch repair factor MutS binds to M1dG:T with similar affinity as to G:T in vitro, and methyl-directed mismatch repair seems to remove PdG from DNA, because a decrease in mutation frequency in uvrA- E. coli is observed only when the PdG is located in the unmethylated strand of a damaged plasmid (25). However, the PdG-induced mutation frequency for mutS- bacteria is lower than that for mismatch repair-proficient cells, indicating that binding of MutS to MDA adducts may occlude them from NER proteins. The induction of frameshift mutations by PdG and M1dG suggests that the adducts alter DNA polymerase elongation (19, 20, 22). PdG does, in fact, interfere with nucleotide insertion and translocation by Klenow fragment and DNA polymerase β in vitro (27, 28); therefore, it is possible that MDA adducts may also impede RNA polymerases. If so, M1dG in expressed genes might be recognized and removed by TCR.
To study the effect of MDA adducts on transcript elongation by T7 RNAP and mammalian RNAPII, templates were constructed containing a site-specific M1dG or PdG in either the transcribed (Xts) or nontranscribed (Xnts) strand template downstream of the T7 promoter and adenovirus major late promoter (AdMLP) (Fig. 2). Additionally, to observe effects of M1dG structure on transcription, the adducts were placed opposite C or T. An M1dG or PdG adduct in the transcribed strand impeded elongation by both RNAPs, with the greatest effects on translocation by RNAPII. The extent of arrest induced by M1dG was dependent on base pairing, whereas PdG completely blocked RNAPII elongation when paired with either C or T. Stable RNAPII complexes persisted at PdG adducts after arrest, as evidenced by RNAPII backup from the adduct and transcript cleavage in response to the elongation factor SII. These data suggest that MDA adducts produced in mammalian cells may block RNAPII translocation and be subject to removal from DNA by TCR.
Fig. 2.

DNA substrates for RNAP transcription. The linear Xts and Xnts DNA templates contain dG, M1dG, or PdG (X) either in the transcribed or nontranscribed strands, respectively, downstream of promoters for T7RNAP (T7P) and RNAPII (AdMLP) transcription. Numbers in parentheses indicate positions in the nontranscribed strand of the DNA plasmid construct. Transcription starts at each promoter (+1) and proceeds in the direction of the bent arrows. Transcripts from the T7 promoter incorporate [α-32P]GTP and from AdMLP incorporate [α-32P]CTP within the labeling cassette. Stalled complexes restart at the end of the cassette (+5 from the T7 promoter and +15 from AdMLP). Transcript lengths expected from runoff (RO) or from arrest at the X are depicted by lines below each template. The XhoI-BamHI 34-bp sequence inserted in the plasmid construct as described in Materials and Methods is shown with the 18-base primer sequence in an enlarged type size.
Materials and Methods
Proteins and Reagents. T7 RNAP and RNasin were purchased from Promega. RNAPII, transcription initiation factors, and TFIIS, purified from rat liver or from recombinant sources (29), were obtained from D. Reines (Emory University, Atlanta). T4 polynucleotide kinase, T4 DNA polymerase, and M13K07 helper phage were purchased from New England Biolabs, and T4 DNA ligase was from Invitrogen. The F′ E. coli strain MV1184 was provided by J. Messing (Rutgers University, Piscataway, NJ). D44 IgG antibody to RNA was purified from rodent ascites fluid and provided by D. Reines (30, 31). Formalin-fixed Staphylococcus aureus was obtained from Calbiochem, and all ribonucleoside triphosphates were purchased from Amersham Biosciences.
Preparation of Oligonucleotides. Oligonucleotides of the 18-base sequence 5′-ATCGGCGCCGXCGGTGTG-3′ containing either a single M1dG or PdG at the position X were synthesized as described (32), and their purity was verified by matrix-assisted laser desorption/ionization mass spectrometry. The undamaged 18-mer (X = dG) and the 34-base oligonucleotides used below were prepared by Qiagen (Valencia, CA).
Construction of Transcription Templates. The transcription templates for T7 RNAP and RNAPII were constructed as described (8). In brief, a 34-bp oligonucleotide (Fig. 2) containing the 18-base sequence above (X = dG or dA) was ligated, at a 20:1 molar ratio of oligonucleotide to vector, into the XhoI-BamHI cloning site downstream of both the T7 promoter and AdMLP (Fig. 2). The constructs were transformed into the F′ E. coli strain MV1184 for replication of the plus strand by the M13K07 phage (33). Plasmids containing dG, M1dG, or PdG in either the transcribed or nontranscribed strand were prepared by T4 DNA polymerase second-strand synthesis with the appropriate 18-mer as a primer. Products were resolved by electrophoresis in TAE (40 mM Tris-acetate/2 mM disodium EDTA, pH 8.5) on 1% agarose containing 0.3 μg/ml ethidium bromide. Plasmids were excised, electroeluted from the gel by using an Amicon Centrilutor (Millipore), and concentrated by centrifugation. The constructs were digested with HindIII, then gel-purified and isolated as described above. The concentration and purity of the linear templates were determined by agarose gel electrophoresis alongside a sample of known DNA concentration.
T7 RNAP Transcription Reactions. T7 RNAP transcription reactions containing 10 ng of linearized DNA template and 20 units of T7 RNAP in 10 μl of T7 transcription buffer (40 mM Tris·HCl, pH 7.9/6 mM MgCl2/2 mM spermidine/10 mM NaCl/10 mM DTT) supplemented with 2.5 μM[α-32P]GTP (400 Ci/mmol; 1 Ci = 37 GBq), 200 μM UTP, and 16 units of RNasin were incubated for 5 min at 37°C for RNAP initiation at the promoter and stalling of transcript elongation at the end of the 4-nt cassette. Heparin was added at 50 ng/μl to inactivate uninitiated T7 RNAP, and elongation by the stalled RNAP complexes was then induced by the addition of 200 μM each of ATP, CTP, and GTP. After 30 min at 37°C (or after times up to 60 min), reactions were stopped with SDS, proteins were digested with proteinase K, and nucleic acids were precipitated with ethanol. Samples were dried under vacuum, resuspended in formamide dye, and resolved by denaturing 5% PAGE in TBE (89 mM Tris-borate/1 mM EDTA, pH 8.0). The gel was dried, and transcripts were then autoradiographed by exposure of Kodak Biomax MR film with intensifying screens at -80°C. The signal intensities from the resolved transcripts were detected with a GS-363 CS phosphorimager screen and were quantified by using a GS-363 imaging system with molecular analyst software (Bio-Rad).
RNAPII Transcription Reactions. For binding of the RNAPII to the AdMLP promoter, 0.5 μg of rat RNAPII and 100 ng of DNA template were incubated with rat liver protein fraction D (2 μg of TFIID and TFIIH) for 30 min at 28°C in 20 μl of RNAPII transcription buffer (3 mM Hepes-NaOH, pH 7.9/20 mM Tris·HCl, pH 7.9/150 mM KCl/2 mM DTT/2.2% polyvinyl alcohol/0.5 mg/ml acetylated BSA/3% glycerol) containing 16 units of RNasin. For preinitiation complex formation, incubation was continued for 20 min after the addition of 33 μl of RNAPII buffer (without KCl) containing fraction B′ (1-μg mixture of TFIIF and TFIIE) and 3 ng of recombinant TFIIB. Transcription was induced by the addition of 7 mM MgCl2, 20 μM ATP, 20 μM UTP, and 0.8 μM [α-32P]CTP (800 Ci/mmol), and transcripts were extended to the end of the 14-nt cassette by incubation of the reactions for 20 min at 28°C. Uninitiated RNAPII molecules were inactivated with heparin, as above, and 800 μM of each ATP, UTP, GTP, and CTP were added for elongation of the stalled transcripts. After 15 min at 28°C, reactions were stopped with SDS, and proteins were digested with proteinase K. To remove unincorporated 32P, samples were filtered by centrifugation through G-25 Sephadex (Amersham Biosciences), and nucleic acids were precipitated with ethanol. Gel electrophoresis, autoradiography, and transcript quantitation were performed as described above.
For SII-mediated transcript cleavage, transcription on 100 ng of DNA template (Xts) containing the PdG:C pair was performed as above to obtain RNAPII complexes arrested at the adduct. Reactions were incubated for 10 min at 4°C after the addition of 10 ng/μl D44 antibody, and the incubation was then repeated after the addition of formalin-fixed S. aureus. The cells, attached by antibody to RNA and RNAPII complexes, were washed three times in complex isolation buffer (3 mM Hepes-NaOH, pH 7.9/20 mM Tris·HCl, pH 7.9/2.2% polyvinyl alcohol/0.2 mg/ml acetylated BSA/60 mM KCl/2 mM DTT/3% glycerol), then resuspended in 56 μl of buffer containing 7 mM MgCl2. Samples were incubated for 1 h at 28°C after the addition of SII, and transcripts were dissociated from the D44 antibody by the addition of SDS. Cells were removed by centrifugation and protein-digested by proteinase K, and RNA transcripts were precipitated and resolved as described above.
Results
M1dG and PdG Adducts Arrest Transcription by T7 RNAP. Transcripts from the Xts and Xnts DNA transcription templates were 32P-labeled by radionucleotide incorporation along a sequence cassette downstream of the transcription start site for T7 RNAP or RNAPII (Fig. 2). By exclusion of the next ribonucleotide required for transcript extension (e.g., CTP from T7 RNAP reactions), an RNAP complex was stalled at the end of the cassette on each template molecule. The stalled RNAP complex interfered with subsequent RNAP binding at the promoter, and heparin addition inactivated free RNAP and RNAP bound nonspecifically to the template. All four ribonucleotides were provided for transcript elongation, and further incorporation of 32P was minimized by the presence of excess cold ribonucleotides. Heparin prevented reinitiation of transcription by RNAPs dissociating during or at the completion of elongation. Therefore, the transcripts produced were the result of a single promoter-initiated transcription event on each active DNA template molecule. Because no repair proteins are present in this reconstituted transcription system, full-length transcripts indicated bypass of the adducted base rather than removal of the adduct, allowing unobstructed translocation of the RNAP. The arrest of transcription by M1dG or PdG was monitored as RNA transcripts of lengths shorter than the length expected for elongation to the end of the DNA template (runoff).
When located in the transcribed strand of the Xts template and correctly paired with C, M1dG arrested 27% of T7 RNAP transcripts (Fig. 3A, lane 3), whereas PdG blocked 68% of elongation (lane 5). T7 RNAP arrest by M1dG increased to 36% when the adduct was paired with T (Fig. 3B, lane 3), whereas the level of T7 RNAP arrest by PdG was unaffected by the base change (lane 5). These results indicate that the exocyclic M1dG is a stronger block to RNAP translocation than the acyclic N2-OPdG formed by cytosine-catalyzed ring opening. Both M1dG and PdG induced the arrest of another 10-15% of transcripts at sites 3′ of the adduct position (MS). The base sequences around these sites (≈10, 24, and 48 bases upstream) have no apparent similarities or features that would be expected to pause the polymerase. As the shorter transcripts are dependent on the presence of the adduct, the arrest sites may arise from similar DNA structural changes induced along the template by M1dG and PdG. Transcription was conducted with two additional Xts templates having different sequences upstream of a PdG:C pair (data not shown). Arrest sites at 10 and 24 bases 3′ from PdG also were observed in both, whereas only one contained the most distal arrest site. The extent of transcriptional arrest at these sites varied between the templates, suggesting that both PdG-induced helical distortions and sequence context contribute to T7 RNAP behavior upstream of the adduct. A pause site (P) was observed in the undamaged sequence ≈15 bases downstream of the adduct position but should have no effect on elongation up to the adduct position (Fig. 3, lane 1). No arrest of T7 RNAP translocation occurred at adducts in the nontranscribed strand of the Xnts template (Fig. 3, lanes 2, 4, and 6).
Fig. 3.

T7RNAP elongation is arrested by M1dG and PdG in the transcribed strand. The 32P-labeled transcripts from T7RNAP elongation on DNA templates with the adduct position paired with C (A) or T (B) are shown. In each panel, lanes 1, 3, and 5 contain transcripts from Xts (T) and lanes 2, 4, and 6 transcripts from Xnts (N) containing dG, M1dG, and PdG, respectively. A 10-base DNA ladder is resolved in lane L. RO, runoff transcripts; P, pause site transcripts; X, adduct-arrested transcripts; MS, minor arrest site transcripts.
To determine the efficiency of adduct bypass and the relationship between T7 RNAP bypass and adduct structure, transcripts from the Xts templates were sampled at times up to 60 min (Figs. 4 and 5). Bypass of M1dG was concurrent with arrest at the adduct, indicating that M1dG caused a transient block to T7 RNAP translocation (lanes 1-7). Runoff transcripts from T7 RNAP bypass of M1dG:T (Fig. 5, lanes 1-7) appeared more slowly than from bypass of M1dG:C (Fig. 4, lanes 1-7). This finding suggests that the closed-ring M1dG impeded the polymerase to a greater extent than did the acyclic N2-OPdG. Because PdG paired with either base blocks nearly 80% of all transcription in the first few minutes (lanes 9-14), the exocyclic ring seems to have a significant role in the arrest of T7 RNAP. The ratio of arrested to runoff transcripts observed after 30 min for M1dG and PdG Xts templates was established within 5 min of NTP addition and remained constant. This finding indicates that all active template molecules are equivalent, both in content of the adduct and in activity of the elongating RNAP. The total amount of transcripts increased during the elongation reaction because of the fact that not all T7 RNAP complexes stalled at the labeling cassette restarted transcription simultaneously upon the addition of NTPs. Therefore, each lane shows RNA products generated from a single round of transcription from elongating T7 RNAP complexes during that reaction time. T7 RNAP efficiently transcribed the undamaged templates within a 5-min elongation period (lane 15). The persistence of shortened transcripts indicates that all forms of the adducts were substantial blocks to T7 RNAP translocation.
Fig. 4.

T7RNAP transcription time course for arrest at C-paired adducts in the transcribed strand. (A) T7RNAP arrest at M1dG (lanes 1-7) or PdG (lanes 8-14) paired with C in the Xts template was observed for 60 min after the initiation of elongation by NTP addition. Transcripts after a 5-min elongation on the template with dG are in lane 15. RO, runoff transcripts; X, adduct-arrested transcripts. (B) A graph of the % of transcripts arrested at X (•, M1dG arrest; ▪, PdG arrest) and the % of full-length transcripts (○, RO past M1dG; □, RO past PdG) for each template over the T7RNAP elongation time course.
Fig. 5.

T7RNAP transcription time course for arrest at T-paired adducts in the transcribed strand. (A) T7RNAP arrest at M1dG (lanes 1-7) or PdG (lanes 8-14) paired with C in the Xts template was observed for 60 min after the initiation of elongation by NTP addition. Transcripts after a 5-min elongation on the template with dG are in lane 15. RO, runoff transcripts; X, adduct-arrested transcripts. (B) A graph of the % of transcripts arrested at X (•, M1dG arrest; ▪, PdG arrest) and the % of full-length transcripts (○, RO past M1dG; □, RO past PdG) for each template over the T7RNAP elongation time course.
M1dG and PdG Adducts Arrest Mammalian RNAPII Translocation. To assess the behavior of RNAPII complexes upon encounter of M1dG and PdG, the Xts and Xnts templates were transcribed from the AdMLP by purified rat liver RNAPII in association with its required initiation and elongation factors (Fig. 6). As observed with T7 RNAP, these adducts had no effect on RNAPII translocation when present in the nontranscribed strand (Fig. 6B, lanes 4-6), and the extent of RNAPII arrest by an M1dG adduct in the transcribed strand depended on whether it was paired with C or T. An M1dG adduct paired with C arrested 60% of RNAPII transcription, whereas M1dG:T caused a 70% arrest of transcript elongation (Fig. 6, lane 2). PdG was a complete block to RNAPII translocation regardless of the opposing base (Fig. 6, lane 3). RNAPII did not arrest at sequences 3′ of the adduct, as was observed with T7 RNAP elongation. This finding indicates that RNAPII may be less sensitive than T7 RNAP to minor distortions of the DNA helix. However, the stronger block of RNAPII transcript elongation by the adducts suggests a mechanistic difference for the T7 and mammalian polymerases when sampling template bases for ribonucleotide addition to the nascent transcript.
Fig. 6.

M1dG and PdG block transcript elongation by RNAPII. 32P-labeled transcripts from RNAPII elongation on the Xts (TS) with X opposite C (A, lanes 1-3) or opposite T (B, lanes 1-3) are shown along with transcripts from RNAPII elongation on Xnts (NTS) opposite T (B, lanes 4-6). The base at X is indicated above each lane (G, dG; M, M1dG; P, for PdG). RO, runoff transcripts; X, adduct-arrested transcripts.
RNAPII Complexes Arrested at PdG Adducts Are Stable. The arrested transcripts produced by RNAPII in the above reactions could be released from stable RNAPII complexes by detergent denaturation or from the dissociation of unstable complexes soon after RNAPII translocation is blocked by the adducts. The stability of RNAPII complexes arrested at sites of DNA damage has been monitored by their response to the elongation factor SII (8, 33). SII activates a cryptic ribonuclease activity of RNAPII and promotes RNAPII read-through of pause sequences during transcription (34). In response to SII, stalled RNAPII complexes reverse translocate on the template, and RNAPII cleaves off a short sequence from the 3′ end of the RNA transcript. This activity repositions the nascent RNA in the active site for elongation. Because PdG paired with C was a complete block to RNAPII elongation, the Xts template containing PdG:C was used to assess the stability of complexes arrested at MDA adducts. PdG-arrested RNAPII complexes were isolated by precipitation with an antibody to the RNA transcript, then incubated with SII in the absence of NTPs. As shown in Fig. 7, a fraction of the arrested transcripts shortened by 10-25 nt from the adduct position, indicating that the transcripts were part of stable RNAPII complexes. These results indicate that RNAPII complexes persist at PdG in an arrested state and, in cells, might initiate a signal for TCR while blocked on the transcribed strand.
Fig. 7.
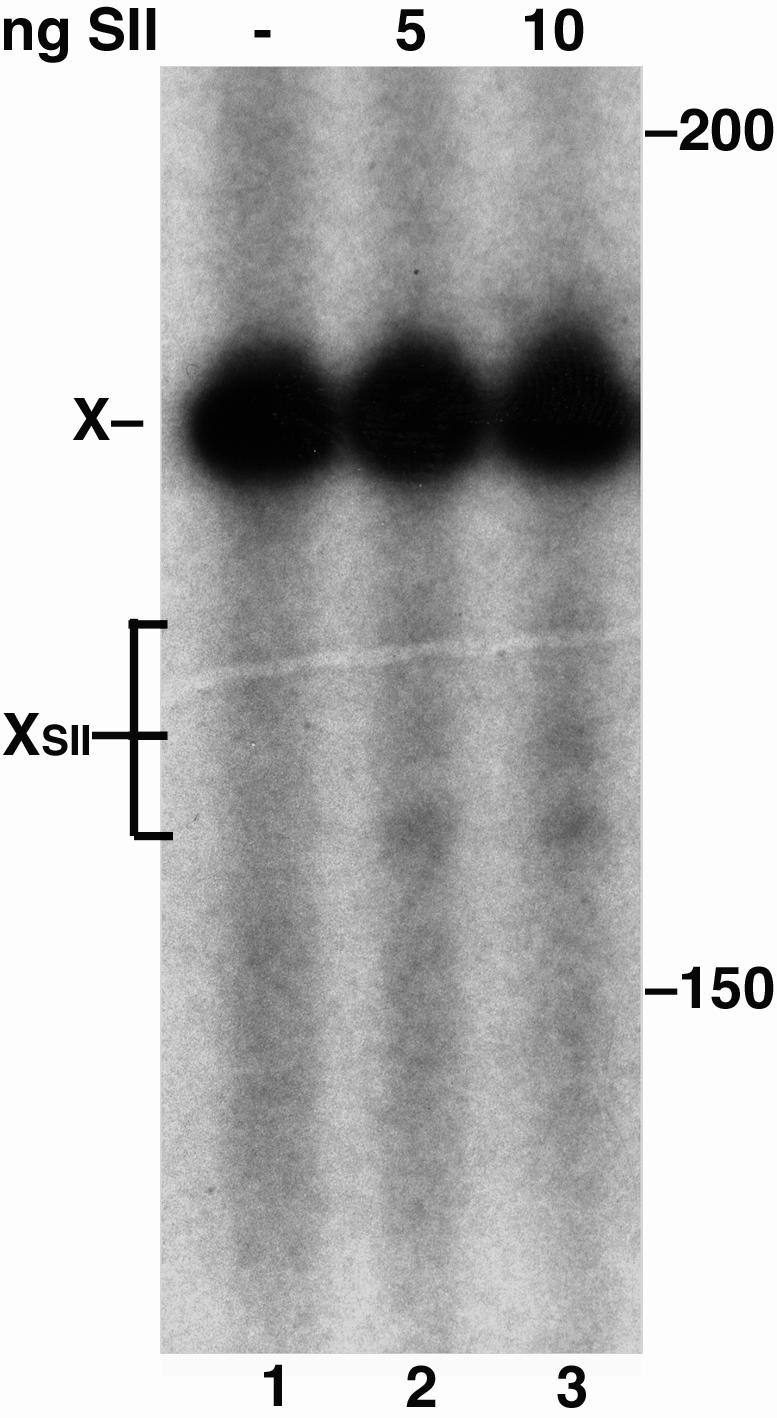
RNAPII complexes arrested at PdG respond to SII. Transcripts arrested at PdG opposite a C in the Xts template (X) were cleaved after SII addition (XSII, lanes 2 and 3).
Discussion
Transcription serves as a sensitive DNA damage recognition mechanism for NER and increases the efficiency of damage removal on the transcribed relative to the nontranscribed strand of active genes. Most forms of damage targeted for repair by TCR also impede the translocation of RNAP complexes along DNA templates in vitro (6, 8, 33, 35, 36). Because bacteria, yeast, and mammals possess TCR, this mechanism may be evolutionarily conserved for efficient repair of DNA modifications that threaten genomic stability.
M1dG adducts, produced during lipid peroxidation, seem to be repaired by NER in both bacteria and human cells (23). Like other adducts, such as CPDs, which arrest RNAPs and are repaired by TCR, PdG, the stable analog of M1dG, also inhibits replication by DNA polymerases (27, 28, 37). Therefore, it was reasoned that M1dG or PdG in the transcribed strand of active genes might impede translocation by RNAPs and potentially initiate TCR.
To gain insight into the transcription-mediated recognition of MDA-induced DNA adducts in cells, site-specific M1dG and PdG adducts in a DNA template were analyzed for their effects on transcript elongation by T7 RNAP and mammalian RNAPII. Transcriptional arrest by both M1dG and PdG was less pronounced for the T7 enzyme, indicating that it is less sensitive to these guanine modifications than RNAPII. The extent of T7 RNAP and RNAPII arrest by M1dG depended on the base opposite the adduct. Both polymerases more readily bypassed N2-OPdG (expected from M1dG opening when paired with C) than the exocyclic M1dG opposite T. The stable, saturated ring of PdG strongly arrested T7 RNAP and completely halted RNAPII translocation, whereas M1dG paired with T was less of an impediment to the polymerases. These results suggest that the relative chemical stability and conformation of M1dG and PdG may play a role in determining polymerase behavior, because the sequence context for transcription was identical. Although the chemistry of M1dG has been studied only in naked DNA in solution (18), this study indicates that M1dG and PdG may interact differently with residues in the active site of the RNAPs to disrupt transcript elongation.
Both M1dG and PdG induce minor arrest sites for T7 RNAP, but not for RNAPII, in the sequence upstream of the adduct position. M1dG-induced arrest at these sites increased proportionally with T7 RNAP arrest at the adduct. The amount of arrest at each of these minor sites relative to the total arrest induced by the adduct remained constant over time as well. This finding suggested that T7 RNAP paused at these sites on all of the actively transcribed molecules and that the sites do not occur because of other damage present in some of the templates. In correlation, T7 RNAP arrest sites were observed upstream of PdG in two additional sequences (data not shown). Minor arrest sites have not been reported in similar studies on T7 RNAP transcription (7) and therefore may reflect DNA structural changes, particular to MDA adducts, that propagate along the helix. Although arrest at these sites was not observed with RNAPII, the changes in DNA structure imposed by M1dG and PdG may have contributed to their intense block of RNAPII.
Thus far, RNAPII arrest has been most prominent at types of DNA damage that cause significant structural distortions in DNA (6, 8, 33, 35, 36). The bends induced in the helical axis by CPDs and cisplatin intrastrand crosslinks may resemble those found in natural pause sequences for RNAPs (38). As the transcription bubble for the polymerase complex reaches the distortion, RNAPII may conform to a less active state that elongates the nascent transcript more slowly and is more sensitive to DNA base modifications that alter pairing with the incoming ribonucleotide.
Depending on the base opposite the adduct, the RNAPs encountered a planar but reactive M1dG, a nonplanar but stable PdG, or an acyclic N2-OPdG. NMR of N2-OPdG, formed upon pairing of M1dG in a short DNA duplex with cytosine, has revealed that the oxopropenyl group of N2-OPdG is accommodated within the minor groove and causes little distortion at the base pair (18). M1dG was predicted to result in distortions similar to those observed for the PdG, which participates in Hoogsteen pairing and rotates to a syn conformation at the glycosylic bond, placing the exocyclic ring in the major groove (39). The altered pairing conformations of the M1dG and PdG adducts as well as the conversion of M1dG to N2-OPdG upon pairing with an incoming CTP in the RNAP active site may pose a problem for templating these adducts for extension of the transcript. Unable to reconcile the pairing anomaly, RNAPII could fall into an arrested state, only to be reactivated for elongation by interaction with SII. RNAPII complexes arrested at CPDs and cisplatin adducts in vitro reverse translocate in response to SII, and the displacement of RNAPII from CPDs allowed access of photolyase to the lesion for photoreversal (33). A fraction of RNAPII complexes arrested at PdG also responded to SII, suggesting that the removal of stably arrested RNAPII from MDA adducts in the transcribed strand may be required before excision of the damage can occur.
M1dG has been reported to occur at levels as high as 6,500 adducts per cell in human tissues, which rivals 7,8 dihydro-8-oxoguanine, the most abundant form of base oxidation (14). 8-Oxo-dG as well as the ethenoadducts produced by lipid peroxidation are recognized by specific base excision repair glycosylases (9). However, M1dG seems to be repaired predominantly by NER and mismatch repair. Because of the frequency of mutations induced by M1dG (500-fold greater than dG in bacteria) (19, 20), this adduct poses a significant threat to genomic stability and must be efficiently removed from DNA. In mammalian cells, M1dG adducts in repetitive CpG sequences cause frameshifts, a type of mutation not commonly observed for other site-specific endogenous lesions (22). This finding points to a potential role for M1dG in the microsatellite instability observed in tumors and some sporadic cancers, most notably colorectal carcinomas. Because inflammation is common in cancer, atherosclerosis, and neurodegeneration, M1dG and other lipid peroxidation adducts may contribute to the progression of these diseases.
Although a role for TCR in the repair of endogenous DNA damage has not been clearly defined, the findings presented here suggest that TCR could be involved in the cellular processing of M1dG. If future experiments reveal that TCR is an important mechanism for M1dG repair in cells, then we may begin to consider whether the inability to repair MDA adducts in transcribed genes underlies the abnormal development of the neural and skeletal systems in the victims of Cockayne's syndrome.
Acknowledgments
We thank the Vanderbilt Mass Spectrometry Research Center for assistance. This work was supported by National Institutes of Health Grants CA87819 (to L.J.M.) and CA77712 (to S.T. and P.C.H.).
Abbreviations: AdMLP, adenovirus major late promoter; CPD, cyclobutane pyrimidine dimer; MDA, malondialdehyde; M1dG, pyrimido[1,2-α]purin-10(3H)-one; N2-OPdG, N2-(3-oxo-1-propenyl)-dG; NER, nucleotide excision repair; PdG, 1,N2-propanodeoxyguanine; RNAP, RNA polymerase; TCR, transcription-coupled repair; Xnts, nontranscribed strand template; Xts, transcribed strand template.
References
- 1.Hanawalt, P. C. (2002) Oncogene 21, 8949-8956. [DOI] [PubMed] [Google Scholar]
- 2.Mellon, I., Spivak, G. & Hanawalt, P. C. (1987) Cell 51, 241-249. [DOI] [PubMed] [Google Scholar]
- 3.Mellon, I. & Hanawalt, P. C. (1989) Nature 342, 95-98. [DOI] [PubMed] [Google Scholar]
- 4.Conconi, A., Bespalov, V. A. & Smerdon, M. J. (2002) Proc. Natl. Acad. Sci. USA 99, 649-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sweder, K. S. & Hanawalt, P. C. (1992) Proc. Natl. Acad. Sci. USA 89, 10696-10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tornaletti, S. & Hanawalt, P. C. (1999) Biochimie 81, 139-146. [DOI] [PubMed] [Google Scholar]
- 7.Jung, Y. & Lippard, S. J. (2003) J. Biol. Chem. 278, 52084-52092. [DOI] [PubMed] [Google Scholar]
- 8.Tornaletti, S., Patrick, S. M., Turchi, J. J. & Hanawalt, P. C. (2003) J. Biol. Chem. 278, 35791-35797. [DOI] [PubMed] [Google Scholar]
- 9.Memisoglu, A. & Samson, L. (2000) Mutat. Res. 451, 39-51. [DOI] [PubMed] [Google Scholar]
- 10.Plosky, B., Samson, L., Engelward, B. P., Gold, B., Schlaen, B., Millas, T., Magnotti, M., Schor, J. & Scicchitano, D. A. (2002) DNA Repair 1, 683-696. [DOI] [PubMed] [Google Scholar]
- 11.Le Page, F., Kwoh, E. E., Avrutskaya, A., Gentil, A., Leadon, S. A., Sarasin, A. & Cooper, P. K. (2000) Cell 101, 159-171. [DOI] [PubMed] [Google Scholar]
- 12.Tornaletti, S., Maeda, L. S., Lloyd, D. R., Reines, D. & Hanawalt, P. C. (2001) J. Biol. Chem. 276, 45367-45371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tornaletti, S., Maeda, L. S., Kolodner, R. D. & Hanawalt, P. C. (2004) DNA Repair 3, 483-494. [DOI] [PubMed] [Google Scholar]
- 14.Marnett, L. J. (2002) Toxicology 182, 219-222. [DOI] [PubMed] [Google Scholar]
- 15.Dedon, P. C., Plastaras, J. P., Rouzer, C. A. & Marnett, L. J. (1998) Proc. Natl. Acad. Sci. USA 95, 11113-11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burger, R. M. (1998) Chem. Rev. 98, 1153-1170. [DOI] [PubMed] [Google Scholar]
- 17.Kadlubar, F. F., Anderson, K. E., Haussermann, S., Lang, N. P., Barone, G. W., Thompson, P. A., MacLeod, S. L., Chou, M. W., Mikhailova, M., Plastaras, J., et al. (1998) Mutat. Res. 405, 125-133. [DOI] [PubMed] [Google Scholar]
- 18.Mao, H., Schnetz-Boutaud, N. C., Weisenseel, J. P., Marnett, L. J. & Stone, M. P. (1999) Proc. Natl. Acad. Sci. USA 96, 6615-6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fink, S. P., Reddy, G. R. & Marnett, L. J. (1997) Proc. Natl. Acad. Sci. USA 94, 8652-8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burcham, P. C. & Marnett, L. J. (1994) J. Biol. Chem. 269, 28844-28850. [PubMed] [Google Scholar]
- 21.Niedernhofer, L. J., Daniels, J. S., Rouzer, C. A., Greene, R. E. & Marnett, L. J. (2003) J. Biol. Chem. 278, 31426-31433. [DOI] [PubMed] [Google Scholar]
- 22.VanderVeen, L. A., Hashim, M. F., Shyr, Y. & Marnett, L. J. (2003) Proc. Natl. Acad. Sci. USA 100, 14247-14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson, K. A., Fink, S. P. & Marnett, L. J. (1997) J. Biol. Chem. 272, 11434-11438. [DOI] [PubMed] [Google Scholar]
- 24.Dosanjh, M. K., Chenna, A., Kim, E., Fraenkel-Conrat, H., Samson, L. & Singer, B. (1994) Proc. Natl. Acad. Sci. USA 91, 1024-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson, K. A., Mierzwa, M. L., Fink, S. P. & Marnett, L. J. (1999) J. Biol. Chem. 274, 27112-27118. [DOI] [PubMed] [Google Scholar]
- 26.Modrich, P. (1997) J. Biol. Chem. 272, 24727-24730. [DOI] [PubMed] [Google Scholar]
- 27.Hashim, M. F. & Marnett, L. J. (1996) J. Biol. Chem. 271, 9160-9165. [DOI] [PubMed] [Google Scholar]
- 28.Hashim, M. F., Schnetz-Boutaud, N. & Marnett, L. J. (1997) J. Biol. Chem. 272, 20205-20212. [DOI] [PubMed] [Google Scholar]
- 29.Gu, W. & Reines, D. (1995) J. Biol. Chem. 270, 11238-11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eilat, D., Hochberg, M., Fischel, R. & Laskov, R. (1982) Proc. Natl. Acad. Sci. USA 79, 3818-3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reines, D. (1991) J. Biol. Chem. 266, 10510-10517. [PMC free article] [PubMed] [Google Scholar]
- 32.Schnetz-Boutaud, N. C., Mao, H., Stone, M. P. & Marnett, L. J. (2000) Chem. Res. Toxicol 13, 90-95. [DOI] [PubMed] [Google Scholar]
- 33.Tornaletti, S., Reines, D. & Hanawalt, P. C. (1999) J. Biol. Chem. 274, 24124-24130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wind, M. & Reines, D. (2000) BioEssays 22, 327-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perlow, R. A., Kolbanovskii, A., Hingerty, B. E., Geacintov, N. E., Broyde, S. & Scicchitano, D. A. (2002) J. Mol. Biol. 321, 29-47. [DOI] [PubMed] [Google Scholar]
- 36.Schinecker, T. M., Perlow, R. A., Broyde, S., Geacintov, N. E. & Scicchitano, D. A. (2003) Nucleic Acids Res. 31, 6004-6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cordeiro-Stone, M., Makhov, A. M., Zaritskaya, L. S. & Griffith, J. D. (1999) J. Mol. Biol. 289, 1207-1218. [DOI] [PubMed] [Google Scholar]
- 38.Shilatifard, A., Conaway, R. C. & Conaway, J. W. (2003) Annu. Rev. Biochem. 72, 693-715. [DOI] [PubMed] [Google Scholar]
- 39.Singh, U. S., Moe, J. G., Reddy, G. R., Weisenseel, J. P., Marnett, L. J. & Stone, M. P. (1993) Chem. Res. Toxicol. 6, 825-836. [DOI] [PubMed] [Google Scholar]