

Abstract
Tumor-induced bone disease is a dynamic process that involves interactions with many cell types. Once metastatic cancer cells reach the bone, they are in contact with many different cell types that are present in the cell-rich bone marrow. These cells include the immune cells, myeloid cells, fibroblasts, osteoblasts, osteoclasts, and mesenchymal stem cells. Each of these cell populations can influence the behavior or gene expression of both the tumor cells and the bone microenvironment. Additionally, the tumor itself can alter the behavior of these bone marrow cells which further alters both the microenvironment and the tumor cells. While many groups focus on studying these interactions, much remains unknown. A better understanding of the interactions between the tumor cells and the bone microenvironment will improve our knowledge on how tumors establish in bone and may lead to improvements in diagnosing and treating bone metastases. This review details our current knowledge on the interactions between tumor cells that reside in bone and their microenvironment.
1. Introduction
Despite recent advances in early detection and therapeutic approaches, metastases still remain the major problem for cancer patients. In particular, bone metastases account for decreased quality of life and ultimately death of prostate, breast, and lung cancer patients. However, current therapeutic approaches are insufficient to effectively cure or prevent bone metastasis. Tumor metastasis is a tightly regulated multistep process, in which specific interactions between disseminating tumor cells and the cells constituting the recipient organ microenvironment play important roles. Increasing evidence supports the prometastatic functions of the microenvironment, with many studies indicating the importance of bone marrow cells in the metastatic niche. Many early studies have shown that these bone marrow cells set up a metastatic niche at the secondary site that allows for cells to establish [1, 2]. Subsequent studies have specifically isolated myeloid-derived suppressor cells [3–6], myofibroblast [7–9], and tumor-associated macrophages [10–12]. Each of these has some overlapping roles in metastasis, but each class of cells is a distinct bone marrow cell type with distinct roles in metastasis (summarized in Figure 1). While these classes of cells were isolated and shown to be important in metastases, many groups are still actively trying to clarify their precise molecular role in the metastatic process. Researchers expect that advanced knowledge on how these cells regulate the tumor microenvironment will allow development of novel therapeutic approaches to alter the niche less hospitable to the cells and therefore reducing or preventing tumor growth. It is also possible that understanding the niche will allow clinicians to better predict which patients may develop secondary disease and which organs may be affected.
Figure 1.
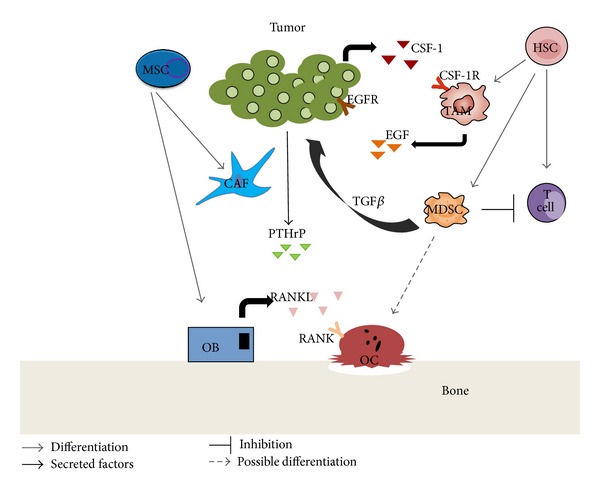
Tumor microenvironment interactions. Tumor cells interact with the cell populations present in the bone marrow. These include cells such as the fibroblasts, osteoblasts, osteoclasts, immune cells, and others as depicted here.
2. Bone Cells
The importance of interactions between tumor cells and other cells in the bone microenvironment was demonstrated in the 1990s by the work of Dr. Greg Mundy and others in the field. Their work strongly showed that there was a vicious cycle between the tumor cells and cells in the bone microenvironment. This work showed that tumor cells secreted factors that stimulated bone destruction, while bone destruction caused the release of growth factors from the bone matrix that further stimulated the tumor cell growth and production of factors that further enhanced bone destruction [13–15].
2.1. Osteoclasts
Osteoclasts are multinucleated cells that are responsible for bone resorption. A functional osteoclast has the ability to resorb mineralized bone matrix as part of normal bone remodeling that occurs during an individual's lifetime [16, 17]. Osteoclasts differentiate from myeloid progenitor cells under the influence of growth factors and cytokines such as macrophage colony stimulating factor (M-CSF) and receptor activator of nuclear factor kappa-B ligand (RANKL) [18, 19]. Physiological bone resorption is a tightly regulated process that involves signals from osteoblasts as well as signals from other cells found in the microenvironment. Osteoclast differentiation, maturation, and activation are dependent on RANK/RANKL/osteoprotegerin (OPG) signaling pathway [20, 21]. OPG is a soluble decoy receptor for RANKL, expressed by osteoblasts and negatively regulates osteoclast activation [19, 21, 22]. Deregulation of this process, such as too much resorption or too little, can lead to increased risk of fracture as well as other bone-related diseases [17].
Overactive osteoclasts can be detrimental and play a role in several diseases such as osteoporosis, pycnodysostosis, and Paget's disease, which occur due to increased bone resorption and bone loss [23]. Primary cancers of breast, lung, and prostate cancer have a propensity to metastasis to bone [22, 24–26]. These cancers cells secrete factors such as parathyroid hormone-related protein (PTHrP) which stimulate osteoclast-mediated bone destruction through the RANK/RANKL/OPG signaling pathway [22, 27]. During bone destruction growth factors including transforming growth factor beta (TGF-β), insulin-like growth factors (IGFs), and others are released from the bone matrix, which can stimulate further tumor growth and the production of tumor-derived factors (such as PTHrP) that can stimulate further bone destruction [15, 22, 28].
Even though much is known about the role of osteoclasts within the vicious cycle, many of their functions are yet to be explored. Increased osteoclast activity can be due to several different factors but the end results seem to be the same, which is that over activation of these cells promotes osteolysis and tumor cell growth, because factors released from bone during resorption stimulate tumor cell proliferation. CXCR4 is found on osteoclast precursors and regulates hematopoietic and tumor cell homing to bone. In studies where mice were reconstituted with Cxcr4 −/− hematopoietic cells had increased bone resorption and bone loss, specifically Cxcr4 −/− osteoclasts had higher resorptive activity and faster differentiation compared to control osteoclasts. The authors concluded that because the reconstituted mice had increased tumor growth in bone compared to control mice that disruption in CXCR4 may increase osteoclastogenesis leading to increased resorption and tumor burden [29]. A recent paper published by Ell et al. showed that mice injected with pre-miR-141 and pre-miR-219 had reduced osteoclastic activity and osteolytic bone metastasis [30]. Src −/− mice have shown impaired osteoclast functions [31, 32], and Src inhibitors have shown to suppress bone resorption effects [33]. These data suggest that Src may be an ideal therapeutic target to suppress tumor cells (frequently expressing high Src activities) and osteoclasts (requiring Src for function) at the same time [34, 35]. However, recent phase III clinical trial results showed that addition of dasatinib (a Src family kinase inhibitor) to docetaxel did not significantly improve overall survival of castration-resistant prostate cancer patients [36]. Araujo et al., the lead investigator of the failed clinical trial, pointed out that further understanding of Src inhibitors' mode of action could identify a better therapeutic role, because the clinical trial included heterogeneous patient population [36].
Inhibition of osteoclast activity by a variety of different factors has shown to decrease tumor burden in a mouse breast cancer bone metastasis model [37]. The most commonly used class of osteoclast inhibitors includes bisphosphonates, which bind to the bone promoting osteoclast apoptosis and inhibiting osteoclast mediated bone resorption [38]. Bisphosphonates (including zoledronate, alendronate, ibandronate, etc.) have been highly successful for reducing skeletal related events in patients with osteoporosis and with tumor-induced bone destruction [39, 40].
Alternatively, RANKL inhibitory antibodies have been promising both clinically and in preclinical models where they can increase time to skeletal related events (SRE) [41, 42]. Denosumab (Prolia, XGEVA), a monoclonal antibody against RANKL, was recently demonstrated to significantly increase time to SRE compared to a bisphosphonate, zoledronic acid, in breast and prostate cancer patients with bone disease [43, 44]. The debate between clinicians regarding which treatment is more efficacious continues, but both options are clearly effective and have their benefits. One concern regarding both treatments is the serious, yet rare, side-effects such as atypical fractures and osteonecrosis of the jaw [25, 37, 38]. Additionally, neither treatment has been shown to cure bone metastases or significantly increase survival in patients with bone metastases.
2.2. Osteoblasts
Osteoblasts are mesenchymal-origin cells lining the endosteal surface of bone and constitute approximately 4–6% of all bone cells. Osteoblasts produce organic matrix of bone and subsequently deposit inorganic components (e.g., calcium and phosphate), resulting in mineralized hard tissue. In addition to their physiologic functions, osteoblasts are important components of the metastatic bone microenvironment. The best-characterized role of osteoblasts in bone metastasis is described in the “vicious cycle hypothesis” where osteoblasts produce M-CSF and RANKL, two essential factors for osteoclastogenesis [14, 27, 45]. Subsequent studies followed to understand how molecular alterations in osteoblasts contribute to create a congenial microenvironment for metastatic tumor cells. Schneider et al. demonstrated that expansion of osteoblasts by administration of bone-anabolic agents such as parathyroid hormone (PTH) increased prostate tumor cell localization and growth in bone [46], suggesting that higher bone turnover rates (i.e., increased activity and number of osteoblasts) are associated with bone metastasis. Other studies have suggested that osteoblasts can function as a prometastatic population of cells. The first experimental evidence to support this come from the physiological phenomena of hematopoietic stem cell (HSC) homing in bone. HSCs migrate and repopulate the bone marrow immediately after birth, while the liver is the primary site of hematopoiesis during feral development. Taichman et al. demonstrated that CXCL12/SDF-1 (expressed by osteoblasts and endothelial cells) and its receptor (CXCR4, expressed by prostate cancer cells) regulate bone-tropism of prostate cancer cells [47]. In addition to the CXCL12/CXCR4/CXCR7 axis [48], Annexin II, expressed by osteoblasts and endothelium, regulates HSC adhesion, homing, and engraftment [49]. More recently, Jung et al. demonstrated that differential levels of growth arrest specific- (GAS-) 6 protein in the bone stromal cells (dominantly osteoblasts) induce metastatic tumor cell dormancy and determine site-specificity (i.e., increased localization in vertebrae and hind limb long bone compared with fore limb bones) of murine experimental metastasis model of human prostate cancer [50]. Furthermore, the same group provided pivotal evidence that osteoblastic niche for HSC is the direct target of tumor cell localization in bone [51]. The authors demonstrated that increasing the HSC niche size (via administration of PTH to induce osteoblast proliferation) promoted skeletal localization of prostate cancer cells, while decreasing the niche size (via conditional ablation of osteoblasts) reduced tumor cell localization. The author further investigated whether HSC compete with metastatic cancer cells for occupancy in the bone marrow. Administration of AMD3100 (a clinical regimen to mobilize HSC) mobilized metastatic cancer cells in the niche back into the circulation, indicating that HSC compete with bone-tropic cancer cells. These data collectively suggest that adhesion molecules and chemokine/chemokine receptors expressed on osteoblasts contribute to localization and subsequent growth of metastatic tumor cells in bone.
Increasing evidence supports that osteoblastic cells contribute to the metastatic progression by releasing cytokines and growth factors in the microenvironment. We have recently demonstrated that primary prostate tumor cells distantly instigate osteoblasts (via PTHrP in the systemic circulation) to increase vascular endothelial growth factor- (VEGF-) A, interleukin- (IL-) 6, and C-C chemokine ligand- (CCL-) 2 in the bone microenvironment and that VEGF-A and IL-6 in turn stimulate myeloid-derived suppressor cells with increased angiogenic potentials [52]. Indeed, hematopoietic lineage cells are dependent on bone cells (predominantly osteoblastic cells) for proliferation, mobilization, and function. This concept of “osteoimmunology” is now expanding to the role of osteoblasts in regulating other adjacent bone marrow cells (e.g., hematopoietic lineage cells with prometastatic functions, such as myeloid-derived suppressor cells). Interestingly, those prometastatic cytokines (in particular, VEGF-A and IL-6) stimulate osteoblasts to produce more VEGF-A and IL-6, suggesting that osteoblastic cells may function as an amplification mechanism of cytokines in the bone microenvironment.
3. Immune Cells
3.1. Myeloid Derive Suppressor Cells
The role and existence of myeloid derived suppressor cells (MDSCs) have been quite controversial among scientists since their initial discovery in 1978 [53]. Initially they were recognized as natural suppressor cells located in the bone marrow and spleen that were able to suppress cell-mediated immunity [54]. These cells did not contain cell surface markers that resembled T cells, B cells, macrophages, or natural killer cells which made it difficult to phenotypically characterize them [55, 56]. MDSCs are a heterogeneous population of myeloid cells that are at different stages of differentiation. This population includes immature macrophages, granulocytes, and dendritic cells as well as myeloid progenitor cells [5, 57, 58]. In mice these cells can be characterized into two major subtypes, monocytic-MDSCs and granulocytic-MDSCs, through lymphocyte antigens Ly6C and Ly6G [59]. Both subtypes have immune suppressive functions that are regulated through distinct mechanisms. Granulocytic-MDSCs have been found to express higher levels of ROS (reactive oxygen species) and low levels of NO (nitric oxide) verses monocytic-MDSCs expressing higher NO and lower ROS expression [59, 60]. Suppressive MDSCs are not found in healthy hosts; only their nonsuppressive counterpart iMCs (immature myeloid cells) are present. MDSCs need to be activated to express suppressive function and are only present at sites of chronic pathological conditions such as infection and cancer [53].
Recently these cells have been recognized to play an important role in tumor progression in many solid tumors by inhibiting antitumor immune responses and by promoting tolerance [58, 61]. These cells have been deemed protumorigenic due to their suppression of T cells, promotion of angiogenesis, invasion, and metastasis [5, 6, 53, 62]. MDSCs have been directly linked to promoting tumor invasion and metastasis through the production and secretion of factors such as MMPs, IFNγ, IL-10, and TGF-β [6, 61]. They have also been known to suppress the immune system by promoting tolerance by accumulating T regulatory cells [58, 61, 63]. In cancer, MDSCs are activated by tumor-secreted factors such as Toll-like receptors (TLRs), IL-4, IL-13, and TGF-β that activate several different signaling pathways [64]. Specific MDSC expansion in the tumor microenvironment is guided through tumor-derived factors and factors from the microenvironment that is context specific dictating which population (monocytic versus granulocytic) is increased [46].
The presence and accumulation of MDSCs has been well reported in several human cancers as well as different disease types in the last several years. A positive correlation between stage and MDSC peripheral density has been reported in both melanoma and head and neck squamous cell carcinoma (HNSCC) patients [65]. A 15 percent increase in circulating CD14+ HLA-DR−/lo cells was correlated with advanced stage (III and IV) as compared to early stage (I and II) HNSCC patients [65]. MDSCs containing the phenotype LIN−HLA-DR−CD33+CD11b+ have been isolated from the blood of patients with glioblastoma, breast, colon, lung, and kidney cancers [58, 62, 66, 67]. MDSCs containing the phenotype CD11b+CD14−HLA-DR−/lowCD33+CD15+ were found in the bone marrow and the peripheral blood of patients with active multiple myeloma compared with healthy donors [68].
The role that MDSCs play in human tumor-induced bone disease is still relatively unknown. With the use of mouse models, several published papers have demonstrated that MDSCs play an important role in bone metastasis. This is consistent with what is known about MDSC's contribution in the primary tumor environment. What is unknown is if MDSCs perform a direct role in promoting tumor establishment or tumor proliferation in bone by assisting the tumor itself or indirectly by secreting protumorigenic factors that prime the bone allowing it to become a hospitable host. Published papers have used mouse models to show that MDSCs can promote tumor growth in bone [52, 57, 69]. In a prostate cancer mouse model, it was demonstrated that tumor-derived PTHrP indirectly increases MDSC's angiogenic potential therefore contributing to tumor growth and angiogenesis [52]. Danilin and colleagues showed that MDSCs contribute to breast cancer osteolysis by inducing expression of Gli2 and PTHrP in tumor-bearing mice. These factors stimulate osteoclast-mediated bone destruction leading to increased bone lesions compared to control mice [57]. This group also showed that MDSCs isolated from tumor-bearing mice had the potential to differentiate into osteoclasts in vitro and in vivo [57]. Sawant et al. published this as well and explained that the reason MDSCs could differentiate into osteoclasts is because they are novel osteoclast progenitors driving bone metastasis during cancer progression [69].
MDSCs as a potential therapeutic target have been the topic of discussion since their identification. Studies have shown that eliminating MDSCs increases immune-surveillance and decreases tumor growth [63, 70, 71]. There are many different ways to target MDSCs including growth factors (anti-VEGF antibodies), chemokines (anti-CCL2 antibodies), cytotoxic drugs (Gemcitabine), enzyme inhibitors (amino-bisphosphonate), signaling inhibitors (sunitinib), and inducing differentiation (ATRA-All-trans retinoic acid) [72]. Src inhibitors have shown promise in targeting MDSCs by inhibiting their recruitment and MMP-9 gene expression in the tumor microenvironment [73]. Gemcitabine is a nucleoside metabolic inhibitor used to treat several types of cancers and has been shown to decrease MDSC levels in tumor-bearing mice by inhibiting expansion [74, 75]; however, its precise mechanism of MDSC inhibition is not fully understood. Bisphosphonates, which are routinely prescribed for cancer patients with bone metastasis, have also been demonstrated to decrease MDSC expansion in tumor-bearing mice through the reduction of MMP-9 expression [76]. Additionally, STAT3 inhibitors have also been successful at targeting MDSC in preclinical models [65]. While more studies are needed to understand the mechanisms of action, it is clear that targeting MDSCs clinically is both possible and promising therapeutically.
3.2. Tumor-Associated Macrophages
Macrophages are professional phagocytes that are differentiated from the myeloid lineage and are identified by the expression of certain markers as well as by the phenotypic differences among them [77, 78]. They have roles in development, homeostasis, tissue repair, and immunity and have been linked to many diseases including cancer [77, 78]. These are plastic cells and their phenotype is consistently modulated by the local microenvironment [79]. Macrophages can be classified by their immunological responses such as classically activated macrophages (M1) that are involved in inflammatory responses and alternatively activated macrophages (M2) that are involved in wound healing [77, 78, 80, 81]. M2 macrophages have been implicated in having protumor properties due to the cytokines, chemokines, and growth factors that they release such as VEGF, IL-10, TGF-β, EGF, and MMPs, among many others [77, 82]. These protumor macrophages are referred to as tumor-associated macrophages (TAMs) and are considered to be phenotypically similar to M2 macrophages [81, 83, 84].
Macrophage growth, chemotaxis, and differentiation are controlled by several chemokines including CCL-2 (also known as monocyte chemoattractant protein [MCP]-1) and growth factors such as CSF-1 [77]. CSF-1 is the regulator of the differentiation, proliferation, and survival of macrophages and their precursors [85]. CSF-1 overexpression has been implicated in the poor prognosis of several cancers and is currently being investigated as a possible therapeutic target [85–89]. In an invasive breast cancer mouse model, macrophages have been implicated in assisting tumor cell motility by participating in an epidermal growth factor- (EGF-) CSF-1 paracrine loop where tumor cells secrete CSF-1 and macrophages contain the corresponding receptor and vice versa [66, 67, 69, 75]. CCL2 is a chemokine that has been implicated in assisting cancer metastasis by mediating a crosstalk between cancer cells and the stromal cells that are present in the tumor microenvironment [90]. CCL2 is expressed by many tumor types as well as by the peripheral myeloid population [91]. Roca and colleagues showed that CCL2 stimulation induces peripheral blood monocytes to differentiate to M2 macrophages compared to unstimulated control monocytes [91].
In several papers macrophages have been reported to promote tumor initiation, progression, invasion, and metastasis [77, 79, 84]. Activated macrophages produce inflammatory factors such as reactive oxygen and nitrogen species in response to signals from other immune cells creating a constant inflamed stromal environment [80]. Chronic inflammation generates a stromal environment susceptible to mutations and has been linked to tumor initiation and growth. Progression of a mass from a neoplasia/adenoma to an early carcinoma is prompted through their secretion of VEGF and other angiogenic factors stimulating angiogenic switch [80]. Several groups have shown that an increase in macrophage density correlates with poor patient prognosis and survival in thyroid, lung, breast, and hepatocellular cancers [84, 89, 92, 93]. However, in other cancers such as stomach, colorectal, and pancreatic cancer, a high macrophage density is correlated with a good patient prognosis [80, 94].
The role of macrophages at the primary site is well established but their function at distant metastatic sites is still being highly investigated. Myeloid derived cells have been found to accumulate at distant sites priming the environment for tumor colonization [1, 2]. This notion of a premetastatic niche has been around for several years and has been found to be important in the primary site but has yet to be proven to exist in bone. This theory encompasses that once there is an established primary tumor site, hematopoietic progenitor cells are signaled to migrate from the bone marrow into secondary metastatic sites, such as the lung, and alter the microenvironment leading to activation of integrins and chemokines that promote attachment, survival, and growth of tumor cells [1]. Proving that this process occurs in bone has been challenging because hematopoietic progenitor cells originate in the bone marrow and do not have to migrate to reach the bone microenvironment. It is more likely that in bone microenvironment stromal cells including macrophages are “reeducated” by tumor-derived factors and begin priming the bone before tumor establishment occurs.
Several therapeutic approaches to target macrophages have been explored. One approach includes the inhibition of TGF-β signaling, which was demonstrated through preclinical studies by deleting TGFβ type 2 receptor (RII) in the macrophages. These studies demonstrated that animals with RII deficient macrophages displayed a reduction in tumor growth due to decreased secretion of myeloid factors that assist in tumor progression [82, 95]. Other therapeutic approaches target macrophage factors such as CSF-1 and its receptor [85, 87]. Currently, in clinical trials are small molecules and monoclonal antibodies that inhibit CSF-1 and prevent its binding, or the tyrosine kinase activity [96]. Other therapeutic strategies include preventing the recruitment of macrophages through inhibition of inflammatory monocyte trafficking with anti-CCL2 or CCR2 antibodies [96]. However, a recent phase II clinical trial for carlumab (anti-CCL2 monoclonal antibody) in metastatic prostate cancer patients did not support antitumoral activity as a single agent (PMID 22907596). Since TAMs, macrophages that have been educated by the tumor cells and assist in cancer progression have been implicated in causing resistance to tamoxifen in breast cancer and to androgen receptor antagonists in prostate cancer; a potential future therapeutic strategy could be to reeducate TAMs to express an antitumor phenotype that would work against the tumor instead of with it [79, 80, 83, 84].
3.3. Other Immune Cells
The bone marrow is a rich environment for many different immune cells including the B-cells, T-cells, and NK-cells, all of which are known to be important in cancer progression and soft tissue metastases [97]. Yet despite their proximity and abundance in bone metastases, relatively few studies have been performed to investigate their role in tumor-induced bone disease. This is in part due to the fact that the vast majority of bone metastasis studies utilize human tumors in immune-deficient mice, most commonly these models of T-cell deficient mice, but other models are also lacking B-cells (SCID, Rag 2−/−, Rag 1−/−). This makes understanding the role of T- and B-cells in bone metastases challenging.
T-cells are well-known to inhibit tumor growth, and in line with this finding it has been shown that stimulating T-cell response in mice reduced tumor burden in bone while reducing it blocks tumor growth in bone [98]. However, a recent study demonstrated that tumor associated T-cells can induce osteolytic bone disease prior to bone colonization. In this study they show that T-cell produced RANKL can induce osteoclastogenesis and bone destruction [99]. These data suggest that T-cells may have a dual role in bone disease in that they can reduce tumor growth but stimulate bone destruction. Regardless, since the majority of cancer and bone studies utilize T-cell deficient mice, it is clear that tumor cells can grow and metastasize to bone in the absence of T-cells.
Much less information exists describing the interactions between tumors in bone and B-cells or NK cells. A few manuscripts describe interactions between NK cells and tumors in bone. Specifically, they show that inhibiting NK cells increases tumor take in animal models of prostate cancer [100]. Other papers describe that NK cells are reduced in prostate cancer [101] but that forced expression of NK associated ligands can reduce tumor growth [102, 103]. Another immune lineage cell that has been implicated in cancer induced bone disease is the Megakaryocytes. Li et al. demonstrated that megakaryocytes could reduce prostate tumor cell growth and increase apoptosis, while their expansion in vivo reduced tumor-induced bone destruction [104].
4. Cancer-Associated Fibroblast (CAF)
Fibroblasts are another cell type that is abundant in the bone marrow microenvironment. CAFs are defined as fibroblast that reside in the tumor mass and are capable of promoting tumor growth. These cells are typically myofibroblast-like cells that express α-Smooth muscle actin (α-SMA), vimentin, and fibroblast specific protein-1 (FSP1) [105]. Some of the early studies showed that these fibroblasts could be recruited from the bone marrow to the tumor [106] and that they could stimulate malignant transformation [7], tumor cell growth, and invasion [107]. These effects on tumor cell growth are thought to be mediated through CXCL12 [108] and TGF-β [109]. Other pathways including Wnt signaling [110], bone morphogenetic proteins [111], and MMPs [112] have also been associated with their invasive potential. Other papers have demonstrated that in addition to factors secreted by fibroblasts that they can induce a more invasive phenotype through physical properties as well. One study showed that the increase in fibroblasts increased the stiffness of the tumor, which can activate pathways within the tumor cells that induces a more invasive phenotype [113]. Interestingly, our previous publications have demonstrated that rigidity influences gene expression in tumor cells [114]. Taken together, this suggests that rigidity may also influence expression in the fibroblast and further contribute to tumor-induced bone disease.
In addition to regulating invasiveness of the primary site, other studies have investigated the role of CAFs in the establishment of secondary sites. For example CAFs have been shown to be recruited to sites of liver metastases in colon cancer [115]. Additionally, CAFs have been associated with bone metastases, in which the loss of TGF-β receptor type II (RII) in the CAFs stimulated prostate cancer cell growth in the bone. More importantly, a recent paper by Joan Massague's group demonstrated that CAF content in triple negative tumors was associated with bone metastases, but not lung, in patient samples [9].
Because of their association with tumor growth, invasion, and metastasis, CAFs make a compelling target for the development of therapeutics. This is also compounded by the fact that CAFs have been associated with chemotherapeutic resistance [116–118]. One group found that CAF-induced resistance to tamoxifen could be reversed using metformin or arsenic trioxide [119]. Other groups tried to target fibroblast activating protein using an anti-FAP antibody (sibrotuzumab) in clinical trials of metastatic colorectal cancer, but these studies showed no significant efficacy [120–122]. However, the use of FAP conjugated therapies has been shown to increase drug efficacy and reduce side-effects associated with chemotherapy [123, 124].
5. Mesenchymal Stem Cell (MSC)
MSCs are a pluripotent population of bone marrow cells that can differentiate with many different cell types, including osteoblasts, adipocytes, chondrocytes, and fibroblasts. Similar to CAFs, MSCs have been shown to associate to sites of tumor in many different tumor types [125] and have been demonstrated to promote proliferation and migration [9, 126, 127]. In Massague's recent paper, they showed that MSCs induced a transcriptional shift in tumors similar to CAFs and that MSCs could recapitulate the CAF phenotype [9]. This suggests that in breast cancer CAFs and MSCs function similarly. However, unlike CAFs, MSC association with tumors has not been completely associated with negative outcomes, and in some cases MSCs may inhibit tumor growth. In fact, in some malignancies, such as multiple myeloma (MM), MSCs are used therapeutically. Some treatments for MM involve cell-based therapies in which patients are given autologous stem cell transplants, under the reasoning that this may recapitulate normal immune cells that may fight the disease [128]. A recent myeloma study suggests that using MSCs with high Fas ligand in multiple myeloma bearing mice increased apoptosis of the myeloma cells [129]. Since MSCs “track” to tumors some groups have developed modified MSCs as cargo for the delivery of therapeutics [125], but due to treatment concerns they have not been tested clinically. Clearly more needs to be understood about MSCs and how to select for more specific populations.
6. Conclusions
The bone microenvironment is a rich milieu of different cell types, with each having a specific role on tumor cells both that metastasize to the bone and to other sites. While the past decade has seen an increase in research devoted to understanding the role of each cell-type in different malignancies, there are still many questions. In reality it is likely that these cells work together to regulate tumor growth, invasion, and metastasis and that new approaches need to be undertaken to study the complex interaction that occur between these multiple cell types. Many groups are beginning to collaborate with systems biologists, engineers, and computer scientists to investigate these interactions in a more comprehensive manner. Once we better understand these interactions, more possibilities of therapeutic interventions will become possible.
Acknowledgments
The authors thank the Department of Veterans Affairs (1I01BX001957-JS), Cellular, Biochemical, and Molecular Science Training Program (5T32GM008554-DB), the National Cancer Institute (1R01CA163499-JS), and the Vanderbilt Center for Bone Biology (JS, SP) for financial support.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Kaplan RN, Rafii S, Lyden D. Preparing the “soil”: the premetastatic niche. Cancer Research. 2006;66(23):11089–11093. doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. Journal of Immunology. 2001;166(1):678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 4.Gabrilovich D, Ishida T, Oyama T, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92(11):4150–4166. [PubMed] [Google Scholar]
- 5.Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 6.Yang L, Huang J, Ren X, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunha GR, Hayward SW, Wang YZ. Role of stroma in carcinogenesis of the prostate. Differentiation. 2002;70(9-10):473–485. doi: 10.1046/j.1432-0436.2002.700902.x. [DOI] [PubMed] [Google Scholar]
- 8.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Research. 1999;59(19):5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang XH, Jin X, Malladi S, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154(5):1060–1073. doi: 10.1016/j.cell.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Research. 1996;56(20):4625–4629. [PubMed] [Google Scholar]
- 11.Lewis CE, Leek R, Harris A, McGee JO. Cytokine regulation of angiogenesis in breast cancer: the role of tumor-associated macrophages. Journal of Leukocyte Biology. 1995;57(5):747–751. doi: 10.1002/jlb.57.5.747. [DOI] [PubMed] [Google Scholar]
- 12.Dutsch-Wicherek M, Kazmierczak W. Creation of a suppressive microenvironment by macrophages and cancer-associated fibroblasts. Frontiers in Bioscience. 2013;18(3):1003–1016. doi: 10.2741/4159. [DOI] [PubMed] [Google Scholar]
- 13.Guise TA, Mundy GR. Cancer and bone. Endocrine Reviews. 1998;19(1):18–54. doi: 10.1210/edrv.19.1.0323. [DOI] [PubMed] [Google Scholar]
- 14.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nature Reviews: Cancer. 2002;2(8):584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 15.Sterling JA, Edwards JR, Martin TJ, Mundy GR. Advances in the biology of bone metastasis: how the skeleton affects tumor behavior. Bone. 2011;48(1):6–15. doi: 10.1016/j.bone.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 16.Martin TJ. Historically significant events in the discovery of RANK/RANKL/OPG. World Journal of Orthopedics. 2013;4(4):186–197. doi: 10.5312/wjo.v4.i4.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crockett JC, Rogers MJ, Coxon FP, Hocking LJ, Helfrich MH. Bone remodelling at a glance. Journal of Cell Science. 2011;124(7):991–998. doi: 10.1242/jcs.063032. [DOI] [PubMed] [Google Scholar]
- 18.Boyce B, Yao Z, Xing L. Osteoclasts have multiple roles in bone in addition to bone resorption. Critical Reviews in Eukaryotic Gene Expression. 2009;19(3):171–180. doi: 10.1615/critreveukargeneexpr.v19.i3.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyce BF, Schwarz EM, Xing L. Osteoclast precursors: cytokine-stimulated immunomodulators of inflammatory bone disease. Current Opinion in Rheumatology. 2006;18(4):427–432. doi: 10.1097/01.bor.0000231913.32364.32. [DOI] [PubMed] [Google Scholar]
- 20.Glass DA, II, Bialek P, Ahn JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Developmental Cell. 2005;8(5):751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 21.Roodman GD. Mechanisms of bone metastasis. The New England Journal of Medicine. 2004;350(16):1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 22.Feller L, Kramer B, Lemmer J. A short account of metastatic bone disease. Cancer Cell International. 2011;11, article 24 doi: 10.1186/1475-2867-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards JR. Osteoclasts: malefactors of disease and targets for treatment. Discovery Medicine. 2012;13(70):201–210. [PubMed] [Google Scholar]
- 24.Zhuang J, Zhang J, Lwin ST, et al. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS ONE. 2012;7(11) doi: 10.1371/journal.pone.0048871.e48871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mercatali L, Ricci M, Scarpi E, et al. RANK/RANK-L/OPG in patients with bone metastases treated with anticancer agents and zoledronic acid: a prospective study. International Journal of Molecular Sciences. 2013;14(6):10683–10693. doi: 10.3390/ijms140610683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin R, Sterling JA, Edwards JR, et al. Activation of NF-kappa B signaling promotes growth of prostate cancer cells in bone. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0060983.e60983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nature Reviews Cancer. 2011;11(6):411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clohisy DR, Ogilvie CM, Carpenter RJ, Ramnaraine MLR. Localized, tumor-associated osteolysis involves the recruitment and activation of osteoclasts. Journal of Orthopaedic Research. 1996;14(1):2–6. doi: 10.1002/jor.1100140103. [DOI] [PubMed] [Google Scholar]
- 29.Hirbe AC, Rubin J, Uluçkan Ö, et al. Disruption of CXCR4 enhances osteoclastogenesis and tumor growth in bone. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(35):14062–14067. doi: 10.1073/pnas.0705203104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ell B, Mercatali L, Ibrahim T, et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell. 2013;24(4):542–556. doi: 10.1016/j.ccr.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR. Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. Journal of Clinical Investigation. 1992;90(4):1622–1627. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64(4):693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Gomez A, Ocio EM, Crusoe E, et al. Dasatinib as a bone-modifying agent: anabolic and anti-resorptive effects. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0034914.e34914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyce B, Xing L. Src inhibitors in the treatment of metastatic bone disease: rationale and clinical data. Clinical Investigation. 2011;1(12):1695–1706. doi: 10.4155/cli.11.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallick GE, Corn PG, Zurita AJ, Lin S. Small-molecule protein tyrosine kinase inhibitors for the treatment of metastatic prostate cancer. Future Medicinal Chemistry. 2012;4(1):107–119. doi: 10.4155/fmc.11.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Araujo JC, Trudel GC, Saad F, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. The Lancet Oncology. 2013;14(13):1307–1316. doi: 10.1016/S1470-2045(13)70479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canon J, Bryant R, Roudier M, Branstetter DG, Dougall WC. RANKL inhibition combined with tamoxifen treatment increases anti-tumor efficacy and prevents tumor-induced bone destruction in an estrogen receptor-positive breast cancer bone metastasis model. Breast Cancer Research and Treatment. 2012;135(3):771–780. doi: 10.1007/s10549-012-2222-2. [DOI] [PubMed] [Google Scholar]
- 38.Boyce BF. Advances in osteoclast biology reveal potential new drug targets and new roles for osteoclasts. Journal of Bone and Mineral Research. 2013;28(4):711–722. doi: 10.1002/jbmr.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khosla S, Bilezikian JP, Dempster DW, et al. Benefits and risks of bisphosphonate therapy for osteoporosis. Journal of Clinical Endocrinology and Metabolism. 2012;97(7):2272–2282. doi: 10.1210/jc.2012-1027. [DOI] [PubMed] [Google Scholar]
- 40.McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. The American Journal of Medicine. 2013;126(1):13–20. doi: 10.1016/j.amjmed.2012.06.023. [DOI] [PubMed] [Google Scholar]
- 41.Canon J, Bryant R, Roudier M, et al. Inhibition of RANKL increases the anti-tumor effect of the EGFR inhibitor panitumumab in a murine model of bone metastasis. Bone. 2010;46(6):1613–1619. doi: 10.1016/j.bone.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Holland PM, Miller R, Jones J, et al. Combined therapy with the RANKL inhibitor RANK-Fc and rhApo2L/TRAIL/dulanermin reduces bone lesions and skeletal tumor burden in a model of breast cancer skeletal metastasis. Cancer Biology & Therapy. 2010;9(7):539–550. doi: 10.4161/cbt.9.7.11266. [DOI] [PubMed] [Google Scholar]
- 43.Stopeck AT, Lipton A, Body J, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. Journal of Clinical Oncology. 2010;28(35):5132–5139. doi: 10.1200/JCO.2010.29.7101. [DOI] [PubMed] [Google Scholar]
- 44.Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. The Lancet. 2011;377(9768):813–822. doi: 10.1016/S0140-6736(10)62344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guise TA, Yin JJ, Taylor SD, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. The Journal of Clinical Investigation. 1996;98(7):1544–1549. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider A, Kalikin LM, Mattos AC, et al. Bone turnover mediates preferential localization of prostate cancer in the skeleton. Endocrinology. 2005;146(4):1727–1736. doi: 10.1210/en.2004-1211. [DOI] [PubMed] [Google Scholar]
- 47.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Research. 2002;62(6):1832–1837. [PubMed] [Google Scholar]
- 48.Sun X, Cheng G, Hao M, et al. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer and Metastasis Reviews. 2010;29(4):709–722. doi: 10.1007/s10555-010-9256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jung Y, Wang J, Song J, et al. Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood. 2007;110(1):82–90. doi: 10.1182/blood-2006-05-021352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jung Y, Shiozawa Y, Wang J, et al. Prevalence of prostate cancer metastases after intravenous inoculation provides clues into the molecular basis of dormancy in the bone marrow microenvironment. Neoplasia. 2012;14(5):429–439. doi: 10.1596/neo.111740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung Y, Shiozawa Y, Wang J, et al. Annexin-2 is a regulator of stromal cell-derived factor-1/CXCL12 function in the hematopoietic stem cell endosteal niche. Experimental Hematology. 2011;39(2):151.e1–166.e1. doi: 10.1016/j.exphem.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park SI, Lee C, Sadler WD, et al. Parathyroid hormone-related protein drives a CD11b+Gr1+ cell-mediated positive feedback loop to support prostate cancer growth. Cancer Research. 2013;73(22):6574–6583. doi: 10.1158/0008-5472.CAN-12-4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nature Reviews Cancer. 2013;13(10):739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bennett JA, Srinivasa Rao V, Mitchell MS. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells. Proceedings of the National Academy of Sciences of the United States of America. 1978;75(10):5142–5144. doi: 10.1073/pnas.75.10.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slavin S, Strober S. Induction of allograft tolerance after total lymphoid irradiation (TLI): development of suppressor cells of the mixed leukocyte reaction (MLR) The Journal of Immunology. 1979;123(2):942–946. [PubMed] [Google Scholar]
- 56.Duwe AK, Singhal SK. The immunoregulatory role of bone marrow. I. Suppression of the induction of antibody responses to T-dependent and T-independent antigens by cells in the bone marrow. Cellular Immunology. 1979;43(2):362–371. doi: 10.1016/0008-8749(79)90180-1. [DOI] [PubMed] [Google Scholar]
- 57.Danilin S, Merkel AR, Johnson JR, Johnson RW, Edwards JR, Sterling JA. Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology. 2012;1(9):1484–1494. doi: 10.4161/onci.21990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood. 2011;117(24):6532–6541. doi: 10.1182/blood-2010-11-317321. [DOI] [PubMed] [Google Scholar]
- 59.Youn J, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. Journal of Immunology. 2008;181(8):5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Movahedi K, Guilliams M, Van Den Bossche J, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell suppressive activity. Blood. 2008;111(8):4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 61.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Research. 2008;68(13):5439–5449. doi: 10.1158/0008-5472.CAN-07-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kusmartsev S, Su Z, Heiser A, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clinical Cancer Research. 2008;14(24):8270–8278. doi: 10.1158/1078-0432.CCR-08-0165. [DOI] [PubMed] [Google Scholar]
- 63.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. The Journal of Immunology. 2007;179(2):977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 64.Delano MJ, Scumpia PO, Weinstein JS, et al. MyD88-dependent expansion of an immature GR-1 +CD11b+ population induces T cell suppression and Th2 polarization in sepsis. Journal of Experimental Medicine. 2007;204(6):1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vasquez-Dunddel D, Pan F, Zeng Q, et al. STAT3 regulates arginase-i in myeloid-derived suppressor cells from cancer patients. The Journal of Clinical Investigation. 2013;123(4):1580–1589. doi: 10.1172/JCI60083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. International Immunopharmacology. 2011;11(7):802–807. doi: 10.1016/j.intimp.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunology, Immunotherapy. 2009;58(1):49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Görgün GT, Whitehill G, Anderson JL, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood. 2013;121(15):2975–2987. doi: 10.1182/blood-2012-08-448548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sawant A, Deshane J, Jules J, et al. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Research. 2013;73(2):672–682. doi: 10.1158/0008-5472.CAN-12-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. The Journal of Immunology. 2001;166(9):5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 71.Najjar YG, Finke JH. Clinical perspectives on targeting of myeloid derived suppressor cells in the treatment of cancer. Frontiers in Oncology. 2013;3, article 49 doi: 10.3389/fonc.2013.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature Reviews Immunology. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liang W, Kujawski M, Wu J, et al. Antitumor activity of targeting Src kinases in endothelial and myeloid cell compartments of the tumor microenvironment. Clinical Cancer Research. 2010;16(3):924–935. doi: 10.1158/1078-0432.CCR-09-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. International Immunopharmacology. 2009;9(7-8):900–909. doi: 10.1016/j.intimp.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 75.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clinical Cancer Research. 2005;11(18):6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 76.Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Research. 2007;67(23):11438–11446. doi: 10.1158/0008-5472.CAN-07-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pollard JW. Trophic macrophages in development and disease. Nature Reviews Immunology. 2009;9(4):259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nature Reviews Cancer. 2004;4(1):71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 80.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rodríguez D, Silvera R, Carrio R, et al. Tumor microenvironment profoundly modifies functional status of macrophages: peritoneal and tumor-associated macrophages are two very different subpopulations. Cellular Immunology. 2013;283(1-2):51–60. doi: 10.1016/j.cellimm.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pang Y, Gara SK, Achyut BR, et al. TGF-β Signaling in myeloid cells is required for tumor metastasis. Cancer Discovery. 2013;3(8):936–951. doi: 10.1158/2159-8290.CD-12-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–1410. doi: 10.1016/j.imbio.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 84.Xuan QJ, Wang JX, Nanding A, et al. Tumor-associated macrophages are correlated with tamoxifen resistance in the postmenopausal breast cancer patients. Pathology Oncology Research. 2014;20(3):619–624. doi: 10.1007/s12253-013-9740-z. [DOI] [PubMed] [Google Scholar]
- 85.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. Journal of Experimental Medicine. 2001;193(6):727–739. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Groblewska M, Mroczko B, Wereszczyńska-Siemiatkowska U, Myśliwiec P, Kedra B, Szmitkowski M. Serum levels of granulocyte colony-stimulating factor (G-CSF) and macrophage colony-stimulating factor (M-CSF) in pancreatic cancer patients. Clinical Chemical Laboratory Medicine. 2007;45(1):30–34. doi: 10.1515/CCLM.2007.025. [DOI] [PubMed] [Google Scholar]
- 87.Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. Journal of Mammary Gland Biology and Neoplasia. 2002;7(2):147–162. doi: 10.1023/a:1020399802795. [DOI] [PubMed] [Google Scholar]
- 88.Sapi E, Kacinski BM. The role of CSF-1 in normal and neoplastic breast physiology. Proceedings of the Society for Experimental Biology and Medicine. 1999;220(1):1–8. doi: 10.1046/j.1525-1373.1999.d01-1.x. [DOI] [PubMed] [Google Scholar]
- 89.Zhu XD, Zhang JB, Zhuang PY, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. Journal of Clinical Oncology. 2008;26(16):2707–2716. doi: 10.1200/JCO.2007.15.6521. [DOI] [PubMed] [Google Scholar]
- 90.Li M, Knight DA, Snyder LA, Smyth MJ, Stewart TJ. A role for CCL2 in both tumor progression and immunosurveillance. OncoImmunology. 2013;2(7) doi: 10.4161/onci.25474.e25474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roca H, Varcos ZS, Sud S, Craig MJ, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. The Journal of Biological Chemistry. 2009;284(49):34342–34354. doi: 10.1074/jbc.M109.042671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ryder M, Ghossein RA, Ricarte-Filho JCM, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocrine-Related Cancer. 2008;15(4):1069–1074. doi: 10.1677/ERC-08-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen JJW, Lin Y, Yao P, et al. Tumor-associated macrophages: the double-edged sword in cancer progression. Journal of Clinical Oncology. 2005;23(5):953–964. doi: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- 94.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Research. 2006;66(2):605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 95.Novitskiy SV, Pickup MW, Chytil A, Polosukhina D, Owens P, Moses HL. Deletion of TGF-β signaling in myeloid cells enhances their anti-tumorigenic properties. Journal of Leukocyte Biology. 2012;92(3):641–651. doi: 10.1189/jlb.1211639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends in Immunology. 2013;34(2):81–89. doi: 10.1016/j.it.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 97.Park SI, Soki FN, Mccauley LK. Roles of bone marrow cells in skeletal metastases: no longer bystanders. Cancer Microenvironment. 2011;4(3):237–246. doi: 10.1007/s12307-011-0081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang K, Kim S, Cremasco V, et al. CD8+ T cells regulate bone tumor burden independent of osteoclast resorption. Cancer Research. 2011;71(14):4799–4808. doi: 10.1158/0008-5472.CAN-10-3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Monteiro AC, Leal AC, Gonçalves-Silva T, et al. T cells induce pre-metastatic osteolytic disease and help bone metastases establishment in a mouse model of metastatic breast cancer. PLoS ONE. 2013;8(7) doi: 10.1371/journal.pone.0068171.e68171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. The Journal of Experimental Medicine. 2005;202(5):583–588. doi: 10.1084/jem.20050994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu G, Lu S, Wang X, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. Journal of Clinical Investigation. 2013;123(10):4410–4422. doi: 10.1172/JCI69369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(20):11521–11526. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413(6852):165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li X, Koh AJ, Wang Z, et al. Inhibitory effects of megakaryocytic cells in prostate cancer skeletal metastasis. Journal of Bone and Mineral Research. 2011;26(1):125–134. doi: 10.1002/jbmr.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Madar S, Goldstein I, Rotter V. 'Cancer associated fibroblasts'—more than meets the eye. Trends in Molecular Medicine. 2013;19(8):447–453. doi: 10.1016/j.molmed.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 106.Direkze NC, Hodivala-Dilke K, Jeffery R, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Research. 2004;64(23):8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 107.Cheng N, Bhowmick NA, Chytil A, et al. Loss of TGf-β type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-α-, MSP-and HGF-mediated signaling networks. Oncogene. 2005;24(32):5053–5068. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 109.Bhowmick NA, Chytil A, Plieth D, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303(5659):848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 110.Placencio VR, Sharif-Afshar A, Li X, et al. Stromal transforming growth factor-β signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Research. 2008;68(12):4709–4718. doi: 10.1158/0008-5472.CAN-07-6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Owens P, Polikowsky H, Pickup MW, et al. Bone morphogenetic proteins stimulate mammary fibroblasts to promote mammary carcinoma cell invasion. PLoS ONE. 2013;8(6) doi: 10.1371/journal.pone.0067533.e67533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eck SM, Côté AL, Winkelman WD, Brinckerhoff CE. CXCR4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Molecular Cancer Research. 2009;7(7):1033–1044. doi: 10.1158/1541-7786.MCR-09-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer and Metastasis Reviews. 2009;28(1-2):113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ruppender NS, Merkel AR, Martin TJ, Mundy GR, Sterling JA, Guelcher SA. Matrix rigidity induces osteolytic gene expression of metastatic breast cancer cells. PLoS ONE. 2010;5(11) doi: 10.1371/journal.pone.0015451.e15451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Suetsugu A, Osawa Y, Nagaki M, et al. Imaging the recruitment of cancer-associated fibroblasts by liver-metastatic colon cancer. Journal of Cellular Biochemistry. 2011;112(3):949–953. doi: 10.1002/jcb.23011. [DOI] [PubMed] [Google Scholar]
- 116.Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. Journal of Clinical Investigation. 2006;116(7):1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer and Metastasis Reviews. 2013;32(1-2):303–315. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tchou J, Conejo-Garcia J. Targeting the tumor stroma as a novel treatment strategy for breast cancer: shifting from the neoplastic cell-centric to a stroma-centric paradigm. Advances in Pharmacology. 2012;65:45–61. doi: 10.1016/B978-0-12-397927-8.00003-8. [DOI] [PubMed] [Google Scholar]
- 119.Martinez-Outschoorn UE, Goldberg A, Lin Z, et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biology & Therapy. 2011;12(10):924–938. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scott AM, Wiseman G, Welt S, et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clinical Cancer Research. 2003;9(5):1639–1647. [PubMed] [Google Scholar]
- 121.Hofheinz RD, Al-Batran S, Hartmann F, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26(1):44–48. doi: 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 122.Togo S, Polanska UM, Horimoto Y, Orimo A. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers. 2013;5(1):149–169. doi: 10.3390/cancers5010149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huang C, Suen C, Lin C, et al. Cleavage-site specificity of prolyl endopeptidase FAP investigated with a full-length protein substrate. Journal of Biochemistry. 2011;149(6):685–692. doi: 10.1093/jb/mvr017. [DOI] [PubMed] [Google Scholar]
- 124.LeBeau AM, Brennen WN, Aggarwal S, Denmeade SR. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Molecular Cancer Therapeutics. 2009;8(5):1378–1386. doi: 10.1158/1535-7163.MCT-08-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chan JK, Lam PY. Human mesenchymal stem cells and their paracrine factors for the treatment of brain tumors. Cancer Gene Therapy. 2013;20(10):539–543. doi: 10.1038/cgt.2013.59. [DOI] [PubMed] [Google Scholar]
- 126.Liang-Kuan B, Nan Z, Cheng L, et al. Kidney cancer cells secrete IL-8 to activate Akt and promote migration of mesenchymal stem cells. Urologic Oncology. 2014;32(5):607–612. doi: 10.1016/j.urolonc.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 127.Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 128.Gasparetto C. Stem cell transplantation for multiple myeloma. Cancer Control. 2004;11(2):119–129. doi: 10.1177/107327480401100218. [DOI] [PubMed] [Google Scholar]
- 129.Atsuta I, Liu S, Miura Y, et al. Mesenchymal stem cells inhibit multiple myeloma cells via the Fas/Fas ligand pathway. Stem Cell Research Therapy. 2013;4(5):p. 111. doi: 10.1186/scrt322. [DOI] [PMC free article] [PubMed] [Google Scholar]