

Abstract
The zebrafish has become an ideal vertebrate animal system for investigating cardiac development due to its genetic tractability, external fertilization, early optical clarity and ability to survive without a functional cardiovascular system during development. In particular, recent advances in imaging techniques and the creation of zebrafish transgenics now permit the in vivo analysis of the dynamic cellular events that transpire during cardiac morphogenesis. As a result, the combination of these salient features provides detailed insight as to how specific genes may influence cardiac development at the cellular level. In this review, we will highlight how the zebrafish has been utilized to elucidate not only the underlying mechanisms of cardiac development and human congenital heart diseases (CHDs), but also potential pathways that may modulate cardiac regeneration. Thus, we have organized this review based on the major categories of CHDs – structural heart, functional heart, and vascular/great vessel defects, and will conclude with how the zebrafish may be further used to contribute to our understanding of specific human CHDs in the future.
Keywords: model organism, high resolution in vivo imaging, cellular mechanisms
Backup keywords: zebrafish, genetics, transgenics, congenital heart disease
INTRODUCTION
The development of the heart is a dynamic and complex process starting with the specification of myocardial, endocardial, and vascular/endothelial precursors and culminating in the morphogenesis of these differentiated cells/tissues into a functional cardiac pumping organ. Because of the exquisite coordination of these events, the slightest cardiac developmental perturbation (genetic or environmental) can easily lead to catastrophic heart defects and subsequent embryonic/fetal demise. Thus, congenital heart diseases (CHDs), including both structural and functional cardiovascular defects, are amongst the most common and most devastating birth defects in humans, occurring in about 5% of live births, and resulting in significant mortality and morbidity [1].
Identifying suitable vertebrate animal model systems that permit the detailed investigations of cardiac development and the mechanisms underlying CHDs can be challenging. However, the zebrafish is particularly well suited to studies of cardiovascular development because, unlike mouse and chick embryos, they do not completely depend on a functional cardiovascular system. The zebrafish’s small embryonic/larval size allows them to receive sufficient oxygen by passive diffusion even when manifesting the most severe cardiovascular defects [2]. Furthermore, zebrafish embryos develop rapidly and are relatively easy to analyze because of their optical transparency and external fertilization [2]. In particular, these specific advantages offer the possibility of live in vivo imaging of the cellular and physiologic processes during cardiac morphogenesis, and recent imaging advances have been developed and utilized to study these dynamic events [3, 4]. Additionally, because of their large offspring numbers and rapid development, the zebrafish is genetically amenable to forward genetic screens, which have led to the identification of a wide range of cardiovascular mutant phenotypes including many that recapitulate known CHDs [5–11]. Conversely, a number of tools, including morpholinos [12], TILLING [13], TALEN [14], and zinc finger nucleases [15], have been created to perturb specific genes of interest (reverse genetics) and subsequently used to model candidate CHD genes. Moreover, the zebrafish is particularly sensitive to small molecule treatment and thus suitable to chemical genetic studies and screens to identify additional cellular and molecular pathways, which may regulate cardiovascular development [16]. Finally, zebrafish and mammalian hearts exhibit fairly well conserved structures including atria, ventricles, cardiac valves and a cardiac conduction system [8, 11, 17–20]. These features are remarkably useful in discovering zebrafish cardiovascular mutants that provide insight into human cardiovascular diseases. Thus, the combination of these advantages makes the zebrafish an attractive vertebrate model organism to complement human and other mammalian studies of CHDs and cardiac development.
STRUCTURAL HEART DEFECTS
A large proportion of CHDs are due to defects in specific structures of the heart, which can lead to hemodynamic compromise and catastrophic clinical outcomes. In order to illuminate the etiologies of these defects, better understanding of the cellular and molecular events of cardiac development is required. To this end, we will review the contribution of zebrafish heart studies to our overall knowledge of cardiac development.
Cardiac specification and differentiation
The heart is one of the first organs to develop in vertebrates. The cardiac progenitor cells that give rise to the heart can be fate mapped as early as the blastula stage in zebrafish [19]. At five hours post-fertilization (hpf), these cell-labeling studies have revealed that two cardiac progenitor populations (atrial and ventricular) exist in the lateral marginal zone on either side of the embryo and are restricted by retinoic acid signaling [21] (Figure 1A). More specifically, the ventricular pool appears to reside more dorsally and closer to the margin than the atrial pool. On the other hand, endocardial progenitor cells appear to be located across the lateral margin without any specific spatial organization [22, 23]. By 6–9 somites, cardiac progenitors are observed within the posterior half of the anterior lateral plate mesoderm (ALPM) and are marked by the transcription factor genes, gata4 and hand2 [24]. As they differentiate into ventricular cardiomyocytes (ventricular myosin heavy chain/vmhc expressing cells) medially at 12 somites and atrial cardiomyocytes (atrial myosin heavy chain/amhc/myh6 expressing cells) laterally at 15 somites [25], these cardiac precursors begin to express both the cardiac transcription factor gene nkx2.5 and the cardiac regulatory myosin light chain gene myl7 [26] (Figure 1B, C). This cardiac field within the ALPM is restricted anteriorly by cranial vascular and hematopoietic precursors [24] and posteriorly by pectoral fin precursors [27]. In cloche mutants, the cardiac field is delimited anteriorly resulting in the expansion of the cardiac field and larger hearts as well as the reduction of the cranial vascular and hematopoietic precursors and subsequent loss of endothelial and blood cells, respectively [24]. Conversely, overexpression of the endothelial/hematopoietic transcription factor genes etsrp and scl in zebrafish embryos leads to the expansion of the cranial vascular and hematopoietic precursors and a reduction of the cardiac progenitors. Furthermore, retinoic acid signaling, which is required for pectoral fin precursor specification, appears to limit the cardiac field posteriorly within the ALPM [27]. Morpholino knockdown of hoxb5b, a downstream target of retinoic acid signaling in the pectoral fin, results in the expansion of atrial cardiac progenitors posteriorly into the forelimb field without perturbing pectoral fin precursors.
Figure 1.
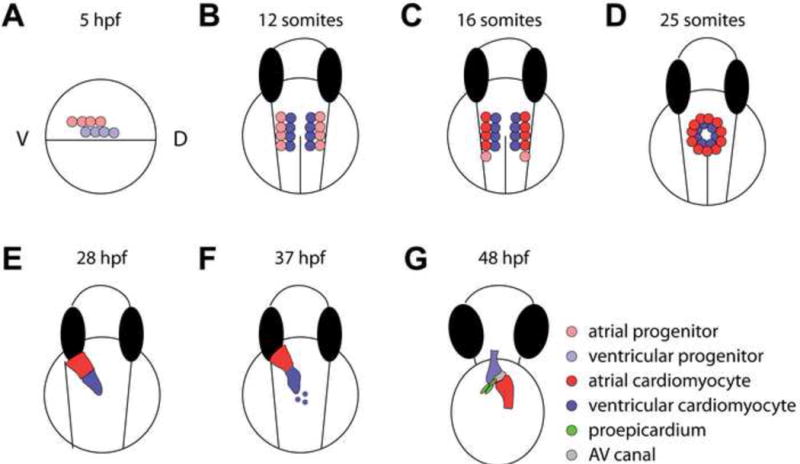
Zebrafish heart developmental stages. (A) Fate mapping experiments identified cardiac progenitors in the lateral marginal zones on either side of the gastrula at ~5 hours post fertilization (hpf), with ventricular progenitors located more dorsally, and closer to the margin than atrial progenitors. (B, C) Cardiac progenitor cells continue to migrate towards the midline into the anterior lateral plate mesoderm (ALPM). From there, (B) the ventricular cardiomyocytes differentiate at the 12 somite stage (~16 hpf) as detected by vmhc expression, whereas (C) the atrial cardiomyocytes differentiate later at the 15 somite stage as detected by amhc/myh6 expression. During 19–26 hpf, additional differentiating atrial cardiomyocytes are added to the venous pole. (D) The progenitor cells further migrate to the midline to form a cardiac disc around the 25 somite stage (~22 hpf), where ventricular and atrial cardiomyocytes are located in the medial and peripheral regions of the disc, respectively. (E) As the cardiac tube extends and turns, the arterial pole is anterior and right-sided, whereas the venous pole is more posterior and left-sided. (F) Late differentiating cardiomyocytes add to the arterial pole from 34–48 hpf, and the heart tube begins to constrict at the future AV canal and loop at ~37 hpf. (G) By 48 hpf, the cardiac chambers have undergone ballooning/expansion, while the AV canal remains constricted. The first proepicardial cells can be observed at this stage near the ventral surface of the looped heart, next to the AV canal. These cells will proliferate and migrate to become the epicardium, which eventually covers the myocardium.
A conserved network of cardiac transcription factors is required for cardiac specification and differentiation in zebrafish and includes HAND, NKX2.5, T-BOX, and GATA cardiac transcription factors as similarly observed in mammalian cardiac development. Recent studies in the zebrafish reveal that Gata 4, 5, and 6 transcription factors may act in combination to control cardiac specification. Furthermore, gata4 is expressed in not only the cardiac progenitors residing early in the ALPM, but also interestingly in regenerating adult cardiomyocytes [24, 28]. The zebrafish faust mutant, which harbors a mutation in gata5, exhibits a reduction in cardiac progenitor cells, and gata5 overexpression can result in the formation of ectopic cardiac progenitor cells [29]. Additionally, Gata5 transcriptional activity appears to be regulated through interactions with the Gridlock/Hey2, Hairy-related basic helix-loop-helix (bHLH), cardiac transcription factor, and gridlock/hey2 overexpression can lead to reduced cardiac progenitors [30]. Though loss of function of Gata 4, 5, and 6 individually can lead to severe cardiac defects, cardiac progenitors can still be generated albeit at reduced numbers. However, combinatorial reductions of these Gata transcription factors reveal that Gata 5 and 6 may act redundantly to specify cardiac progenitors as depletion of both these factors results in zebrafish embryos without hearts [31, 32]. In contrast to mammals where two HAND transcription factors, HAND1 [33] and HAND2 [34], exist to regulate cardiogenesis, only Hand2 is present in the zebrafish to control myocardial differentiation [24, 35]. hand2 mutants still develop nkx2.5 expressing cardiac progenitor cells; however, it appears that these cells fail to efficiently differentiate into mature myl7 expressing cardiomyocytes, suggesting that hand2 may primarily act to regulate cardiac differentiation. Interestingly, recent studies reveal that Nkx2.5 may act redundantly with Nkx2.7 to promote heart tube extension early but chamber size later in cardiac development, thus suggesting that Nkx2.5/2.7 may not be necessary for early cardiac specification [36, 37]. Furthermore, Hand2 also appears to regulate tbx5 expression in the zebrafish, and similar to Holt-Oram patients, who harbor TBX5 mutations, the tbx5/heartstring zebrafish mutants also display cardiac and pectoral fin (limb) defects [38]. In addition to Tbx5, other T-box transcription factors, including Tbx2 and Tbx20, have been shown to be crucial in zebrafish cardiac development as well. Tbx2 is duplicated in the zebrafish genome, but its homologues appear to have redundant functions in regulating the formation of non-chambered myocardium as well as controlling cardiac proliferation possibly through Tbx5 regulation [39–41]. Additionally, tbx5 expression appears to be regulated by Tbx20, and tbx20 morpholino knockdown in zebrafish embryos results in not only tbx5 misexpression throughout the heart tube, but also cardiac looping and chamber formation defects as observed in patients with TBX20 mutations [42–44]. Though future studies will be required to elucidate the organization of this cardiac transcription factor network, recent studies have begun to reveal the importance of epigenetic regulation through the Brg1/Brm-associated factor (BAF) chromatin remodeling complexes in order to coordinate the activities of many of these cardiac transcription factors [45].
Induction of cardiac precursors and their differentiation into specific cardiac lineages is dependent on several signaling pathways. As previously mentioned, retinoic acid signaling appears to restrict the cardiac progenitor pool during the blastula stage and its reduction can lead to an expansion of cardiac precursors [21]. However, retinoic acid signaling appears to play a different role at later stages by restricting the cardiac field in the ALPM posteriorly [27]. Furthermore, retinoic acid produced by the endocardium and epicardium appears to be necessary for the cardiomyocyte proliferation during zebrafish heart regeneration [46]. Thus, retinoic acid signaling plays an essential role in both cardiac development and regeneration but through context dependent mechanisms. Similarly, Wnt signaling can also temporally regulate cardiac progenitor numbers. Inhibiting Wnt signaling by Dkk1 overexpression prior to gastrulation leads to reduced cardiac progenitors; however, Wnt inhibition one hour after the onset of gastrulation results in increased cardiac progenitors [47]. Furthermore, Hedgehog (Hh) signaling plays a cell-autonomous role during early cardiac specification to control the numbers of cardiac progenitors [48]. Reduced Hh signaling leads to decreased cardiac precursors, whereas increased Hh signaling produces more cardiac precursors. Though reduced Nodal [22, 49], Bmp [50], and Fgf signaling [51] decreases both atrial and ventricular precursors depending on the developmental stage, it appears that the Nodal and Fgf pathways may impact ventricular progenitors more than atrial progenitors [22, 49, 51], whereas BMP may conversely influence atrial progenitors more than ventricular progenitors [42, 49, 50, 52]. Finally, in order for early cardiac progenitor cells to receive additional instructive cues for cardiac specification, these cells must migrate to the correct location within the ALPM. This migration is dependent on the Apelin G-protein coupled receptor signaling pathway and, the grinch mutant, which harbors a mutation in the Apelin receptor agtrl1b, fails to develop the cardiac fields within the ALPM due to the inability of these mutant cardiac progenitors to migrate properly [53, 54]. As a result, these mutants frequently develop without a heart. Overall, although these studies reveal that these cardiac signaling pathways act in a developmental stage-specific manner, further studies will be necessary to elucidate the downstream pathways that are activated by these signals to control cardiac specification and differentiation.
Heart tube formation
After forming in the ALPM, the bilateral myocardial progenitors migrate towards the embryonic midline to assemble into a heart tube (Figure 1D, E). This event appears to be regulated by not only cell autonomous but also cell non-autonomous mechanisms from the surrounding tissues including the overlying anterior endoderm, underlying yolk syncytial layer (YSL), and the endocardium. Cardia bifida mutants identified from zebrafish forward genetic screens have been particularly useful in revealing the cellular and molecular pathways that control these processes [5, 7, 55]. Mutants with endodermal formation defects, such as casanova, bonnie and clyde, and faust, exhibit cardia bifida [29, 56, 57]; however, the endodermal signals that regulate myocardial progenitor cell migration remain unclear. Two particularly interesting cardia bifida mutants, miles apart (mil) and two of heart (toh)/sphingolipid transporter (spns2), have illuminated the importance of the sphingolipid signaling in myocardial progenitor cell migration, and moreover were found to genetically interact [58–60]. mil encodes the Sphingosine-1-phosphate receptor 2 (S1P2) protein, which transmits Sphingosine-1-phosphate (S1P) signaling and is located in the endoderm, whereas Spns2 may regulate the release or trafficking of S1P in cells. Interestingly, YSL specific expression of wildtype spns2 results in rescue of the toh/spns2 mutants, suggesting that the YSL may provide the S1P ligand to signal to the endoderm. One possible mechanism as to how the endoderm and YSL may modulate myocardial cell midline migration is through the extracellular matrix (ECM) surrounding myocardial progenitor cells. Cardia bifida is observed in not only the fibronectin mutant, natter (nat) [61], but also embryos where syndecan 2 (sdc2), a proteoglycan gene, is morpholino knocked down in the YSL [62]. Furthermore, though hand2 mutants fail to produce adequate numbers of cardiomyocytes, they appear to also have a midline migration defect due to increased fibronectin levels. Interestingly, the hand2 midline defect can be partially rescued in the nat heterozygous background [63]. These data suggest that a proper balance of ECM is necessary to control myocardial migration and heart tube assembly.
Myocardial precursors retain an apical-basal polarity during their midline migration, which may be regulated by their interactions with fibronectin. However, epithelial polarity mutants including heart and soul (has/prkci) [64, 65], mosaic eyes (moe/epb41l5) [66], nagie oko (nok/mpp5a) [67], oko meduzy (ome/crb2) [68, 69], and pard6gb [70], appear to still migrate to the midline, but fail to properly assemble into a linear heart tube [64, 65]. More detailed analysis using time-lapse imaging reveals that defects in has/prkci or nok/mpp5a leads to disruption of myocardial precursor epithelial sheets as well as uncoordinated migration of these cells, subsequently resulting in failure to create a linear heart tube [71]. Thus, these studies highlight the importance of cell polarity to maintain myocardial precursor epithelial sheet coherence and migration to form the linear heart tube [72, 73].
Finally, the endocardial cells, the inner lining of the myocardial heart tube, is required for the formation of the heart tube. These progenitor cells, which are more anteriorly located in the ALPM [24], migrate medially and posteriorly to line the interior of the developing heart cone [74]. Embryos with mutations in the Scl/Tal1 transcription factor exhibit defective endocardial cell migration, resulting in these cells localizing primarily in the ventricular pole of the heart. Though the myocardial precursor cells are able to migrate to the midline in these mutants, their heart tubes fail to form properly. Using cloche mutants, more recent studies have revealed that the endocardium may act to more specifically control myocardial progenitor cell movements that are necessary to close the cardiac cone [75]. Interestingly, the myocardial progenitor cells of mil/s1p2 cardia bifida mutants are able to undergo these particular cellular movements despite the absence of their midline migration. Future studies will be required to elucidate the signals emanating from the endocardium to control myocardial progenitor cell movements.
Cardiac growth and proliferation
As the heart continues to develop and undergo morphologic changes, additional cardiomyocytes are required to create the final adult cardiac structures. The relatively small size of the developing zebrafish heart is particularly conducive towards elucidating how the vertebrate heart may derive these new cardiomyocytes. Taking advantage of fluorescent protein folding differences of GFP and dsRed as well as the photoconvertible kaede protein, recent developmental-timing studies have used a combination of cardiac-specific transgenic lines including Tg(cmlc2:GFP), Tg(cmlc2:nuc-dsRed) and Tg(cmlc2:kaede), to reveal two late-differentiating cardiomyocyte populations that may extend the ends of the heart at the arterial and venous poles [76] (Figure 1C, F). Late differentiating atrial cardiomyocyte progenitors appear to add to the venous pole from 16 to 26 somites and are created through an islet1 dependent mechanism, suggesting a possible secondary heart field in the zebrafish [76] (Figure 2A–C). After the linear heart tube forms by 30 hpf, a second wave of new ventricular cardiomyocytes is added to the arterial pole and regulated through Fgf signaling (Figure 1F, 2D–F). As a result, islet1 [76] and fgf8/ace mutants [51, 77] exhibit reduced numbers of atrial and ventricular cardiomyocytes at later cardiac developmental stages. Using genetic fate-mapping approaches, more recent studies have revealed that the latent TGF-β binding protein 3 (ltbp3) gene not only marks these late cardiac progenitors at the arterial pole, but also may regulate their proliferation [78]. Although zebrafish hearts have only two cardiac chambers, these studies suggest that the events driving cardiac development, including second heart field formation, appears to be highly conserved amongst vertebrates.
Figure 2.
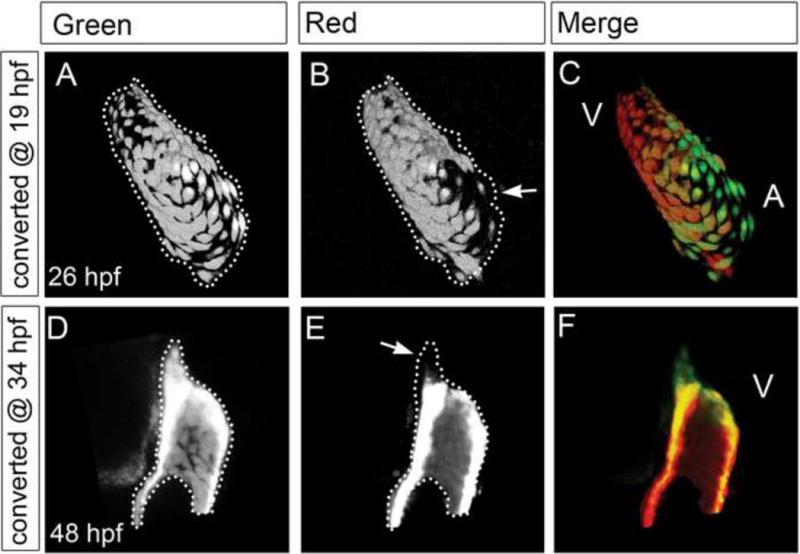
Late differentiating cardiomyocytes are added to the cardiac poles. Using Tg(cmlc2:kaede), cardiomyocytes expressing the kaede fluorophore can be photoconverted from green to red to label preexisting cardiomyocytes for developmental timing assays. (A–C) As a result, Tg(cmlc2:kaede) hearts photoconverted to red at 19 hpf displayed atrial cardiomyocytes that are green only at 26 hpf (arrows), suggesting that new green kaede atrial cardiomyocytes are added after 19 hpf. (D–F) Additionally, Tg(cmlc2:kaede) hearts photoconverted to red at 34 hpf displayed ventricular cardiomyocytes that are green only at 48 hpf (arrows), suggesting that new green kaede ventricular cardiomyocytes are added after 34 hpf. The arterial poles are positioned to the top in all confocal images. Dotted line marks the heart region containing green myocardium (A, D). When this area is compared to the original photoconverted red myocardium area (B, E), the addition of new cardiomyocytes to the cardiac poles can be appreciated (C, F). A: atrium, V: ventricle. Figure: Courtesy of Yelon D. [76]
During the addition of new cardiomyocytes from 24–48 hpf, pre-existing cardiomyocytes within the developing zebrafish heart appears to slowly divide [41, 76]. However, between 48 to 72 hpf, growth of the heart may be dependent on endogenous cardiomyocytes proliferating. The liebeskummer (lik) mutant, which harbors an activating mutation in the DNA-stimulated ATPase reptin, displays a dramatic increase in ventricular cardiomyocyte proliferation between 48 and 72 hpf [79]. Morpholino knockdown of pontin, another DNA-stimulated ATPase known to be a reptin antagonist, can recapitulate the lik phenotype. Furthermore, the island beat (isl) mutant, which contains a mutation in the calcium channel, voltage-dependent, L type, alpha 1C subunit (cacna1c) gene, interestingly also exhibits a significant reduction of cardiomyocytes at 72 hpf despite normal cardiac development up to 48 hpf, thus suggesting that calcium signaling may regulate cardiomyocyte proliferation [80]. Because of the recent observations that postnatal mouse and adult zebrafish injured hearts can regenerate by cardiomyocyte proliferation [28, 81, 82], future studies illuminating the underlying mechanisms of early cardiomyocyte proliferation may prove particularly useful towards understanding how adult/mature cardiomyocytes may divide.
Finally, a series of endocardial mutants, heart of glass (heg), santa (san), and valentine (val), have shed light on the role of the endocardium to regulate cardiac growth [83, 84]. The mutants exhibit similar cardiac phenotypes including enlarged hearts and thin chamber walls. Positional cloning studies reveal that these affected genes encode proteins associated with familial cerebral cavernous malformations (CCM1/KRIT/san and CCM2/val) in humans [83, 84]. Additional genetic and biochemical studies show that these genes and their encoded proteins interact with each other, and their combinatorial morpholino knockdowns at low doses can phenocopy the enlarged heart phenotype [85–87]. Though it remains unclear as to how the endocardium regulates myocardial growth in the zebrafish, perhaps similar cardiac events in mouse, where the endocardium signals to the myocardium to control myocardial trabeculae development [88], could also be involved in zebrafish cardiac growth.
Cardiac looping and Chamber formation
In zebrafish, the first morphological differences between the two cardiac chambers can be observed after the formation of the linear heart tube although molecular differences between atrium and ventricle are apparent much earlier [89]. At 24 hpf, the heart tube begins to extend and turn such that the ventricle becomes anterior and right-sided, whereas the atrium becomes more posterior and left-sided (Figure 1E). These initial morphogenetic events lead to the looping of the heart, and the subsequent followed by the ballooning of the cardiac chambers as well as the development of the atrioventricular canal (see below) [90] (Figure 1F). Molecular events that may transpire to guide these morphogenetic events include the specification of chambered and non-chambered myocardium [39, 41, 91–93]. Similar to amniotes, Bmp signaling and Tbx2 appear to control non-chamber myocardial specification, possibly through Wnt signaling in the zebrafish [40]. Furthermore, the expansion of tbx2 expression in zebrafish gridlock/hey2 mutants suggests that this non-chamber myocardial BMP-Tbx2 pathway may be regulated through a negative feedback loop with the chamber myocardial Notch/Hrt pathways as observed in mouse and chick [92]. Moreover, chambered myocardial specific markers, such as Anf, appear to be expressed in the chambered myocardium both in zebrafish and amniotes [94], thus suggesting that chamber and non-chamber myocardial specification as well as cardiac looping and chamber formation may be a conserved process amongst vertebrates.
In order for the chambers to balloon and expand, cardiomyocytes in each chamber must differentially undergo cell shape changes. As a result, by 48–56 hpf, the ventricular concave inner curvature (IC) cardiomyocytes nearest the non-chambered myocardium (i.e. atrioventricular canal, inflow/outflow tract) remain more rounded, whereas the ventricular convex outer curvature (OC) cardiomyocytes become more elongated (Figure 3A) [11, 94]. These cellular morphogenetic events appear to be regulated by not only mechanical but also electrical forces [95]. In weak atrium (wea/myh6) mutants, which lack atrial contractions, the OC ventricular cardiomyocytes fail to elongate thus resulting in a smaller ventricular chamber and reduced anf expression, whereas in half-hearted (haf/vmhc) mutants, which lack ventricular contraction due to failure to form ventricular sarcomeres, their OC ventricular cardiomyocytes become more elongated leading to larger ventricular chambers and increased anf expression [94]. Though these studies suggest mechanical forces are required for chamber formation, future studies will be required to elucidate whether myocardial stretch, myocardial contractility and/or hemodynamic flow is responsible for these cardiomyocyte cell shape changes. Additionally, recent studies have also shown that electrical conduction can also influence chamber cardiomyocyte morphogenesis. The dococ (dco) mutant, which exhibits disorganized conduction because of a mutation in gap junction protein alpha 3 (gja3), fails to undergo proper cardiac morphogenesis to develop expanded cardiac chambers [95] (Figure 3B). To determine whether this morphogenetic defect was due to physical and/or electrical forces, dco hearts were further examined in non-contractile hearts using silent heart (sih) mutants (Figure 3C, D). As a result, these dco;sih double mutants also exhibited similar chamber defects, suggesting that electrical forces independent of physical forces can influence cardiomyocyte morphogenesis and cardiac chamber ballooning (Figure 3D). Though cellular shape changes are crucial for cardiac chamber formation, future studies are required to elucidate the underlying molecular mechanisms that translate these biophysical forces to cell morphogenesis.
Figure 3.
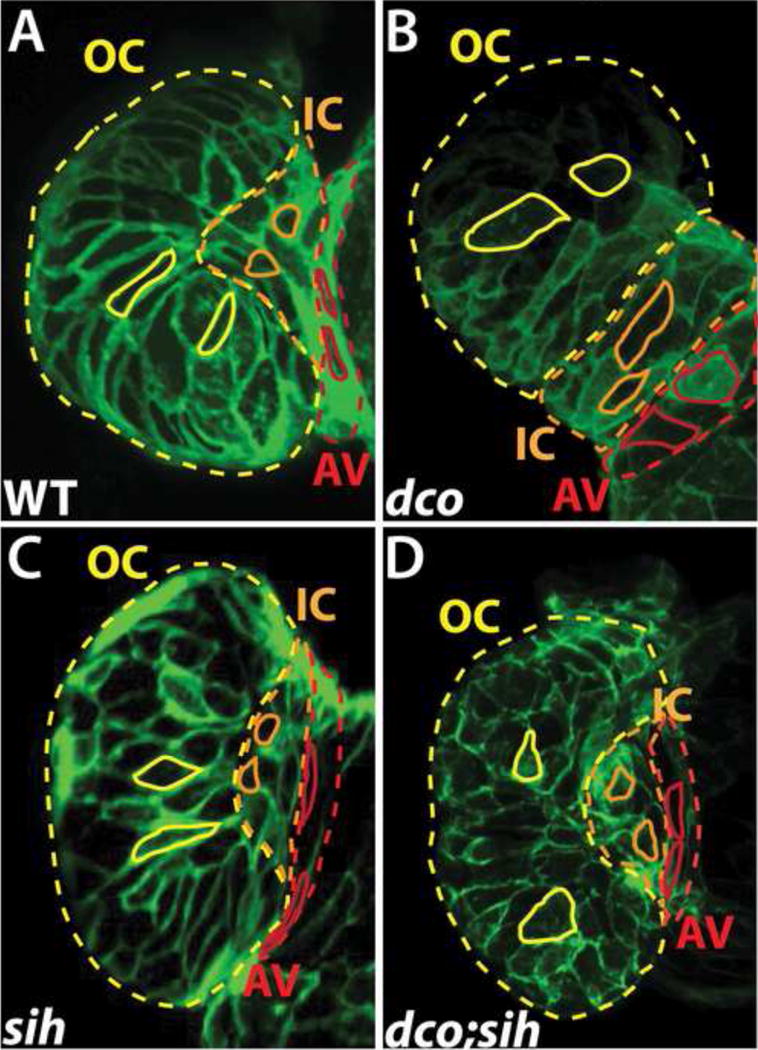
Cardiac conduction, independent of biomechanical forces, can influence cardiomyocyte morphogenesis. (A–D) Confocal images of the heart at 60 hpf, using Tg(cmlc2:ras-eGFP) to visualize the shape of individual cardiomyocytes in (A) wildtype (WT), (B) dco mutant (C) sih mutant, and (D) dco;sih double mutant. Solid yellow, orange, and red lines outline cardiomyocytes in the outer curvature (OC), inner curvature (IC), and atrioventricular canal (AV) of the ventricle, respectively. Dashed yellow, orange, and red lines outline the OC, IC and AV regions of the ventricle, respectively. Note ventricular OC cardiomyocytes in dco and dco;sih hearts are more circular compared to those in WT and sih hearts. [95]
Atrioventricular Canal and Valve Development
Division of the heart into chambers represents an essential evolutionary milestone in vertebrates that allows for the heart to receive (atrium) and pump (ventricle) blood throughout a closed circulatory system [96]. The atrioventricular (AV) canal has developed several specialized functions to regulate forward blood flow through these chambers. Despite programming a second cardiac chamber in the invertebrate chordate, Ciona intestinalis, bidirectional blood flow is still observed in these animals due to uncoordinated beating of the chambers and lack of cardiac valve leaflets [97]. Thus, vertebrate animals have evolved a distinct genetic and cellular program to form these specialized AV structures, which include the AV cushions that give rise to the AV valves and part of the septal walls, as well as the AV myocardial conduction delay (AV node) between chambers. Here, we will specifically focus on the development of the AV canal and AV valve leaflets, and AV conduction development will be further discussed in detail in the conduction/arrhythmia section.
Although AV canal and valve development has been extensively studied in mouse and chick [98], the zebrafish system presents several advantages, including the ability to carry out forward genetic screens [8] and study vertebrate gene function at the single cell level [8, 39, 99]. The AV canal forms at the border between the atrium and ventricle, and the first molecular indication of AV canal specification in the zebrafish occurs at approximately 37 hpf with the restriction of bmp4, tbx2b, and versican expression to the AV myocardium [18, 39] (Figure 1F). At ~45 hpf, the expression of notch1b, hyaluronan synthase 2 and neuregulin becomes restricted to the AV endocardium [83, 100–102]. These molecular changes are accompanied by AV myocardial cellular changes resulting in cardiac looping, constriction of the AV canal, and the initiation of AV endocardial morphologic changes [8] (Figure 4C, E, F). These cellular changes can be observed as early as ~40 hpf where AV cardiomyocytes extend their basolateral surface and constrict their apical surface while the AV endocardial cells become cuboidal [39] and express the cell adhesion molecule DM-grasp [8]. Though the differentiated AV canal begins to block some retrograde blood flow in the 48 hpf zebrafish, recent high-speed imaging studies have revealed that AV endocardial cells continue to remodel to create primitive valve leaflets allowing for complete block of retrograde blood flow at 72 hpf [99]. During this developmental period, these AV endocardial cells not only undergo cell polarity and cell shape changes [8], but also become two cell layer thick [99]. As a result, AV valve leaflet endocardial cells nearest to the AV canal remain cuboidal whereas those closest to the chambered myocardium become rounded. Although regulated by similar signaling pathways that occur during mammalian AV valve development, these AV endocardial cellular events do not require epithelial-to-mesenchymal transformation (EMT) to form these valve structures. However, at later developmental stages (12–16 day postfertilization/dpf), the AV valve leaflets appear to thicken and lengthen by downregulating epithelial markers and upregulating mesenchymal markers, thus suggesting that EMT may occur during later stages of zebrafish development [103]. In addition to these AV myocardial and endocardial cellular events, the extracellular matrix (ECM) also participates in AV canal morphogenesis [18, 102]. The jekyll mutant, which harbors a mutation in the UDP-glucose dehydrogenase (udgh) gene, exhibits blood flow toggling between the atrium and ventricle [18]. This enzyme is crucial for the synthesis of the ECM components, Hyaluronic acid, Heparan sulfate, and Chondroitin sulfate glycosaminoglycans. Consistent with these findings, chondroitin sulfate inhibition by reverse and chemical genetics in zebrafish results in the lack of AV canal formation [104]. Interestingly, recent human AV canal defect genetic studies have discovered mutations in the UGDH gene [105].
Figure 4.
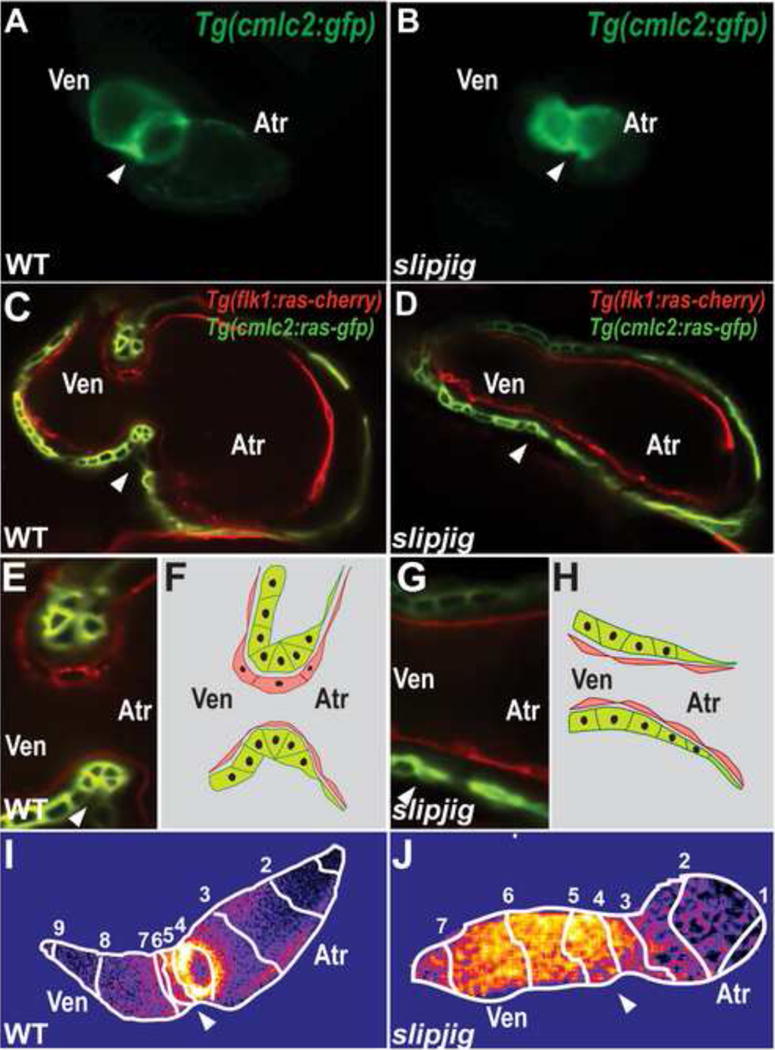
slipjig/foxn4 mutant fails to form the AV canal and develop an AV conduction delay. (A, B) Epifluorescence micrographs show that Tg(cmlc2:GFP) (A) wildtype (WT) hearts loop by 48 hpf but (B) slipjig/foxn4 mutant hearts do not. (C, E) WT and (D, G) slipjig Tg(flk1:ras-cherry); Tg(cmlc2:ras-GFP) hearts, which have endocardial and myocardial cells outlined in red and green, respectively, were imaged by confocal microscopy at 40 hpf. AV myocardial and endocardial cell morphologies become triangular and cuboidal, respectively in (C, E) WT but not (D, G) slipjig/foxn4 hearts. These cell morphology changes are further illustrated in (F, H). (I, J) Optical mapping by calcium imaging using Tg(cmlc2:gCaMP) shows that AV conduction delay as observed in (I) WT is absent in (J) the slipjig mutant. Isochronal lines (numbered white lines) indicate temporal calcium activation every 60 ms from venous to arterial pole. White arrowhead: AV canal; Atr: atrium; Ven: ventricle. Numbers: calcium activation sequence. [39]
Several zebrafish studies have begun to elucidate the signaling pathways that may regulate AV canal formation. One of the earliest events of AV canal formation is the specification of non-chambered myocardium [106]. BMP2/4 signaling participates in AV canal formation by inducing Tbx2 expression in the non-chambered myocardium between the atrial and ventricular chambers [107], and loss of function of tbx2b, bmp4, or activin A receptor, type I like/lost a fin (laf) results in AV canal defects in the zebrafish [39–41]. Recent studies have revealed several factors that may regulate this signaling pathway including the ECM protein Nephronectin [108], the single pass transmembrane protein Tmem2 [109, 110], and Wnt signaling [40]. Loss of Nephronectin and Tmem2 function leads to expansion of AV specific genes, including BMP4, throughout the heart as well as ectopic AV canal like cardiac cells in the heart chambers, whereas increased Tmem2 expression through loss of microRNA-23 function may result in endocardial cushion defects [111]. Furthermore, recent studies have shown that Wnt signaling may regulate bmp4 and tbx2b expression in the AV canal [40]. Interestingly, the zebrafish adenomatous polyposis coli (apc) mutant, which maintains constitutively active β-catenin signaling, exhibits a profuse endocardial layer at the AV boundary at 72 hpf, whereas inhibition of Wnt signaling by dkk1 RNA injection leads to a lack of AV canal differentiation and AV blood toggling [102]. Despite the lack of EMT requirement in zebrafish early AV valve leaflet formation, signaling pathways implicated in mammalian valve development, such as Notch and NFATc/calcineurin, have been shown to also regulate zebrafish AV valve formation [112, 113]. Detailed studies in the zebrafish suggest that the Notch signaling pathway may control specific stages of AV canal development. Constitutive Notch activation at 24 hpf in the endocardium results in lack of AV endocardial cell differentiation whereas Notch inhibition leads to the presence of ectopic cuboidal, DM-grasp positive (AV-like) endocardial cells in the ventricle; however, inhibition of Notch signaling at 36 hpf results in disorganized endocardial cells at the AV canal [8]. Using cyclosporine A (CsA) to block calcineurin signaling, recent studies have shown that NFAT signaling is also a conserved signaling pathway for vertebrate AV valve formation [8, 113], although the cellular effects of CsA treatment on cushion and valve formation remain to be fully examined. Taking advantage of chemical genetics in the zebrafish, recent studies have shown that ErbB/neuregulin, TGF β, and prostaglandin signaling pathways may also regulate specific developmental stages of zebrafish AV canal and valve leaflet formation.
Finally, combining its optical clarity, genetic tractability, and external fertilization, the developing zebrafish embryo/larvae has been used to show in vivo that hemodynamic fluid forces can regulate AV canal and valve development. Cardiac contractile mutants, such sih and cardiofunk (cfk), display not only reduced cardiac contraction, but also impaired AV valve formation [114]. Furthermore, physical occlusion of either inflow or outflow tracts with glass beads at 37 hpf results in failure to develop AV valves as well, supporting the requirement of blood flow during AV endocardial cell differentiation [115]. Additional detailed studies using high speed imaging of blood flow, have further investigated the specific hemodynamic components that regulate AV valve leaflet formation [116]. Using a combination of blood morpholino knockdowns, these studies revealed that AV endocardial cell differentiation was dependent on oscillatory blood flow but not on wall shear stress. These findings are consistent with the observation that many zebrafish contractility mutants, which have low oscillatory blood flow, exhibit AV valve formation defects. Furthermore, klf2a, an oscillatory flow-sensitive gene, appears to be expressed in the wildtype AV canal, whereas it is significantly downregulated in zebrafish cardiac contractility mutants. Consistent with these findings, klf2a morpholino knockdown results in failure to form AV valve leaflets despite not changing oscillatory flow.
Cardiac Trabeculae
Cardiac trabecuale are highly-organized muscular protrusions present in the ventricular lumen. They likely serve to not only increase myocardial surface area for blood oxygenation but also regulate cardiac function. As a result, both hypotrabeculation and hypertrabeculation (non-compaction in humans) depending on the vertebrate species can lead to both developmental and functional defects [117, 118]. Additionally, the ventricular cardiac conduction system may also initially develop from the ventricular trabeculae (see below, Conduction/Arrhythmia section).
After cardiac looping, chamber ballooning, and AV canal formation have occurred, zebrafish ventricular cardiomyocytes invaginate into the ventricular lumen at 60 hpf to initiate cardiac trabeculae formation [119]. By 72 hpf, the ventricular chambers develop distinguishable trabeculae, which predominantly line the ventricle outer curvature by 5 dpf [119, 120]. Though the trabeculae continue to expand and mature up to 15 dpf, the compact myocardium still remains one cell layer thick [120], which is consistent with the relatively thin compact myocardium observed in adult zebrafish ventricles [121]. Similar to mammalian hearts [122–125], the Neuregulin/ErbB pathway appears to also regulate ventricular trabeculation in zebrafish hearts [119, 120]. Functional loss of this pathway using a combination of small molecules and erbb2 mutants/morphants resulted in failure to form trabeculae, progressive cardiac failure, and cardiac conduction defects. Additionally, the reduction of blood flow as observed in silent heart or weak atrium mutants can lead to reduced trabeculae, suggesting the requirement of proper blood flow for progression of trabeculation [11, 119] (Figure 5D). Finally, lineage tracing and transplantation studies further revealed that through an ErbB2 cell-autonomous manner, cardiac trabeculation is initiated by directional cardiomyocyte migration instead of oriented cell division. Because Notch can regulate the Neuregulin pathway through EphrinB2 during mammalian ventricle trabeculation [88], future studies will be required to investigate whether these pathways are also required during zebrafish cardiac trabeculation.
Figure 5.
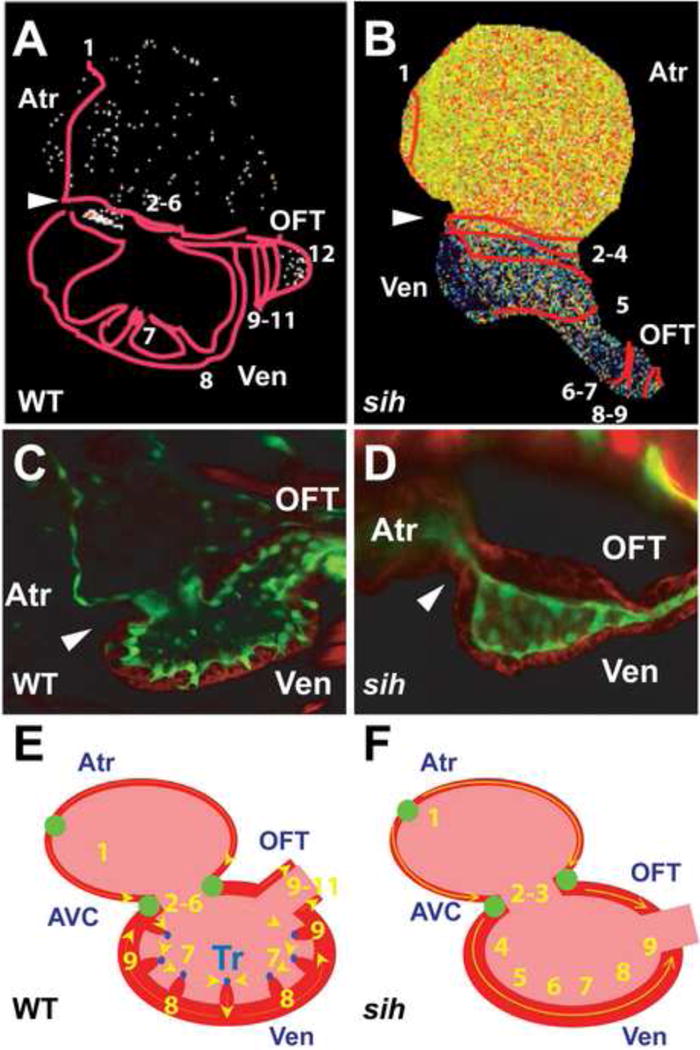
Cardiac conduction in wildtype (WT) and sih mutant hearts. (A, B) Optical mapping of calcium activation in 100 hpf (A) WT and (B) sih mutant hearts, using Tg(cmlc2:gCaMP). Isochronal lines (numbered red lines) indicate temporal calcium activation every 60 ms from venous to arterial pole. Note that the initial ventricular calcium activation within the WT ventricle begins in the trabeculae, whereas it initiates in the ventricular wall in sih ventricles. (C, D) Confocal images of rhodamine phalloidin stainied Tg(flk1:EGFP) (C) WT and (D) sih hearts. Green: endocardial cells; Red: cardiomyocytes; White arrowhead: AV canal. Though both WT and sih hearts have completed cardiac looping, trabeculae has formed only in WT hearts. (E, F) Schematic representation of WT and sih hearts as seen in (A, B). Numbers indicate calcium activation sequence. Yellow arrows show the direction of conduction. Green circles represent slow conduction nodes. Atr: atrium; Ven: ventricle; OFT: outflow track; AVC: AV canal. [11]
Epicardium
The epicardium is an epithelial layer located on the outer surface of the myocardium (reviewed by Gittenberger de Groot et al in this issue) [126]. It has gained extensive interest because of its ability to transform into multiple cardiac cell-types and to secrete factors that may contribute to the developing and regenerating vertebrate heart [46, 127–133]. In the zebrafish, the first remnants of the epicardium is observed as the proepicardium which is located near the ventral surface of the looped heart at 48 hpf (Figure 1G). By 72 hpf, cells emanating from the proepicardium appear to spread across the myocardium, fully covering the heart by 96 hpf. Similar to the mammalian heart, wt1, tbx18, and tcf21 appear to be expressed in the proepicardium and epicardial cells [134, 135]. Furthermore, tbx5 and BMP signaling is required for the formation of the epicardium [134]. Because of its recent roles in cardiac regeneration, future developmental studies of the epicardium may illuminate the mechanisms of how the epicardium may contribute to heart regeneration.
FUNCTIONAL HEART DEFECTS
The main function of the heart is to receive and deliver blood throughout the animal. This is primarily achieved through coordinated pumping of the cardiac chambers and is thus dependent on cardiomyocyte contractile function and electrical activation. Defects in these biophysical processes can lead to cardiomyopathies and arrhythmias, respectively. We will highlight recent zebrafish studies that have further elucidated the cellular and molecular pathways regulating cardiac function.
Contraction/Cardiomyopathies
Cardiac contractile function is primarily modulated through actin-myosin filament units (i.e. sarcomeres) sliding across each other to allow for cardiomyocyte force contraction and subsequent ejection of blood from the heart. Mutations in genes responsible for sarcomeric function and organization can lead to reduced contraction (dilated cardiomyopathies) and/or altered sarcomeric/cardiomyocyte/cardiac structures (hypertrophic cardiomyopathies). Several mutants exhibiting such cardiomyopathies have been recovered from large forward genetic screens in the zebrafish [5, 7, 8]. Recent positional cloning studies on many of these mutants have identified mutations in sarcomeric genes, such as amhc, cmlc1, myl7, vmhc, titin, and tnnt2 [94, 136–139].
Interestingly, positional cloning of other cardiac contraction mutants has revealed that additional non-sarcomeric proteins may regulate contractile function. For example, the lost-contact (loc)/main squeeze (msq)/integrin-linked kinase (ilk) and phospholipase C γ 1 (plc γ 1) mutants have provided insight as to how cardiomyocyte stretch-sensing is crucial for cardiomyocyte function. Though sarcomeric organization is intact in these mutants, contractile function is severely reduced due to the inability to respond to stretch [140, 141]. ILK, which co-localizes with α-actinin and PINCH proteins at the Z disks, may sense stretch through its interactions with β -parvin, an integrin binding protein, and the PINCH proteins [142]. This ILK pathway in turn is thought to activate PLC γ 1 through Vegf signaling via the Flt-1 receptor, which then results in increased calcium transients and subsequent excitation-contraction coupling. Further supporting this cardiac-sensing signaling pathway, zebrafish morpholino knockdown of laminin α 4, which is present in the extracellular matrix and interacts with integrins, not only results in cardiac dysfunction but also genetically interacts with the loc/ilk mutants [143]. Moreover, recent positional cloning studies have revealed proteins important for sarcomere assembly and maintenance. The futka (ftk) mutant, which harbors a mutation in connexin36.7, has elucidated a novel pathway in which this connexin may regulate sarcomeric organization through maintaining nkx2.5 expression during heart tube formation (16 hpf) [144]. Notably, overexpression of nkx2.5 in ftk mutants can rescue not only the sarcomere structure defects, but also the reduced contractile function in these mutants. The flatline (fla) mutant, which contains a nonsense mutation within the SET- and MYND-domain-containing protein gene (smyd1), exhibits disrupted sarcomere assembly in cardiac and fast-twitch skeletal muscle [145]. Though the Smyd1 histone methyltransferase activity appears to be dispensable for sarcomerogenesis, more recent studies have shown that Smyd2 may control myofilament organization through cytoplasmic lysine methylation of Hsp90 [146].
Cardiac contractile defects originally discovered from zebrafish forward genetic screen mutants, such as ilk and titin, have also now been detected in human cardiomyopathies from human genome sequencing and genetic studies [143, 147]. Conversely, zebrafish reverse genetics has also been employed to identify and confirm new human cardiomyopathy genes. Because of the epistatic relationship between laminin α 4 and ilk genes, mutations were screened in these genes from a cohort of patients with severe dilated cardiomyopathies and two mutations were subsequently identified in the laminin α 4 gene [143]. Similar to this candidate cardiomyopathy gene strategy, Nexilin, a highly enriched cardiac protein in the sarcomeric Z disk, was morpholino knocked down in the zebrafish to investigate its function [148, 149]. As a result, these nexilin morphants exhibited severely reduced cardiac contractile function, which led to the sequencing and identification of NEXILIN mutations in a registry of human dilated cardiomyopathy patients. Overall, as additional candidate cardiomyopathy genes are discovered through future human genetic studies (Mendelian, genome wide association studies, Next-Generation sequencing), there will be a great need to validate that mutations in these genes can result in cardiomyopathies. Because of its ability to rapidly assess protein function by morpholino knockdown, the zebrafish presents an opportunity to efficiently and cost-effectively confirm the role of candidate cardiac genes in contractile function.
Conduction/Arrhythmias
Vertebrate hearts have evolved into multi-chambered hearts requiring coordinate beating of their chambers in order to achieve forward blood flow throughout the vasculature. This electrical synchronization is accomplished through the combination of specialized cardiomyocytes comprising the cardiac conduction system (CCS) as well as intrinsic ion channels for all cardiomyocytes. In the vertebrate CCS, the initial cardiac electrical impulses are generated in the sino-atrial (SA) node and are propagated across the atrium through specialized atrial cardiomyocytes. The electrical impulse is next delayed at the atrioventricular (AV) node, a set of highly specialized slowly conducting cardiomyocytes residing in the AV canal [106, 150], and then travels from the AV node through the rapidly conducting fast ventricular CCS otherwise known as the His-Purkinje system. This ventricular activation allows for conduction to travel from apex to base [151] resulting in efficient ejection of blood from the ventricles (apex to outflow tracts) [152]. Thus, disruptions in these electrical pathways can lead to arrhythmias including re-entrant arrhythmias, heart blocks, and atrial and ventricular fibrillation/sudden cardiac death (further reviewed by Jongbloed et. al. in this issue) [153]. Though the zebrafish heart is comprised of only two chambers (one atrium and one ventricle), recent physiologic studies in the zebrafish using optical mapping (calcium and voltage sensitive dyes or transgenics), electrophysiologic analysis, and optogenetics have discovered that a cardiac electrical wiring system, similar to four-chambered vertebrate hearts, is required to coordinate the contractions of the zebrafish atrial and ventricular chambers [11, 17, 154] (Figure 4I, J and 5A, B). Because of the external fertilization and development of zebrafish embryos, these tools can be employed in vivo to examine CCS development in the intact developing vertebrate embryo.
As a result, SA node/pacemaker activity has been established as early as 24 hpf in the zebrafish heart tube using both calcium and voltage imaging/optical mapping studies [11, 101, 155]. Additional optogenetic studies utilizing a cardiac expressing light-activated hyperpolarizing ion channel transgenic line revealed that pacemaker activity is diffusely present at the venous pole of the heart tube at 24 hpf, but eventually becomes restricted to the dorsal right quadrant of the SA ring by 72 hpf [154], which is similar to the electrical mapping of the SA node in developing chick hearts [156]. Recent reverse genetic studies have revealed that Wnt signaling as well as the shox2 and isl1 transcription factors may be involved in regulating SA node development [157, 158].
Optical mapping studies have revealed that the development of the AV conduction delay occurs as early as 48 hpf when atrial and ventricular chambers are morphologically formed, and patch clamping experiments of atrial, ventricular and AV cardiomyocytes have further confirmed these findings [11]. Recent studies have shown that specific AV signaling and transcriptional pathways are required to establish this AV conduction delay. For example, cloche mutants fail to develop AV conduction delay, suggesting that the endocardium may be necessary for the development of the conduction delay in AV myocardial tissue [101]. Consistent with these findings, morpholino knockdown of notch1b and neuregulin, which are expressed in the AV endocardium, leads to loss of AV conduction delay. Additional studies have revealed that the BMP-Tbx2 AV myocardial signaling pathway may be also required to establish AV conduction delay. AV conduction delay fails to develop in not only morpholino knockdowns of tbx2b and foxn4, a transcription factor that regulates tbx2b expression [39] (Figure 4J), but also laf mutants, which have impaired BMP signaling [40]. Finally, from a CCS forward genetic screen, a tcf2 mutant was identified to have AV conduction delay defects [11]. The crosstalk between AV endocardium and myocardium to establish AV conduction delay remains to be further elucidated in the future. Combining forward, reverse, and opto-genetic studies with in vivo physiologic analysis may provide novel approaches towards dissecting how these pathways regulate the AV conduction development.
Though lacking a ventricular septum, optical mapping studies have revealed that adult zebrafish cardiac ventricles also display similar ventricular electrical activation as adult mammalian ventricles [17]. PSA-NCAM positive trabecular myocardial bands connecting the AV canal with the ventricle may be responsible for this mature ventricular conduction as severing these bands results in ventricular conduction block. Development of the ventricular conduction can be observed as early as 72 hpf when trabeculae start forming [11]. Hemodynamic flow may be required to initiate ventricular CCS development as sih mutants, which lack blood flow, also fail to develop the ventricular CCS (Figure 5B, F). Interestingly, this lack of the ventricular CCS in sih mutants may be due to failure to develop trabeculae [11, 120] and is consistent with previous findings in mouse that the trabeculae may differentiate into the ventricular CCS [159–161]. Interestingly, recent studies have suggested that the ventricular CCS may be established earlier than 72 hpf prior to the formation of trabeculae. Optical mapping studies in 48 hpf zebrafish hearts reveal a difference in conduction velocity within the ventricle where ventricular conduction is slower in the inner curvature but is faster in the outer curvature [155]. This ventricular conduction disparity appears to be due to a differential L-type calcium channel activation gradient across the ventricular myocardium that is established by Wnt11 signaling. In contrast to the more mature ventricular CCS development, this early ventricular conduction difference appears to be not affected in sih mutants.
All cardiomyocytes are highly excitable thus allowing them to become electrically activated for excitation-contraction coupling. This excitability is established by a diverse set of ion channels that regulate cardiomyocyte action potentials. Moreover, specific cardiac lineages exhibit their own individual set of ion channels leading to distinctive action potentials and excitation susceptibility. Thus, disruptions in these ion channels and their regulatory proteins can lead to a vast array of human cardiac arrhythmias. Because of their similar heart rates to humans, zebrafish cardiac conduction studies may have particular relevance to human cardiac arrhythmias, especially those that may be affected by ion channel/action potential defects. For example, positional cloning of zebrafish conduction mutants has revealed that similar genes are affected in both zebrafish and human cardiac arrhythmias [80, 162]. In some cases, the affected gene, such as cacna1c, was identified in a zebrafish conduction mutant (island beat) [80] before it was discovered in a human cardiac arrhythmia (Timothy syndrome) [163]. Conversely, recent studies have exploited the ability to rapidly knockdown candidate genes in the zebrafish by morpholinos. For instance, morpholino knockdown of gins3, the zebrafish ortholog of a candidate gene discovered in a human genome wide association study on cardiac repolarization, resulted in repolarization abnormalities in zebrafish hearts [164]. Finally, because the zebrafish is sensitive to chemical treatments, small molecule screens can be easily performed to test their effects on cardiac conduction. To this end, the zebrafish has been recently used to test the ability of drugs to cause cardiac repolarization defects [165]. In the future, the zebrafish could potentially be used to screen drugs for their effects on cardiac repolarization prior to human use.
VASCULAR/CARDIAC OUTFLOW TRACT DEFECTS
Because the zebrafish possesses one atrial and ventricular chamber and no pulmonary circulatory system, the zebrafish outflow tract does not septate to form systemic and pulmonary great vessels but does consist of a simple bulbus arteriosus (BA), which is comprised of smooth muscle cells [166]. Recent lineage tracing studies indicate that epicardial cells can give rise to a subset of BA smooth muscle cells [130]; yet, it remains unclear as to the source of the remaining BA smooth muscle cells. One possibility is that cardiac neural crest cells, which normally give rise to vascular smooth muscle cells of the proximal outflow tract in mammalian hearts, may also contribute to the zebrafish BA. However, zebrafish fate-mapping studies have revealed that zebrafish cardiac neural crest may actually give rise to myocardial cells [166–169]. Thus, additional studies are warranted to elucidate the origins of the outflow tract including whether latent cardiovascular progenitor cells at the arterial pole may also contribute to not only cardiomyocytes but also BA smooth muscle cells [76, 78]. Despite this unresolved BA origin issue, zebrafish outflow tract and proximal aorta mutants discovered from both forward and reverse cardiac and vascular genetic studies which recapitulate similar defects observed in mammalian outflow tract defects [10, 170], may still provide insight into human CHDs. For example, the gridlock/hey2 vascular mutant exhibits a similar phenotype to patients with coarctation of the aorta as well as the mouse hey2 knockout vascular defect [171]. Furthermore, from a chemical screen on the gridlock mutants, two modulators of VEGF signaling were discovered to restore blood flow in gridlock mutants, thus highlighting how the combination of zebrafish mutants and small molecule screens could lead to potential therapeutic compounds [172].
Summary and Future Directions
With the rapid rise of new technologies to execute human genetic studies (i.e. Genome Wide Association Studies/GWAS, Whole Exomic Sequencing/WES, Whole Genomic Sequencing/WGS, Array-comparative Genomic Hybridization/aCGH), a plethora of potentially disease causing genetic variants/genes will need to be rapidly tested for their ability to cause the disease phenotypes. Because of its relative ease and speed to morpholino knockdown genes or inject the RNA of human genes, and then to analyze phenotype, the zebrafish has become an ideal vertebrate animal model system to validate potential deleterious sequence variants (PDSVs) discovered from these human genetic studies. As a result, a number of groups have used a combination of morpholino knockdown studies and RNA expression of human genes harboring PDSVs to confirm causal effect [143, 148, 173–175]. Furthermore, the ability to titrate the amount of morpholino or RNA injected into the zebrafish also permits the examination of gene dosage effects as well [85, 176]. Thus, it is likely that in the future, the zebrafish will be used more frequently as a complementary animal model system to confirm and model human PDSVs causing CHDs.
Though these genetic studies may reveal genes critical for cardiac development, the mechanisms as to how they control cardiac morphogenesis may not be fully elucidated. Traditionally, most of our understanding of cardiac development has come from the analysis of static cardiac and embryonic samples. However, cardiac development is a highly dynamic process starting with the convergence of the myocardial and endocardial precursors to the midline and culminating in the generation of a complex organ both morphologically and functionally. Thus, the zebrafish embryo offers the possibility of further investigating in vivo the role of these candidate genes during cardiac development using fluorescent cardiac-specific transgenics and new microscopy techniques. Furthermore, because of the zebrafish heart’s relatively small size, these dynamic developmental events may be observed at not only single cell but also subcellular resolution [177]. The ultimate goal of such cellular and physiology studies is to understand with high temporal and spatial resolution the dynamics of each cardiac cell, the location of various signals, and the cellular response to these signals. This understanding, combined with the genetic identification of critical heart regulators, will likely provide greater mechanistic insight into cardiac development and CHDs.
Acknowledgments
We thank Deborah Yelon for generously providing Tg(cmlc2:kaede) developmental timing images. N.C.C. is supported by grants from the NIH (HD069305, HL104239, DP2OD07464).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pierpont ME, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):3015–38. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 2.Stainier DY. Zebrafish genetics and vertebrate heart formation. Nat Rev Genet. 2001;2(1):39–48. doi: 10.1038/35047564. [DOI] [PubMed] [Google Scholar]
- 3.Huisken J, et al. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305(5686):1007–9. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- 4.Huisken J, Stainier DY. Selective plane illumination microscopy techniques in developmental biology. Development. 2009;136(12):1963–75. doi: 10.1242/dev.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stainier DY, et al. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development. 1996;123:285–92. doi: 10.1242/dev.123.1.285. [DOI] [PubMed] [Google Scholar]
- 6.Sehnert AJ, Stainier DY. A window to the heart: can zebrafish mutants help us understand heart disease in humans? Trends Genet. 2002;18(10):491–4. doi: 10.1016/s0168-9525(02)02766-x. [DOI] [PubMed] [Google Scholar]
- 7.Chen JN, et al. Mutations affecting the cardiovascular system and other internal organs in zebrafish. Development. 1996;123:293–302. doi: 10.1242/dev.123.1.293. [DOI] [PubMed] [Google Scholar]
- 8.Beis D, et al. Genetic and cellular analyses of zebrafish atrioventricular cushion and valve development. Development. 2005;132(18):4193–204. doi: 10.1242/dev.01970. [DOI] [PubMed] [Google Scholar]
- 9.Weinstein BM, et al. Hematopoietic mutations in the zebrafish. Development. 1996;123:303–9. doi: 10.1242/dev.123.1.303. [DOI] [PubMed] [Google Scholar]
- 10.Jin SW, et al. A transgene-assisted genetic screen identifies essential regulators of vascular development in vertebrate embryos. Dev Biol. 2007;307(1):29–42. doi: 10.1016/j.ydbio.2007.03.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi NC, et al. Genetic and physiologic dissection of the vertebrate cardiac conduction system. PLoS Biol. 2008;6(5):e109. doi: 10.1371/journal.pbio.0060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bill BR, et al. A primer for morpholino use in zebrafish. Zebrafish. 2009;6(1):69–77. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wienholds E, et al. Efficient target-selected mutagenesis in zebrafish. Genome Res. 2003;13(12):2700–7. doi: 10.1101/gr.1725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang P, et al. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011;29(8):699–700. doi: 10.1038/nbt.1939. [DOI] [PubMed] [Google Scholar]
- 15.Meng X, et al. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol. 2008;26(6):695–701. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson RT, Fishman MC. Discovery and use of small molecules for probing biological processes in zebrafish. Methods Cell Biol. 2004;76:569–91. doi: 10.1016/s0091-679x(04)76026-4. [DOI] [PubMed] [Google Scholar]
- 17.Sedmera D, et al. Functional and morphological evidence for a ventricular conduction system in zebrafish and Xenopus hearts. Am J Physiol Heart Circ Physiol. 2003;284(4):H1152–60. doi: 10.1152/ajpheart.00870.2002. [DOI] [PubMed] [Google Scholar]
- 18.Walsh EC, Stainier DY. UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science. 2001;293(5535):1670–3. doi: 10.1126/science.293.5535.1670. [DOI] [PubMed] [Google Scholar]
- 19.Stainier DY, Lee RK, Fishman MC. Cardiovascular development in the zebrafish. I. Myocardial fate map and heart tube formation. Development. 1993;119(1):31–40. doi: 10.1242/dev.119.1.31. [DOI] [PubMed] [Google Scholar]
- 20.Yelon D, Stainier DY. Patterning during organogenesis: genetic analysis of cardiac chamber formation. Semin Cell Dev Biol. 1999;10(1):93–8. doi: 10.1006/scdb.1998.0278. [DOI] [PubMed] [Google Scholar]
- 21.Keegan BR, et al. Retinoic acid signaling restricts the cardiac progenitor pool. Science. 2005;307(5707):247–9. doi: 10.1126/science.1101573. [DOI] [PubMed] [Google Scholar]
- 22.Keegan BR, Meyer D, Yelon D. Organization of cardiac chamber progenitors in the zebrafish blastula. Development. 2004;131(13):3081–91. doi: 10.1242/dev.01185. [DOI] [PubMed] [Google Scholar]
- 23.Lee RK, et al. Cardiovascular development in the zebrafish. II. Endocardial progenitors are sequestered within the heart field. Development. 1994;120(12):3361–6. doi: 10.1242/dev.120.12.3361. [DOI] [PubMed] [Google Scholar]
- 24.Schoenebeck JJ, Keegan BR, Yelon D. Vessel and blood specification override cardiac potential in anterior mesoderm. Dev Cell. 2007;13(2):254–67. doi: 10.1016/j.devcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berdougo E, et al. Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development. 2003;130(24):6121–9. doi: 10.1242/dev.00838. [DOI] [PubMed] [Google Scholar]
- 26.Glickman NS, Yelon D. Cardiac development in zebrafish: coordination of form and function. Semin Cell Dev Biol. 2002;13(6):507–13. doi: 10.1016/s1084952102001040. [DOI] [PubMed] [Google Scholar]
- 27.Waxman JS, et al. Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev Cell. 2008;15(6):923–34. doi: 10.1016/j.devcel.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kikuchi K, et al. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 464(7288):601–5. doi: 10.1038/nature08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiter JF, et al. Gata5 is required for the development of the heart and endoderm in zebrafish. Genes Dev. 1999;13(22):2983–95. doi: 10.1101/gad.13.22.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia H, et al. Vertebrate heart growth is regulated by functional antagonism between Gridlock and Gata5. Proc Natl Acad Sci U S A. 2007;104(35):14008–13. doi: 10.1073/pnas.0702240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holtzinger A, Evans T. Gata5 and Gata6 are functionally redundant in zebrafish for specification of cardiomyocytes. Dev Biol. 2007;312(2):613–22. doi: 10.1016/j.ydbio.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peterkin T, Gibson A, Patient R. Redundancy and evolution of GATA factor requirements in development of the myocardium. Dev Biol. 2007;311(2):623–35. doi: 10.1016/j.ydbio.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riley P, Anson-Cartwright L, Cross JC. The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat Genet. 1998;18(3):271–5. doi: 10.1038/ng0398-271. [DOI] [PubMed] [Google Scholar]
- 34.Srivastava D, Cserjesi P, Olson EN. A subclass of bHLH proteins required for cardiac morphogenesis. Science. 1995;270(5244):1995–9. doi: 10.1126/science.270.5244.1995. [DOI] [PubMed] [Google Scholar]
- 35.Yelon D, et al. The bHLH transcription factor hand2 plays parallel roles in zebrafish heart and pectoral fin development. Development. 2000;127(12):2573–82. doi: 10.1242/dev.127.12.2573. [DOI] [PubMed] [Google Scholar]
- 36.Targoff KL, Schell T, Yelon D. Nkx genes regulate heart tube extension and exert differential effects on ventricular and atrial cell number. Dev Biol. 2008;322(2):314–21. doi: 10.1016/j.ydbio.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tu CT, Yang TC, Tsai HJ. Nkx2.7 and Nkx2.5 function redundantly and are required for cardiac morphogenesis of zebrafish embryos. PLoS ONE. 2009;4(1):e4249. doi: 10.1371/journal.pone.0004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garrity DM, Childs S, Fishman MC. The heartstrings mutation in zebrafish causes heart/fin Tbx5 deficiency syndrome. Development. 2002;129(19):4635–45. doi: 10.1242/dev.129.19.4635. [DOI] [PubMed] [Google Scholar]
- 39.Chi NC, et al. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008;22(6):734–9. doi: 10.1101/gad.1629408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verhoeven MC, et al. Wnt signaling regulates atrioventricular canal formation upstream of BMP and Tbx2. Birth Defects Res A Clin Mol Teratol. 2011;91(6):435–40. doi: 10.1002/bdra.20804. [DOI] [PubMed] [Google Scholar]
- 41.Ribeiro I, et al. Tbx2 and Tbx3 Regulate the Dynamics of Cell Proliferation during Heart Remodeling. PLoS ONE. 2007;2:e398. doi: 10.1371/journal.pone.0000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandel EM, et al. The BMP pathway acts to directly regulate Tbx20 in the developing heart. Development. 2010;137(11):1919–29. doi: 10.1242/dev.043588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szeto DP, Griffin KJ, Kimelman D. HrT is required for cardiovascular development in zebrafish. Development. 2002;129(21):5093–101. doi: 10.1242/dev.129.21.5093. [DOI] [PubMed] [Google Scholar]
- 44.Shen T, et al. Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. J Clin Invest. 2011;121(12):4640–54. doi: 10.1172/JCI59472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takeuchi JK, et al. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat Commun. 2011;2:187. doi: 10.1038/ncomms1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kikuchi K, et al. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell. 2011;20(3):397–404. doi: 10.1016/j.devcel.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueno S, et al. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104(23):9685–90. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas NA, et al. Hedgehog signaling plays a cell-autonomous role in maximizing cardiac developmental potential. Development. 2008;135(22):3789–99. doi: 10.1242/dev.024083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reiter JF, Verkade H, Stainier DY. Bmp2b and Oep promote early myocardial differentiation through their regulation of gata5. Dev Biol. 2001;234(2):330–8. doi: 10.1006/dbio.2001.0259. [DOI] [PubMed] [Google Scholar]
- 50.de Pater E, et al. Bmp signaling exerts opposite effects on cardiac differentiation. Circ Res. 2012;110(4):578–87. doi: 10.1161/CIRCRESAHA.111.261172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marques SR, et al. Reiterative roles for FGF signaling in the establishment of size and proportion of the zebrafish heart. Dev Biol. 2008;321(2):397–406. doi: 10.1016/j.ydbio.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marques SR, Yelon D. Differential requirement for BMP signaling in atrial and ventricular lineages establishes cardiac chamber proportionality. Dev Biol. 2009;328(2):472–82. doi: 10.1016/j.ydbio.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scott IC, et al. The g protein-coupled receptor agtrl1b regulates early development of myocardial progenitors. Dev Cell. 2007;12(3):403–13. doi: 10.1016/j.devcel.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 54.Zeng XX, et al. Apelin and its receptor control heart field formation during zebrafish gastrulation. Dev Cell. 2007;12(3):391–402. doi: 10.1016/j.devcel.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 55.Yelon D. Cardiac patterning and morphogenesis in zebrafish. Dev Dyn. 2001;222(4):552–63. doi: 10.1002/dvdy.1243. [DOI] [PubMed] [Google Scholar]
- 56.Kikuchi Y, et al. casanova encodes a novel Sox-related protein necessary and sufficient for early endoderm formation in zebrafish. Genes Dev. 2001;15(12):1493–505. doi: 10.1101/gad.892301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kikuchi Y, et al. The zebrafish bonnie and clyde gene encodes a Mix family homeodomain protein that regulates the generation of endodermal precursors. Genes Dev. 2000;14(10):1279–89. [PMC free article] [PubMed] [Google Scholar]
- 58.Kupperman E, et al. A sphingosine-1-phosphate receptor regulates cell migration during vertebrate heart development. Nature. 2000;406(6792):192–5. doi: 10.1038/35018092. [DOI] [PubMed] [Google Scholar]
- 59.Osborne N, et al. The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr Biol. 2008;18(23):1882–8. doi: 10.1016/j.cub.2008.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kawahara A, et al. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science. 2009;323(5913):524–7. doi: 10.1126/science.1167449. [DOI] [PubMed] [Google Scholar]
- 61.Trinh LA, Stainier DY. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev Cell. 2004;6(3):371–82. doi: 10.1016/s1534-5807(04)00063-2. [DOI] [PubMed] [Google Scholar]
- 62.Arrington CB, Yost HJ. Extra-embryonic syndecan 2 regulates organ primordia migration and fibrillogenesis throughout the zebrafish embryo. Development. 2009;136(18):3143–52. doi: 10.1242/dev.031492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garavito-Aguilar ZV, Riley HE, Yelon D. Hand2 ensures an appropriate environment for cardiac fusion by limiting Fibronectin function. Development. 2010;137(19):3215–20. doi: 10.1242/dev.052225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horne-Badovinac S, et al. Positional cloning of heart and soul reveals multiple roles for PKC lambda in zebrafish organogenesis. Curr Biol. 2001;11(19):1492–502. doi: 10.1016/s0960-9822(01)00458-4. [DOI] [PubMed] [Google Scholar]
- 65.Peterson RT, et al. Convergence of distinct pathways to heart patterning revealed by the small molecule concentramide and the mutation heart-and-soul. Curr Biol. 2001;11(19):1481–91. doi: 10.1016/s0960-9822(01)00482-1. [DOI] [PubMed] [Google Scholar]
- 66.Jensen AM, Westerfield M. Zebrafish mosaic eyes is a novel FERM protein required for retinal lamination and retinal pigmented epithelial tight junction formation. Curr Biol. 2004;14(8):711–7. doi: 10.1016/j.cub.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 67.Wei X, Malicki J. nagie oko, encoding a MAGUK-family protein, is essential for cellular patterning of the retina. Nat Genet. 2002;31(2):150–7. doi: 10.1038/ng883. [DOI] [PubMed] [Google Scholar]
- 68.Malicki J, Driever W. oko meduzy mutations affect neuronal patterning in the zebrafish retina and reveal cell-cell interactions of the retinal neuroepithelial sheet. Development. 1999;126(6):1235–46. doi: 10.1242/dev.126.6.1235. [DOI] [PubMed] [Google Scholar]
- 69.Omori Y, Malicki J. oko meduzy and related crumbs genes are determinants of apical cell features in the vertebrate embryo. Curr Biol. 2006;16(10):945–57. doi: 10.1016/j.cub.2006.03.058. [DOI] [PubMed] [Google Scholar]
- 70.Munson C, et al. Regulation of neurocoel morphogenesis by Pard6 gamma b. Dev Biol. 2008;324(1):41–54. doi: 10.1016/j.ydbio.2008.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rohr S, Bit-Avragim N, Abdelilah-Seyfried S. Heart and soul/PRKCi and nagie oko/Mpp5 regulate myocardial coherence and remodeling during cardiac morphogenesis. Development. 2006;133(1):107–15. doi: 10.1242/dev.02182. [DOI] [PubMed] [Google Scholar]
- 72.Rohr S, Otten C, Abdelilah-Seyfried S. Asymmetric involution of the myocardial field drives heart tube formation in zebrafish. Circ Res. 2008;102(2):e12–9. doi: 10.1161/CIRCRESAHA.107.165241. [DOI] [PubMed] [Google Scholar]
- 73.Smith KA, et al. Rotation and asymmetric development of the zebrafish heart requires directed migration of cardiac progenitor cells. Dev Cell. 2008;14(2):287–97. doi: 10.1016/j.devcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 74.Bussmann J, Bakkers J, Schulte-Merker S. Early endocardial morphogenesis requires Scl/Tal1. PLoS Genet. 2007;3(8):e140. doi: 10.1371/journal.pgen.0030140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Holtzman NG, et al. Endocardium is necessary for cardiomyocyte movement during heart tube assembly. Development. 2007;134(12):2379–86. doi: 10.1242/dev.02857. [DOI] [PubMed] [Google Scholar]
- 76.de Pater E, et al. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development. 2009;136(10):1633–41. doi: 10.1242/dev.030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reifers F, et al. Fgf8 is mutated in zebrafish acerebellar (ace) mutants and is required for maintenance of midbrain-hindbrain boundary development and somitogenesis. Development. 1998;125(13):2381–95. doi: 10.1242/dev.125.13.2381. [DOI] [PubMed] [Google Scholar]
- 78.Zhou Y, et al. Latent TGF-beta binding protein 3 identifies a second heart field in zebrafish. Nature. 2011;474(7353):645–8. doi: 10.1038/nature10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rottbauer W, et al. Reptin and pontin antagonistically regulate heart growth in zebrafish embryos. Cell. 2002;111(5):661–72. doi: 10.1016/s0092-8674(02)01112-1. [DOI] [PubMed] [Google Scholar]
- 80.Rottbauer W, et al. Growth and function of the embryonic heart depend upon the cardiac-specific L-type calcium channel alpha1 subunit. Dev Cell. 2001;1(2):265–75. doi: 10.1016/s1534-5807(01)00023-5. [DOI] [PubMed] [Google Scholar]
- 81.Jopling C, et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 464(7288):606–9. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Porrello ER, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–80. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mably JD, et al. santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development. 2006;133(16):3139–46. doi: 10.1242/dev.02469. [DOI] [PubMed] [Google Scholar]
- 84.Mably JD, et al. heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol. 2003;13(24):2138–47. doi: 10.1016/j.cub.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 85.Gore AV, et al. Combinatorial interaction between CCM pathway genes precipitates hemorrhagic stroke. Dis Model Mech. 2008;1(4–5):275–81. doi: 10.1242/dmm.000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kleaveland B, et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat Med. 2009;15(2):169–76. doi: 10.1038/nm.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yoruk B, et al. Ccm3 functions in a manner distinct from Ccm1 and Ccm2 in a zebrafish model of CCM vascular disease. Dev Biol. 2012;362(2):121–31. doi: 10.1016/j.ydbio.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 88.Grego-Bessa J, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12(3):415–29. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yelon D, Horne SA, Stainier DY. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev Biol. 1999;214(1):23–37. doi: 10.1006/dbio.1999.9406. [DOI] [PubMed] [Google Scholar]
- 90.Bakkers J, Verhoeven MC, Abdelilah-Seyfried S. Shaping the zebrafish heart: from left-right axis specification to epithelial tissue morphogenesis. Dev Biol. 2009;330(2):213–20. doi: 10.1016/j.ydbio.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 91.Christoffels VM, et al. T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev Dyn. 2004;229(4):763–70. doi: 10.1002/dvdy.10487. [DOI] [PubMed] [Google Scholar]
- 92.Rutenberg JB, et al. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development. 2006;133(21):4381–90. doi: 10.1242/dev.02607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kokubo H, et al. Hesr1 and Hesr2 regulate atrioventricular boundary formation in the developing heart through the repression of Tbx2. Development. 2007;134(4):747–55. doi: 10.1242/dev.02777. [DOI] [PubMed] [Google Scholar]
- 94.Auman HJ, et al. Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 2007;5(3):e53. doi: 10.1371/journal.pbio.0050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chi NC, et al. Cardiac conduction is required to preserve cardiac chamber morphology. Proc Natl Acad Sci U S A. 107(33):14662–7. doi: 10.1073/pnas.0909432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313(5795):1922–7. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davidson B, et al. FGF signaling delineates the cardiac progenitor field in the simple chordate, Ciona intestinalis. Genes Dev. 2006;20(19):2728–38. doi: 10.1101/gad.1467706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105(5):408–21. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Scherz PJ, et al. High-speed imaging of developing heart valves reveals interplay of morphogenesis and function. Development. 2008;135(6):1179–87. doi: 10.1242/dev.010694. [DOI] [PubMed] [Google Scholar]
- 100.Kortschak RD, Tamme R, Lardelli M. Evolutionary analysis of vertebrate Notch genes. Dev Genes Evol. 2001;211(7):350–4. doi: 10.1007/s004270100159. [DOI] [PubMed] [Google Scholar]
- 101.Milan DJ, et al. Notch1b and neuregulin are required for specification of central cardiac conduction tissue. Development. 2006;133(6):1125–32. doi: 10.1242/dev.02279. [DOI] [PubMed] [Google Scholar]
- 102.Hurlstone AF, et al. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003;425(6958):633–7. doi: 10.1038/nature02028. [DOI] [PubMed] [Google Scholar]
- 103.Martin RT, Bartman T. Analysis of heart valve development in larval zebrafish. Dev Dyn. 2009;238(7):1796–802. doi: 10.1002/dvdy.21976. [DOI] [PubMed] [Google Scholar]
- 104.Peal DS, et al. Chondroitin sulfate expression is required for cardiac atrioventricular canal formation. Dev Dyn. 2009;238(12):3103–10. doi: 10.1002/dvdy.22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smith KA, et al. Dominant-negative ALK2 allele associates with congenital heart defects. Circulation. 2009;119(24):3062–9. doi: 10.1161/CIRCULATIONAHA.108.843714. [DOI] [PubMed] [Google Scholar]
- 106.Christoffels VM, Burch JB, Moorman AF. Architectural plan for the heart: early patterning and delineation of the chambers and the nodes. Trends Cardiovasc Med. 2004;14(8):301–7. doi: 10.1016/j.tcm.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 107.Yamada M, et al. Expression of chick Tbx-2, Tbx-3, and Tbx-5 genes during early heart development: evidence for BMP2 induction of Tbx2. Dev Biol. 2000;228(1):95–105. doi: 10.1006/dbio.2000.9927. [DOI] [PubMed] [Google Scholar]
- 108.Patra C, et al. Nephronectin regulates atrioventricular canal differentiation via Bmp4-Has2 signaling in zebrafish. Development. 2011;138(20):4499–509. doi: 10.1242/dev.067454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smith KA, et al. Transmembrane protein 2 (Tmem2) is required to regionally restrict atrioventricular canal boundary and endocardial cushion development. Development. 2011;138(19):4193–8. doi: 10.1242/dev.065375. [DOI] [PubMed] [Google Scholar]
- 110.Totong R, et al. The novel transmembrane protein Tmem2 is essential for coordination of myocardial and endocardial morphogenesis. Development. 2011;138(19):4199–205. doi: 10.1242/dev.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lagendijk AK, et al. MicroRNA-23 restricts cardiac valve formation by inhibiting Has2 and extracellular hyaluronic acid production. Circ Res. 2011;109(6):649–57. doi: 10.1161/CIRCRESAHA.111.247635. [DOI] [PubMed] [Google Scholar]
- 112.Timmerman LA, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18(1):99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chang CP, et al. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118(5):649–63. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 114.Bartman T, et al. Early myocardial function affects endocardial cushion development in zebrafish. PLoS Biol. 2004;2(5):E129. doi: 10.1371/journal.pbio.0020129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hove JR, et al. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature. 2003;421(6919):172–7. doi: 10.1038/nature01282. [DOI] [PubMed] [Google Scholar]
- 116.Vermot J, et al. Reversing blood flows act through klf2a to ensure normal valvulogenesis in the developing heart. PLoS Biol. 2009;7(11):e1000246. doi: 10.1371/journal.pbio.1000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jenni R, Rojas J, Oechslin E. Isolated noncompaction of the myocardium. N Engl J Med. 1999;340(12):966–7. doi: 10.1056/NEJM199903253401215. [DOI] [PubMed] [Google Scholar]
- 118.Weiford BC, V, Subbarao D, Mulhern KM. Noncompaction of the ventricular myocardium. Circulation. 2004;109(24):2965–71. doi: 10.1161/01.CIR.0000132478.60674.D0. [DOI] [PubMed] [Google Scholar]
- 119.Peshkovsky C, Totong R, Yelon D. Dependence of cardiac trabeculation on neuregulin signaling and blood flow in zebrafish. Dev Dyn. 2011;240(2):446–56. doi: 10.1002/dvdy.22526. [DOI] [PubMed] [Google Scholar]
- 120.Liu J, et al. A dual role for ErbB2 signaling in cardiac trabeculation. Development. 2010;137(22):3867–75. doi: 10.1242/dev.053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hu N, et al. Structure and function of the developing zebrafish heart. Anat Rec. 2000;260(2):148–57. doi: 10.1002/1097-0185(20001001)260:2<148::AID-AR50>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 122.Gassmann M, et al. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378(6555):390–4. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- 123.Lee KF, et al. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378(6555):394–8. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- 124.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378(6555):386–90. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 125.Lai D, et al. Neuregulin 1 sustains the gene regulatory network in both trabecular and nontrabecular myocardium. Circ Res. 2010;107(6):715–27. doi: 10.1161/CIRCRESAHA.110.218693. [DOI] [PubMed] [Google Scholar]
- 126.Gittenberger-de Groot AC, et al. The arterial and cardiac epicardium in development, disease and repair. Differentiation. 2012 doi: 10.1016/j.diff.2012.05.002. (this issue) [DOI] [PubMed] [Google Scholar]
- 127.Lepilina A, et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 2006;127(3):607–19. doi: 10.1016/j.cell.2006.08.052. [DOI] [PubMed] [Google Scholar]
- 128.Cai CL, et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008;454(7200):104–8. doi: 10.1038/nature06969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou B, et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454(7200):109–13. doi: 10.1038/nature07060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kikuchi K, et al. tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development. 2011;138(14):2895–902. doi: 10.1242/dev.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim J, et al. PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc Natl Acad Sci U S A. 2010;107(40):17206–10. doi: 10.1073/pnas.0915016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Smart N, et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011;474(7353):640–4. doi: 10.1038/nature10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bock-Marquette I, et al. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature. 2004;432(7016):466–72. doi: 10.1038/nature03000. [DOI] [PubMed] [Google Scholar]
- 134.Liu J, Stainier DY. Tbx5 and Bmp signaling are essential for proepicardium specification in zebrafish. Circ Res. 2010;106(12):1818–28. doi: 10.1161/CIRCRESAHA.110.217950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Serluca FC. Development of the proepicardial organ in the zebrafish. Dev Biol. 2008;315(1):18–27. doi: 10.1016/j.ydbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 136.Sehnert AJ, et al. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat Genet. 2002;31(1):106–10. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 137.Xu X, et al. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat Genet. 2002;30(2):205–9. doi: 10.1038/ng816. [DOI] [PubMed] [Google Scholar]
- 138.Meder B, et al. A single serine in the carboxyl terminus of cardiac essential myosin light chain-1 controls cardiomyocyte contractility in vivo. Circ Res. 2009;104(5):650–9. doi: 10.1161/CIRCRESAHA.108.186676. [DOI] [PubMed] [Google Scholar]
- 139.Rottbauer W, et al. Cardiac myosin light chain-2: a novel essential component of thick-myofilament assembly and contractility of the heart. Circ Res. 2006;99(3):323–31. doi: 10.1161/01.RES.0000234807.16034.fe. [DOI] [PubMed] [Google Scholar]
- 140.Bendig G, et al. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 2006;20(17):2361–72. doi: 10.1101/gad.1448306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rottbauer W, et al. VEGF-PLCgamma1 pathway controls cardiac contractility in the embryonic heart. Genes Dev. 2005;19(13):1624–34. doi: 10.1101/gad.1319405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Meder B, et al. PINCH proteins regulate cardiac contractility by modulating integrin-linked kinase-protein kinase B signaling. Mol Cell Biol. 2011;31(16):3424–35. doi: 10.1128/MCB.05269-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Knoll R, et al. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation. 2007;116(5):515–25. doi: 10.1161/CIRCULATIONAHA.107.689984. [DOI] [PubMed] [Google Scholar]
- 144.Sultana N, et al. Zebrafish early cardiac connexin, Cx36.7/Ecx, regulates myofibril orientation and heart morphogenesis by establishing Nkx2.5 expression. Proc Natl Acad Sci U S A. 2008;105(12):4763–8. doi: 10.1073/pnas.0708451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Just S, et al. The myosin-interacting protein SMYD1 is essential for sarcomere organization. J Cell Sci. 2011;124(Pt 18):3127–36. doi: 10.1242/jcs.084772. [DOI] [PubMed] [Google Scholar]
- 146.Donlin LT, et al. Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev. 2012;26(2):114–9. doi: 10.1101/gad.177758.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gerull B, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30(2):201–4. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 148.Hassel D, et al. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat Med. 2009;15(11):1281–8. doi: 10.1038/nm.2037. [DOI] [PubMed] [Google Scholar]
- 149.Wang H, et al. Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2010;87(5):687–93. doi: 10.1016/j.ajhg.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Challice CE, Viragh S. The architectural development of the early mammalian heart. Tissue Cell. 1974;6(3):447–62. doi: 10.1016/0040-8166(74)90037-8. [DOI] [PubMed] [Google Scholar]
- 151.Challice CE, Viragh S. The phylogenetic and ontogenetic development of the mammalian heart: some theoretical considerations. Acta Biochim Biophys Acad Sci Hung. 1974;9(1–2):131–40. [PubMed] [Google Scholar]
- 152.Fishman GI. Understanding conduction system development: a hop, skip and jump away? Circ Res. 2005;96(8):809–11. doi: 10.1161/01.RES.0000165653.83279.20. [DOI] [PubMed] [Google Scholar]
- 153.Jongbloed MRM, et al. Normal and abnormal development of the cardiac conduction system; implications for conduction and rhuthm disorders in the child and adult. Differentiation. Differentiation. 2012 doi: 10.1016/j.diff.2012.04.006. (this issue) [DOI] [PubMed] [Google Scholar]
- 154.Arrenberg AB, et al. Optogenetic control of cardiac function. Science. 2010;330(6006):971–4. doi: 10.1126/science.1195929. [DOI] [PubMed] [Google Scholar]
- 155.Panakova D, Werdich AA, Macrae CA. Wnt11 patterns a myocardial electrical gradient through regulation of the L-type Ca(2+) channel. Nature. 466(7308):874–8. doi: 10.1038/nature09249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kamino K, Hirota A, Fujii S. Localization of pacemaking activity in early embryonic heart monitored using voltage-sensitive dye. Nature. 1981;290(5807):595–7. doi: 10.1038/290595a0. [DOI] [PubMed] [Google Scholar]
- 157.Sun Y, et al. Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev Biol. 2007;304(1):286–96. doi: 10.1016/j.ydbio.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Blaschke RJ, et al. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation. 2007;115(14):1830–8. doi: 10.1161/CIRCULATIONAHA.106.637819. [DOI] [PubMed] [Google Scholar]
- 159.Gourdie RG, et al. Endothelin-induced conversion of embryonic heart muscle cells into impulse-conducting Purkinje fibers. Proc Natl Acad Sci U S A. 1998;95(12):6815–8. doi: 10.1073/pnas.95.12.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hall CE, et al. Hemodynamic-dependent patterning of endothelin converting enzyme 1 expression and differentiation of impulse-conducting Purkinje fibers in the embryonic heart. Development. 2004;131(3):581–92. doi: 10.1242/dev.00947. [DOI] [PubMed] [Google Scholar]
- 161.Rentschler S, et al. Neuregulin-1 promotes formation of the murine cardiac conduction system. Proc Natl Acad Sci U S A. 2002;99(16):10464–9. doi: 10.1073/pnas.162301699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Arnaout R, et al. Zebrafish model for human long QT syndrome. Proc Natl Acad Sci U S A. 2007;104(27):11316–21. doi: 10.1073/pnas.0702724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Splawski I, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 164.Milan DJ, et al. Drug-sensitized zebrafish screen identifies multiple genes, including GINS3, as regulators of myocardial repolarization. Circulation. 2009;120(7):553–9. doi: 10.1161/CIRCULATIONAHA.108.821082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Milan DJ, et al. Drugs that induce repolarization abnormalities cause bradycardia in zebrafish. Circulation. 2003;107(10):1355–8. doi: 10.1161/01.cir.0000061912.88753.87. [DOI] [PubMed] [Google Scholar]
- 166.Grimes AC, et al. Solving an enigma: arterial pole development in the zebrafish heart. Dev Biol. 2006;290(2):265–76. doi: 10.1016/j.ydbio.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 167.Sato M, Yost HJ. Cardiac neural crest contributes to cardiomyogenesis in zebrafish. Dev Biol. 2003;257(1):127–39. doi: 10.1016/s0012-1606(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 168.Li YX, et al. Cardiac neural crest in zebrafish embryos contributes to myocardial cell lineage and early heart function. Dev Dyn. 2003;226(3):540–50. doi: 10.1002/dvdy.10264. [DOI] [PubMed] [Google Scholar]
- 169.Keyte A, Hutson MR. The neural crest in cardiac congenital anomalies. 2012 doi: 10.1016/j.diff.2012.04.005. (this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lawson ND, Wolfe SA. Forward and reverse genetic approaches for the analysis of vertebrate development in the zebrafish. Dev Cell. 2011;21(1):48–64. doi: 10.1016/j.devcel.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 171.Zhong TP, et al. gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science. 2000;287(5459):1820–4. doi: 10.1126/science.287.5459.1820. [DOI] [PubMed] [Google Scholar]
- 172.Peterson RT, et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nat Biotechnol. 2004;22(5):595–9. doi: 10.1038/nbt963. [DOI] [PubMed] [Google Scholar]
- 173.Zaghloul NA, Katsanis N. Zebrafish assays of ciliopathies. Methods Cell Biol. 2011;105:257–72. doi: 10.1016/B978-0-12-381320-6.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Valente EM, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 42(7):619–25. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lee JE, et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet. 2012;44(2):193–9. doi: 10.1038/ng.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fink M, et al. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques. 2009;46(2):101–13. doi: 10.2144/000113078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Trinh le A, et al. A versatile gene trap to visualize and interrogate the function of the vertebrate proteome. Genes Dev. 2011;25(21):2306–20. doi: 10.1101/gad.174037.111. [DOI] [PMC free article] [PubMed] [Google Scholar]