

Abstract
Presenilin conditional double knockout (PScDKO) mice have been used as animal models to study the development of Alzheimer’s disease (AD) phenotypes. Studies to date indicate that these animals exhibit memory dysfunction and decreased synaptic plasticity before the onset of neurodegeneration. Therefore, the current study sought to examine how the loss of presenilin expression leads to these defects. Drebrin A, a neuron-specific actin-binding protein, has been shown to play an important role in the activity-dependent redistribution of the NMDA type of glutamate receptors at the synapse which, in turn, is a critical step for enabling synaptic plasticity. It is hypothesized that defects in the activity dependent redistribution of NMDA receptors in PScDKO mice may be due to loss of drebrin A. In this study, electron microscopic immunocytochemistry (EM-ICC) was used to quantify and locate drebrin A in the CA1 field of the hippocampus of PScDKO mice. The high resolution of EM-ICC allowed for differentiation between drebrin A at the synapse and at nonsynaptic sites, the latter of which would reflect the protein’s role in regulating the reserve or degradative pool of NMDA receptors. The results here demonstrate that loss of function of presenilin in mice leads to a decrease in immunoreactivity for drebrin A at both synaptic (54% decrease, P < 0.01) and nonsynaptic areas (40% decrease, P < 0.01) and overall (44% decrease, P < 0.01). The reduction of drebrin A in both synaptic and non-synaptic locations of the spine may implicate impairment in glutamate receptor trafficking to and from the postsynaptic plasma membrane. In addition, because of reduced drebrin A at nonsynaptic sites, the regulation of the reserve and degradative pools of glutamate receptors may also be impaired, leading to further synaptic dysfunction. Therefore, this study provides evidence to the theory that drebrin A has a causal role in compromising activity-dependent glutamate receptor trafficking in PScDKO mice.
Keywords: electron microscopy, immunocytochemistry, drebrin A, Alzheimer’s disease, CA1, hippocampus, mouse, animal model
INTRODUCTION
Many studies on Alzheimer’s disease (AD) have focused on the accumulation of amyloid beta plaques (Aβ) and neurofibrillary tangles (NFT). Correlations are often made between Aβ/NFT markers and the severity of dementia, but the best correlate is arguably synaptic strength and density (Walsh and Selkoe, 2004). Not only does impaired synaptic function provide a better correlate to severity of dementia, but it also manifests itself long before the accumulation of these markers and neuronal loss (Bertoni-Freddari et al., 1996). Therefore, deciphering the molecular basis of synaptic dysfunction remains a crucial problem in understanding memory failure in Alzheimer’s disease.
Critical to learning and memory is the ability of healthy synapses of the cortex and hippocampus to convert synaptic activity levels over time into the maintenance and up/downregulation of receptors at the synapse (Rao and Craig, 1997). This mechanism is necessary for the steady maintenance of receptors (homeostatic plasticity) as well as long-term potentiation (LTP) and depression (LTD) (Malenka and Bear, 2004; Rumpel et al., 2005). If such mechanisms are impaired, the acquisition of new memory and updating of old memory would be impaired as well. These findings on memory formation and storage have added evidence to why and how synaptic dysfunction plays a major role in the AD brain. An important tool in the examination of these impairments is electron microscopy-immunocytochemistry (EM-ICC), which allows for the ability to locate and quantify changes in synaptic proteins at high spatial resolution.
The source of many genetic models that are used to investigate AD come from the study of mutations linked to familial AD; more than 90% of these mutations are located on genes encoding the proteins presenilin 1 and presenilin 2 (PS1 and PS2) (Hutton and Hardy, 1997). Presenilins form part of the protease complex, γ-secretase, which carries out the intramembraneous cleavage of amyloid precursor protein (APP) and the Notch receptors (De Strooper et al., 1998; 1999). PS mutations have been demonstrated to increase the production of the pathogenic 42-amino acid variant of Aβ (Moehlmann et al., 2002).
Loss of PS1 and PS2 function by conditional double knockout (PScDKO) results in AD phenotypes in mice (Shen and Kelleher, 2007). By two months of age, these PScDKO mice exhibit reduction of long-term potentiation and paired-pulse facilitation in the stratum radiatum of the CA1 hippocampus, as well as impairment of spatial memory acquisition (Saura et al., 2004). Moreover, by six months of age, the cortex and hippocampus of these mice begin to demonstrate deposits of glial fibrillary acidic proteins, neurofibrillary tangles, and hyperphosphorylated tau. Also present at six months are markers of inflammation and neurodegeneration, as well as reduced dendritic branching complexity and spine loss (Saura et al., 2004). Many months before these abnormalities occur, however, PScDKO mice demonstrate increased levels of the NR2A subunit of the N-methyl-D-aspartic acid receptor (NMDAR) at the synapse, reflecting a dysfunction of receptor trafficking at the synaptic level (Aoki et al., 2009b). In this study, the goal was to determine how the loss of PS function leads to increased levels of NMDARs at the synapse and the subsequent decrease in synaptic plasticity.
Among thousands of synaptic proteins to examine (Kennedy and Ehlers, 2006), drebrin A was chosen because of its central role in synaptic plasticity. Although F-actin occurs both pre- and postsynaptically, this neuron-specific protein is the only F-actin-binding molecule found exclusively on the postsynaptic side of excitatory synapses (Aoki et al., 2005). Drebrin A has properties suitable for modulating the trafficking of receptors into and out of spines (Shirao and Sekino, 2001). In addition, drebrin A knockout in hippocampal neurons leads to impaired homeostatic synaptic plasticity (HSP); specifically, drebrin A knockout causes impaired NMDAR activity-dependent upregulation of NMDARs within spine heads (Aoki et al., 2009a). Drebrin A, through its association with actin, has also been implicated in the regulation of the size, number, and morphology of dendritic spines (Hayashi and Shirao, 1999; Takahashi et al., 2006). Lastly, patients diagnosed with AD or mild cognitive impairment demonstrate decreased levels of drebrin A (Counts et al., 2006; Harigaya et al., 1996; Hatanpaa et al., 1999). Thus, the objective of this study was to determine whether the loss of PS function leads to synaptic impairments through a mechanism involving drebrin A. To accomplish this goal, EM-ICC was used to quantify and localize the distribution of drebrin A within synapses of intact brain tissue of PScDKO mice.
MATERIAL AND METHODS
Animals
Generation of PScDKO mice, of C57BL6/129 hybrid background, was achieved at the animal facility at Harvard Medical School. Floxed PS1 (fPS1), αCaMKII-Cre transgenic (Yu et al., 2001), and PS2−/− (Steiner et al., 1999) mice were crossed to obtain fPS1/fPS1; αCaMKII-Cre; PS2−/− mice. Four PScDKO mice, derived from two litters, were used for the EM analysis. For controls, two genotypes of mice were used: one was the fPS1 (three mice), which were floxed but otherwise of the same genotype as wildtype (WT) and expressed both PS1 and PS2. The other control was the PS2−/− (two mice), from which the PS2 gene was deleted but the PS1 gene was retained. Neither of these controls exhibited histological abnormalities, as assessed by light microscopy. Their memory was also intact, based on the performance in the Morris water maze to assess spatial memory or contextual fear conditioning, used to assess associative memory (Saura et al., 2004). Importantly, none of the animals that underwent EM-ICC analyses had been used for behavioral studies ante-mortem, to avoid inducing any change in synaptic proteins because of experience.
Preparation of brain tissue for electron microscopy
All procedures were in accordance with the protocol approved by the University Animal Welfare Committee of Harvard Medical School and in full conformance with the recommendations of the 1996 edition of the NIH Guide for the Care and Use of Laboratory Animals and all applicable federal, state, and local laws, and as outlined in the NIH the principles for use of animals (NIH Manual Chapter 4206).
Brains were fixed in the laboratory of Jie Shen, Harvard Medical School, and then shipped to the laboratory of Chiye Aoki, New York University, where tissue was prepared for EM-ICC. At the age of 8 weeks postnatal, mice were deeply anesthetized using Nembutal (50 mg/kg i.p.) and brains fixed by sequential transcardial perfusion using a peristaltic pump (10 mL/min) with: (1) 10–50 mL of saline containing heparin (1000 U/mL) and (2) 200 mL of 4% paraformaldehyde in 0.1M phosphate buffer (PB, pH 7.4). No other aldehydes were added to the perfusate. This omission was intentional to avoid overfixation of the tissue during the shipment from Harvard to NYU.
Following perfusion, brains were removed from the skull, and then immersed in the same fixative while shipped on ice to the Aoki laboratory at NYU. Within one day following the perfusion, the brains were cut into 40-μm thick coronal sections using a vibratome. The sections were subsequently stored at 4°C in 0.01M PB with 0.9% sodium chloride (PBS, set to pH 7.4) containing 0.05% sodium azide.
Antibody and immmunoreagents immunoreagents information
The drebrin A antibody, DAS2, was obtained from IBL international (Hamburg, Germany, Cat. No. JP28023). DAS2 was generated using the immunogen, Phe-Ile-Lys-Ala Ser-Asp-Ser-Gly-Pro-Ser-Ser-Ser, which corresponds to a sequence of residues 325–336 of drebrin that is unique to the adult form, drebrin A (Shirao et al., 1992). DAS2 was produced in rabbits and purified by epitope selection, using the above polypeptide. The specificity of the DAS2 antibody was previously confirmed using Western blot analysis (Aoki et al., 2009a). Brain homogenates from wildtype mice exhibited a single band corresponding to drebrin A, whereas homogenates from drebrin A knockout mice lacked the immunoreactive band. Furthermore, the antibody was able to discriminate drebrin A from its isomer, drebrin E. These results were verified by light microscopy of the entire forebrain and electron microscopy analysis of the cortex.
Secondary antibodies used for the described studies were biotinylated antirabbit IgG produced in goat (from Vector Laboratories, Burlingame, CA, Elite kit, Cat. No. PK 6200) or gold-conjugated antirabbit IgG, produced in goat, for which the colloidal gold particles were 10 nm in diameter (Aurion, EM-Science, Fort Washington, PA, Cat. No. 810.011). Bovine serum albumin (BSA) was purchased from Sigma Chemicals (St. Louis, MO). Glutaraldehyde, osmium tetroxide, paraformaldehyde, and EMBED-812 were purchased from EMSciences. The silver-intensification kit was purchased from KPL (Gaithersburg, MD, Cat. No. 50-22-01).
Drebrin A immunocytochemistry
Immunocytochemistry was performed to visualize drebrin A using two EM-ICC procedures as described previously (Aoki et al., 2000). The horseradish peroxidase-diaminobenzidine (HRP-DAB) procedure was used to optimize detection of drebrin A within spines while the silver-intensified gold (SIG) procedure was chosen to optimize subcellular localization of drebrin A within spines.
For both labeling procedures, sections were first immersed in a solution of 1% hydrogen peroxide in 0.1M PB at room temperature for 30 minutes in order to enhance tissue antigenicity. The sections were then incubated in a solution containing 0.01M PB, saline (0.9% NaCl), and 1% BSA to minimize background immunolabeling and 0.05% sodium azide to minimize bacterial growth in the buffer. After pre-incubating sections for a minimum of 30 minutes, the sections were then incubated in DAS2 antibody (diluted 1:1000) in PBS-BSA-azide. The incubation length was three days at room temperature with constant, gentle agitation. Following incubation, sections were rinsed in PBS for one hour at room temperature, and then immersed in secondary antibody solution (1:200 dilution in PBS-BSA azide).
Previous studies have shown that tissue postfixed with glutaraldehyde after the immunocytochemical procedure but before fixation with osmium tetroxide provide adequate preservation of ultrastructure and antigenicity for most immunocytochemical procedures (Aoki et al., 2000). In this procedure, sections are postfixed in a phosphate buffered saline (PBS) solution containing 2% glutaraldehyde for 30 minutes under a ventilation hood, and then rinsed in PBS three times at five-minute intervals. For immunolabeling using the HRP-DAB reaction product as the label, the standard ABC Elite kit from Vector was used. For immunolabeling using the SIG reaction, sections were incubated overnight at room temperature in a solution of colloidal gold-conjugated goat anti-rabbit IgG (1:100 dilution in PBS-BSA azide). Sections were then incubated for 10 minutes using the KPL silver-intensification kit at room temperature. Sections labeled by HRP-DAB were further fixed using 1% osmium tetroxide for 60 minutes. SIG sections were post-fixed without osmium tetroxide and were instead incubated in solutions of tannic acid (1%, 40 minutes), uranyl acetate (1%, 40 minutes), and iridium tetrabromide (0.5%, 20 minutes). Further details were as described previously (Aoki et al., 2000; Phend et al., 1995).
Imaging of immunocytochemically stained sections
Sections were flat-embedded in Embed 812, and then capsule-embedded in order to perform ultrathin sectioning. HRP-DAB labeled sections were not counterstained to optimize the ability to detect low levels of reaction products. SIG-labeled sections were counterstained with Reynold’s lead citrate, because SIG particles could still be identified easily against contrast-enhanced images. Ultrathin sections were viewed under a JEOL 1200XL electron microscope. Images were captured digitally using a 1.2 megapixel Hamamatsu CCD camera from AMT (Boston, MA).
All persons capturing or analyzing images were kept blind to the genotype of the animal to avoid any sampling bias. Micrographs for quantitative analysis were taken from the stratum radiatum of CA1 of the hippocampus. Images were digitally captured at a magnification of ×40,000, with each micrograph spanning 9.87 μm2; image capturing was performed in systematic sweeps across the neuropil, such that no image overlapped.
Image analysis of synapses
Images were analyzed using Adobe Photoshop software CS5 (version 12.0). Within 2D digitized EM images, asymmetric and Gray type 1 synapses were identified based on the presence of thick postsynaptic densities (PSDs) along the intracellular surfaces of dendritic spines, as well as the presence of an opposing vesicle-filled axon terminal (Peters et al., 1991). In each animal, 200 excitatory synapses were analyzed, requiring an average of 40 images for both control and PScDKO mice. Thus, the amount of synapses found per unit area was not different, consistent with the finding that spine density in PScDKO mice is not yet decreased at two months of age (Saura et al., 2004). For DAB-labeled tissue, four control mice and four PScDKO mice were analyzed. Spines of excitatory synapses were characterized as either labeled or not labeled. For SIG labeled tissue, three control mice and four PScDKO mice were compared. Labeling was characterized as either synaptic (occurring at the PSD or near the PSD) or non-synaptic (Fig. 1).
Fig. 1.

Cartoon depiction indicating the location profiles for the SIG and HRP-DAB analyses. Both techniques confirmed that drebrin A occurs exclusively postsynaptically. For SIG analysis, any particles found within the PSD or a distance equal to the thickness of two PSDs were categorized “Synaptic” and other particles found within the spine head were categorized as “Nonsynaptic”. To minimize variability, the location of each particle was designated as its centermost point. For HRP-DAB analysis, spines were categorized as “Labeled” or “Unlabeled.”
Statistical analyses
For DAB-labeled tissue, the mean proportion of immunolabeling per 10 spines was obtained for each animal, and these mean values were compared across the two genotypes. For SIG-labeled tissue, the mean proportion of immunolabeling per 10 spines and the mean number of SIG particles per 10 spines were obtained for each animal, and these mean values were compared across the two genotypes. Normality of distribution of the mean values of the data were evaluated using the Kolmogorov-Smirnov normality test, the Lilliefors test, and Shapiro-Wilk’s W test, with the N-value reflecting the means of randomly the divided groups of 10 spines. The normality tests indicated that both the proportion of spines labeled and the number of SIG particles were not normally distributed. Therefore the nonparametric Mann-Whitney’s U test was used for subsequent statistical comparisons.
All data from the electron microscopic immunocytochemical quantification were analyzed using the software Statistica (version 10.0, released from StatSoft). Experimenters performing the analysis were kept blind to the genotype of each animal. Differences were accepted as significant for P values <0.01.
Figure preparation
Electron micrographs representative of each genotype were selected from the original image data. Adobe Photoshop software (version 12.0) was used to adjust the brightness and contrast of the images and to add visual markers. For images in which HRP-DAB was the immunolabel, color-coded lines were added to indicate dendritic shafts and labeled/unlabeled spines. For images in which SIG was the immunolabel, arrows were added to indicate the location of gold colloidal particles. In addition, lettering was added to show the locations of spine heads and axon terminals.
RESULTS
The HRP-DAB labeling technique was first used to identify the presence or absence of drebrin A within dendritic spines. The SIG labeling technique was then used to localize and quantify labeling within individual dendritic spine profiles. EM micrographs were sampled from the stratum radiatum of CA1 in four control mice and four PScDKO mice for the HRP-DAB method, and three control mice and four PScDKO mice for the SIG method. From each animal, 200 synapses were sampled. In both methods, EM analysis demonstrated the occurrence of drebrin A labeling in both control and PScDKO mice. Moreover, immunoreactivity to drebrin A was confined exclusively to spines and dendritic shafts, which has been observed previously (Aoki et al., 2005; Shirao and Sekino, 2001). Labeling was not detectable within axon terminals (Figs. 2 and 4).
Fig. 2.
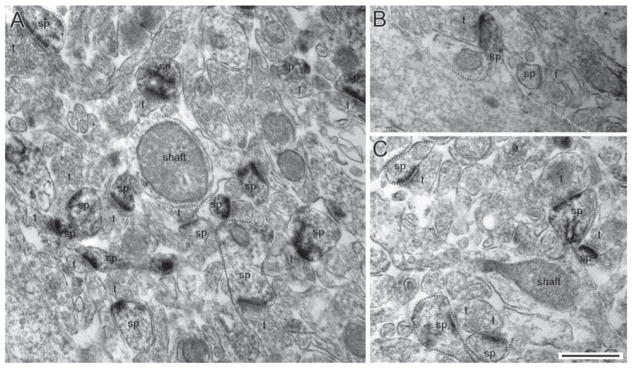
EM micrographs taken from the CA1 hippocampus of control (panel A) and PScDKO (panels B and C) brains. In all panels, drebrin A was immunolabeled by the HRP-DAB procedure. Spines (sp) that are positively labeled for drebrin A are outlined in red and form synapses with axon terminals (t). Unlabeled spines are outlined in blue and dendritic shafts outlined in yellow. Labeling occurred exclusively postsynaptically at excitatory synapses. Calibration bar = 500 nm, applicable to all micrographs. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Fig. 4.

EM micrographs taken from the CA1 hippocampus of control (panels A and B) and PScDKO (panels C and D) brains. In all panels, drebrin A was immunolabeled by the SIG procedure. Spines (sp) form excitatory asymmetric synapses with axon terminals (t). SIG particles occurring within spines are labeled with solid arrows. Labeling also occurred at nonsynaptic areas or within dendritic shafts, indicated by hollow arrows. Plus signs (+) indicate regions in which the ultrathin tissue is missing; this is due to the sampling region being near the vibratome sectioning surface. Calibration bar = 500 nm, applicable to all micrographs.
Comparison of the proportion of spines labeled across genotypes, as assessed by using HRP-DAB and SIG labels
EM analysis of tissue labeled by the HRP-DAB procedure revealed that the proportion of spines containing drebrin A immunoreactivity in PScDKO mice was decreased compared with that in control mice (Fig. 2). Control mice exhibited 5.8 ± 0.20 spines labeled per 10 compared with 3.36 ± 0.25 spines labeled per 10 in PScDKO mice (Fig. 3). This 42% decrease in drebrin A labeling in the CA1 hippocampus of PScDKO brains relative to control brains was statistically significant (P < 0.001, z-value = 6.532, Mann-Whitney U test). In addition, the proportion of spines labeled by SIG was determined, similar to the analysis of the HRP-DAB labeled tissue. In this comparison, spines were designated labeled if they contained one or more SIG particles (Fig. 4). Control mice exhibited 2.82 ± 0.23 spines labeled per 10, whereas PScDKO mice exhibited 1.68 ± 0.13 spines labeled per 10 (Fig. 3). This 40% decrease in labeling by SIG was similar to the previous 42% decrease observed in the HRP-DAB analysis and was statistically significant (P < 0.001, z-value = 3.897, Mann-Whitney U test). Expectedly, the more sensitive HRP-DAB procedure labeled roughly twice as many spines as the SIG procedure.
Fig. 3.
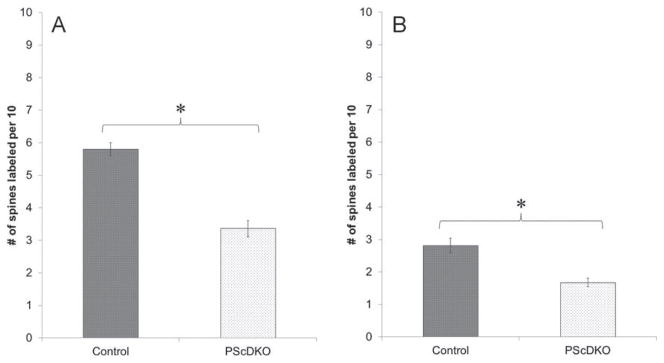
Proportion of spines labeled by drebrin A. The HRP-DAB analysis (A) revealed a 42% decrease in the CA1 hippocampus of PScDKO brains relative to control. The SIG analysis (B) revealed a 40% decrease in the CA1 hippocampus of PScDKO brains relative to control. Asterisk denotes P value <0.01. Values are reported as mean ± SEM.
The effect of PScDKO upon the localization of drebrin A within spines
The previous results demonstrated a significant decrease in the proportion of spines immunoreactive to drebrin A in the hippocampus of PScDKO mice relative to control. The precise location of this decrease could be determined by the localization of SIG particles. Quantification of SIG particles revealed a decrease in drebrin A immunolabeling in the stratum radiatum of CA1 in PScDKO brains compared with that in control brains (Figs. 4 and 5). This decrease occurred at both synaptic sites (at and near the PSD) and at non-synaptic sites (within spines but not at or near the PSD, see Fig. 1). At synaptic locations, control mice exhibited 1.01 ± 0.13 SIG particles per 10 spines compared with 0.46 ± 0.09 particles per 10 spines in PScDKO mice. This 54% decrease was statistically significant (P < 0.01, z-value = 3.185, Mann-Whitney U test). At non-synaptic locations, control mice had 2.75 ± 0.19 particles per 10 spines compared with 1.66 ± 0.14 particles per 10 spines in PScDKO. This 40% decrease was significant (P < 0.01, z-value = 2.920, Mann-Whitney U test). Overall, at both synaptic and non-synaptic sites, control mice had 3.77 ± 0.34 particles per 10 spines compared with 2.14 ± 0.19 particles per 10 spines in PScDKO mice, reflecting a 44% decrease. This decrease was also statistically significant (P < 0.001, z-value = 3.756, Mann-Whitney U test). The difference between the synaptic and nonsynaptic decreases (54% and 40%, respectively) was not significant (P > 0.05, z-value = 1.129, Mann-Whitney U Test).
Fig. 5.

Quantitative analysis of SIG labeling of drebrin A. Analysis revealed 54% and 40% decreases at synaptic and nonsynaptic locations respectively in the CA1 hippocampus of PScDKO brains relative to control. At all locations there was a 44% decrease. Asterisks denote P value <0.01. Values are reported as mean ± SEM.
Pattern distribution of drebrin A across the spine population
Lastly, a histogram analysis was performed on the number of SIG particles per 10 spines (Fig. 6), to examine the distribution of particle labeling across spines. The histogram revealed that the decrease in SIG particle labeling in PScDKO mice is due to the presence of more spines with fewer SIG particles as well as fewer spines with more SIG particles.
Fig. 6.

Frequency histogram analysis of the distribution of the number of SIG particles per 10 spines. Control mice have more synapses containing more SIG particles, whereas PScDKO mice have more synapses containing fewer SIG particles.
DISCUSSION
The present study demonstrates an overall reduction of drebrin A at postsynaptic spines within the stratum radiatum of the CA1 hippocampus of PScDKO mice relative to control. This reduction was present both at synaptic locations (at and near the PSD) and at non-synaptic locations (away from the PSD). To obtain these results, two separate and unique labeling techniques were used. The HRP-DAB technique diffusely labeled postsynaptic spinous profiles containing drebrin A; this technique was used to optimize detection. Meanwhile, the SIG technique labeled the location of drebrin A within spinous profiles. These two techniques demonstrate that the proportion of spines with drebrin A as well as the number of drebrin A particles within each spine is decreased in PScDKO mice.
Role of drebrin A in PScDKO mice
At two months of age, PScDKO mice exhibit normal brain morphology, including normal neuron number, volume, and spine density (Saura et al., 2004). However, at this age these mice exhibit reduced spatial memory, contextual fear learning, LTP, and NMDAR response. Past studies on PScDKO mice have attempted to explain these results on a molecular level by demonstrating increased levels of NR2A sub-units of NMDARs at synapses compared with control (Aoki et al., 2009b). This result is thought to reflect impaired regulation of NDMARs. Taken together, these observations are especially interesting given that the impairments are seen at two months of age, whereas other symptoms such as neurodegeneration and decreased spine density do not appear until six months of age. Therefore, elucidating the mechanisms by which these upstream impairments occur remains an important question in the study of AD.
Drebrin A was an ideal candidate to answer this question because of its role in homeostatic synaptic plasticity (HSP) and its ability to modulate receptor trafficking (Aoki et al., 2009a; Shirao and Sekino, 2001). HSP is involved in the maintenance of synaptic strength and is necessary for healthy brain function (Turrigiano, 2008). In normal brains, the blockade of NMDARs by the antagonist D-APV causes activity-dependent upregulation of additional NMDARs to the synapse (Aoki et al., 2003). However, knockout of drebrin A (DAKO) impairs the activity-dependent redistribution of the NR2A subunit of NMDARs (Aoki et al., 2009a). Downregulation of drebrin A also causes impaired synaptic targeting of the NR1 subunit (Takahashi et al., 2006). Therefore, drebrin A is likely to play a role in the regulation of NMDAR distribution within spines.
In the present study, these previous studies are linked and show that loss of presenilin function results in reduction of drebrin A at the synapse at two months of age. The current findings support the theory that the loss of PS function leads to impaired regulation of NMDARs through a mechanism involving drebrin A. It is therefore plausible that these early events may eventually lead to the phenotypes exhibited at six months of age: neurodegeneration, decreased dendritic branching, and decreased spine density (Saura et al., 2004). At six months of age, PScDKO animals also exhibit severely impaired spatial memory and contextual fear learning, as well as hyperphosphorylated tau. These morphological abnormalities at six months of age may very well be a result of drebrin A decrease that occurs at a younger age, since the protein has been shown to regulate spine size, number, and morphology (Hayashi and Shirao, 1999; Kobayashi et al., 2007).
Mechanism of drebrin A in spines of PScDKO mice
Through its interaction with the actin cytoskeleton network, drebrin A has been linked to many other properties of dendritic spines, including spine number, size, and morphology (Hayashi and Shirao, 1999). Drebrin A may also be able to modulate trafficking of receptors into and out of spines (Shirao and Sekino, 2001), because these receptors are associated with F-actin through α-actinin (Krupp et al., 1999; Wyszynski et al., 1997). Therefore, it is possible that these glutamate receptors can be anchored or liberated from the inner postsynaptic membrane surface through the interaction of drebrin A and F-actin. Through this mechanism, a drastic reduction in drebrin A, such as that seen here, may lead to the abnormal NMDAR regulation seen in previous studies. Moreover, because drebrin A is also reduced at nonsynaptic locations within spines, the regulation of the reserve and degradative pools of NMDARs may be impaired as well.
Recently, drebrin A has been associated with the regulation of Ca2+ entry into the cell via a mechanism involving store-operated calcium channels (Mercer et al., 2010). In this pathway, the reduction of intracellular Ca2+ causes store-operated calcium channels to allow the influx of Ca2+. Mercer et al. discovered a compound, 3,5-bis(trifluoromethyl)pyrazole (BTP), which disrupted this process; interestingly, they found that BTP accomplishes this disruption by targeting and inhibiting drebrin A function. The role of drebrin A in this process may be to favor actin filament depolymerization; in turn, this would allow actin filament turnover, which is necessary for activation of store-operated calcium channels (Hao and August, 2005). Therefore, through its interaction with the actin cytoskeleton, drebrin A may also play a crucial role in the regulation of Ca2+ influx into the cell.
CONCLUDING REMARKS
Aβ plaques and neurofibrillary tangles are often the focal point of much research on the Alzheimer’s disease brain, but synaptic dysfunction clearly plays an important role as well. The finding that plasticity impairments appear long before many others is especially interesting and promising. However, the task of elucidating the molecular mechanics of this dysfunction is staggering. In the current study, the finding of a decrease in drebrin A in the PScDKO mouse reveals many more questions to be answered, such as the relationship between PS and drebrin A and why this decrease occurred; for example, drebrin A may be decreased due to lower expression, higher levels of degradation, or impaired trafficking of the molecule itself. Regardless, this finding provides one more element in the understanding of Alzheimer’s disease and synaptic dysfunction.
Acknowledgments
Contract grant sponsors: Dean’s Undergraduate Research Fund; New York University’s Challenge Fund; Contract grant sponsor: National Institutes of Health; Contract grant numbers: 2R01DA009618-10A1, NS41783; Contract grant sponsor: Alzheimer’s Association; Contract grant number: IIRG 07-58479.
The authors thank Dr. Angela Ho and Dr. Jie Shen of Harvard University for the crucial generation of all the wild-type and PScDKO mice that were used in this study.
References
- Aoki C, Rodrigues S, Kurose H. Use of electron microscopy in the detection of adrenergic receptors. Methods Mol Biol. 2000;126:535–563. doi: 10.1385/1-59259-684-3:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki C, Sekino Y, Hanamura K, Fujisawa S, Mahadomrongkul V, Ren Y, Shirao T. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. J Comp Neurol. 2005;483:383–402. doi: 10.1002/cne.20449. [DOI] [PubMed] [Google Scholar]
- Aoki C, Fujisawa S, Mahadomrongkul V, Shah PJ, Nader K, Erisir A. NMDA receptor blockade in intact adult cortex increases trafficking of NR2A subunits into spines, postsynaptic densities, and axon terminals. Brain Res. 2003;963:139–149. doi: 10.1016/s0006-8993(02)03962-8. [DOI] [PubMed] [Google Scholar]
- Aoki C, Kojima N, Sabaliauskas N, Shah L, Ahmed TH, Oakford J, Ahmed T, Yamazaki H, Hanamura K, Shirao T. Drebrin a knockout eliminates the rapid form of homeostatic synaptic plasticity at excitatory synapses of intact adult cerebral cortex. J Comp Neurol. 2009a;517:105–121. doi: 10.1002/cne.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki C, Lee J, Nedelescu H, Ahmed T, Ho A, Shen J. Increased levels of NMDA receptor NR2A subunits at pre- and postsynaptic sites of the hippocampal CA1: An early response to conditional double knockout of presenilin 1 and 2. J Comp Neurol. 2009b;517:512–523. doi: 10.1002/cne.22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni-Freddari C, Fattoretti P, Casoli T, Caselli U, Meier-Ruge W. Deterioration threshold of synaptic morphology in aging and senile dementia of Alzheimer’s type. Analytical and quantitative cytology and histology/the International Academy of Cytology. American Society of Cytology. 1996;18:209–213. [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:592–601. doi: 10.1097/00005072-200606000-00007. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of pre-senilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Hao S, August A. Actin depolymerization transduces the strength of B-cell receptor stimulation. Mol Biol Cell. 2005;16:2275–2284. doi: 10.1091/mbc.E04-10-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigaya Y, Shoji M, Shirao T, Hirai S. Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J Neurosci Res. 1996;43:87–92. doi: 10.1002/jnr.490430111. [DOI] [PubMed] [Google Scholar]
- Hatanpaa K, Isaacs KR, Shirao T, Brady DR, Rapoport SI. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:637–643. doi: 10.1097/00005072-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Shirao T. Change in the shape of dendritic spines caused by overexpression of drebrin in cultured cortical neurons. J Neurosci. 1999;19:3918–3925. doi: 10.1523/JNEUROSCI.19-10-03918.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Hardy J. The presenilins and Alzheimer’s disease. Hum Mol Genet. 1997;6:1639–1646. doi: 10.1093/hmg/6.10.1639. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi C, Aoki C, Kojima N, Yamazaki H, Shirao T. Drebrin a content correlates with spine head size in the adult mouse cerebral cortex. J Comp Neurol. 2007;503:618–626. doi: 10.1002/cne.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci. 1999;19:1165–1178. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mercer JC, Qi Q, Mottram LF, Law M, Bruce D, Iyer A, Morales JL, Yamazaki H, Shirao T, Peterson BR, August A. Chemicogenetic identification of drebrin as a regulator of calcium responses. Int J Biochem Cell Biol. 2010;42:337–345. doi: 10.1016/j.biocel.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci USA. 2002;99:8025–8030. doi: 10.1073/pnas.112686799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster Hd. The fine structure of the nervous system: Neurons and their supporting cells. New York: Oxford University Press; 1991. p. xviii.p. 494. [Google Scholar]
- Phend KD, Rustioni A, Weinberg RJ. An osmium-free method of epon embedment that preserves both ultrastructure and antigenicity for post-embedding immunocytochemistry. J Histochem Cytochem J Histochem Soc. 1995;43:283–292. doi: 10.1177/43.3.7532656. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, III, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., III The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirao T, Kojima N, Obata K. Cloning of drebrin A and induction of neurite-like processes in drebrin-transfected cells. Neurore-port. 1992;3:109–112. doi: 10.1097/00001756-199201000-00029. [DOI] [PubMed] [Google Scholar]
- Shirao T, Sekino Y. Clustering and anchoring mechanisms of molecular constituents of postsynaptic scaffolds in dendritic spines. Neurosci Res. 2001;40:1–7. doi: 10.1016/s0168-0102(01)00209-7. [DOI] [PubMed] [Google Scholar]
- Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, Citron M, Kopan R, Pesold B, Keck S, Baader M, Tomita T, Iwatsubo T, Baumeister R, Haass C. A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and notch signaling. J Biol Chem. 1999;274:28669–28673. doi: 10.1074/jbc.274.40.28669. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Mizui T, Shirao T. Down-regulation of drebrin A expression suppresses synaptic targeting of NMDA receptors in developing hippocampal neurones. J Neurochem. 2006;97(Suppl 1):110–115. doi: 10.1111/j.1471-4159.2005.03536.x. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: Synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Yu H, Saura CA, Choi SY, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, Younkin S, Kandel ER, Kirkwood A, Shen J. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]