

Abstract
This review focuses on a relatively new class of microtubule stabilizers, the taccalonolides. The taccalonolides are highly oxygenated pentacyclic steroids isolated from plants of the genus Tacca. Originally identified in a cell-based phenotypic screen, the taccalonolides have many properties similar to other microtubule stabilizers. They increase the density of interphase microtubules, causing microtubule bundling, and form abnormal multipolar mitotic spindles leading to mitotic arrest and ultimately, apoptosis. However, the taccalonolides differ from other microtubule stabilizers in that they retain efficacy in taxane resistant cell lines and in vivo models. Binding studies with the newly identified, potent taccalonolide AJ demonstrated covalent binding to β-tubulin at or near the luminal and/or pore taxane binding site(s) which stabilizes microtubule protofilaments in a unique manner as compared to other microtubule stabilizers. The isolation and semi-synthesis of 21 taccalonolides helped to identify key structure activity relationships and the importance of multiple regions across the taccalonolide skeleton for optimal biological potency.
1. Introduction
Nature has provided a wide range of compounds that affect the mammalian cytoskeleton, including compounds that bind to tubulin and disrupt the structure and function of cellular microtubules. Microtubule disrupting agents have been isolated from wide variety of sources, including marine sponges, mycobacteria, cyanobacteria, and plants. Moreover, new microtubule active compounds with significant biological activities continue to be discovered from nature. Plant-derived compounds that destabilize microtubules include the vinca alkaloids, colchicine and the combretastatins. The first microtubule stabilizer identified, taxol, was isolated from the bark of the Pacific Yew. Plants of the genus Tacca have a history of use in traditional medicines for a wide variety of ailments, and while the chemical structures of the taccalonolides were elucidated in the 1980’s, their microtubule stabilizing and antitumor activities were only discovered in the last 10 years.
2. Chemical isolation and structure elucidation of natural taccalonolides A-Y
A compound named taccalin was initially isolated from the tubers of Tacca leontopetaloides by the Scheuer laboratory in 1963, but the structure was not solved.1 Twenty-five years later, Chen’s group isolated and elucidated the structures of the taccalonolides A and B from the rhizomes of Tacca plantaginea.2 The taccalonolide backbone consists of a highly oxygenated pentacyclic steroidal skeleton which contains a C2-C3 epoxide group and an enol-γ-lactone. Other natural taccalonolides were isolated by various laboratories from 1987-2008, including the taccalonolides A – M2-6 and W – Y7 from Tacca plantaginea, taccalonolides N8 and R – V9 from Tacca paxiana, and taccalonolides O – Q from Tacca subflabellata.10, 11 Only the taccalonolides A and G – K were reported to exhibit weak cytotoxic activity against P-388 leukemia cells in vitro.
3. Identification of the taccalonolides A and E as microtubule stabilizers
The ability of Tacca chantrieri extracts to cause bundling of interphase microtubules was discovered in a cell-based screen of crude extracts. Bioassay-guided fractionation led to the isolation of taccalonolides A and E, the most abundantly occurring secondary metabolites, as new microtubule stabilizers.12 The highly acetylated pentacyclic skeleton of the taccalonolides makes them structurally distinct from all other microtubule stabilizers. The only structural difference between the taccalonolides A and E is the lack of an acetoxy group at C11 in taccalonolide E (Figure 1).12 Taccalonolides A and E cause bundling of interphase microtubules and mitotic arrest of cancer cells with multiple aberrant spindles that initiate apoptosis in a manner similar to paclitaxel.12 However, the taccalonolides retain efficacy in cells containing mutations in the paclitaxel binding site as well as those expressing P-glycoprotein (Pgp),12 a drug efflux pump which contributes to the clinical resistance of paclitaxel and docetaxel.13, 14 Additional studies with the taccalonolides A and E, as well as the semi-synthetic derivatives B and N, demonstrated their ability to overcome resistance due to expression of Pgp, the βIII-isotype of tubulin or the MRP7 drug efflux transporter.15 Most importantly, the taccalonolides A and E were found to have excellent in vivo antitumor activity in a Pgp-expressing, paclitaxel and doxorubicin resistant syngeneic mammary tumor model.15 Although the in vitro antiproliferative potencies of the taccalonolides A, E and N were at least 100-fold lower than paclitaxel, antitumor trials demonstrated that they had unexpectedly high in vivo potency comparable to or better than paclitaxel.15, 16 Thus, in spite of the low in vitro potencies of taccalonolides A and E, their excellent in vivo potency and efficacy coupled with their ability to overcome taxane resistance prompted further interest in this class of stabilizers.
Figure 1.
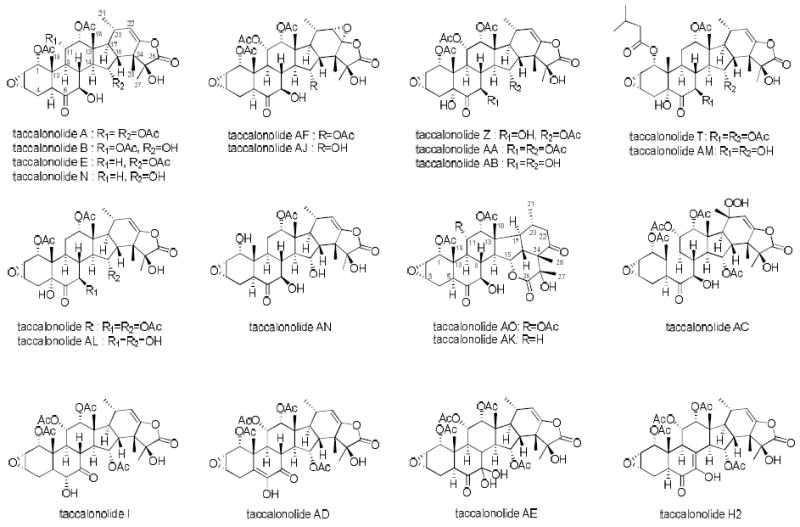
The structures of taccalonolides described in this study
Studies were conducted to identify the mechanism of microtubule stabilization of the taccalonolides A and E. Surprisingly, in a comprehensive study of 19 structurally diverse agents that were reported to have microtubule stabilizing activity, Buey and colleagues found that the taccalonolides A and E were unable to interact directly with microtubules or to enhance the polymerization of purified tubulin.17 Furthermore, taccalonolide A was unable to enhance the polymerization of tubulin even in cellular extracts that contained a full complement of cytosolic proteins.18 These initial studies suggested that the taccalonolides A and E stabilize microtubules in cells independently of direct microtubule binding. However, later studies showed that the significantly more potent taccalonolides, AF and AJ, directly interacted with microtubules.19 It is interesting to speculate that the taccalonolides A and E are prodrugs that are converted into binding-competent taccalonolides in the intact cell where they exert microtubule stabilizing effects and in vivo antitumor efficacy.
4. Mechanistic uniqueness of the taccalonolides
In addition to the inability of taccalonolides A and E to stimulate tubulin polymerization in vitro, several other properties were noted that distinguished this class of microtubule stabilizers from classic taxane-site binding agents. Another major difference is that taccalonolide A causes bundling of interphase microtubules at low antiproliferative concentrations while paclitaxel does not cause gross microtubule bundling until concentrations over 30-fold higher than its IC50 are used.18 This finding is interesting in light of recent evidence that the interphase effects of microtubule targeted agents likely play an important role in their anticancer actions.20, 21 Taccalonolide A also has a high degree of cellular persistence based on the observation that exposure of cells to antiproliferative concentrations for only 4 hours effectively eliminated any subsequent colony formation.18 The strong interphase effects of the taccalonolides combined with their high degree of cellular persistence were proposed to partially explain why they are much more potent in vivo than would be expected from cell-based studies.18
The mitotic spindles formed by the taccalonolides are also quite distinct from those initiated by other classes of microtubule stabilizers, including paclitaxel and laulimalide.12,22 Studies evaluating the mitotic signaling pathways leading from microtubule stabilization to initiation of mitotic arrest and subsequent apoptosis showed differences among microtubule stabilizers.22 At antiproliferative concentrations, the taccalonolides cause the formation of multipolar mitotic spindles, often with 5-6 punctate mitotic spindle asters, whereas paclitaxel treatment causes the appearance of 2-3 diffuse spindle asters.12,22 Unlike other classes of microtubule stabilizers, the taccalonolides also inhibit centrosome disjunction and separation at antiproliferative concentrations. These centrosomal defects initiated by the taccalonolides appear to be due to their ability to inhibit Polo-like kinase 1 (Plk1), a mitotic kinase that regulates centrosome disjunction and separation.22 Additionally, the taccalonolides cause premature and amplified expression of Aurora A and Aurora A interacting proteins while the opposite is seen in paclitaxel-treated cells.22 These studies demonstrated the significant differences in mitotic signaling events that occur after treatment of cells with the taccalonolides and other microtubule stabilizers.
5. Isolation and semi-synthesis of the potent new taccalonolides AF and AJ and their direct effects on tubulin polymerization
The distinct effects of the major taccalonolides A and E led to a search for additional minor taccalonolides to define structure activity relationships. Taccalonolide AF was isolated as a rare constituent from one fraction from Tacca plantaginea.23 This new taccalonolide has potent antiproliferative and microtubule stabilizing activities with an IC50 of 23 nM. Interestingly, the only difference between AF and the major plant metabolite taccalonolide A is the presence of an epoxide group at C22-23, which increased the potency over 200-fold (Figure 1).23 Due to its low abundance and similarity to taccalonolide A, a semi-synthetic approach to produce AF was explored. Taccalonolide AF was generated from taccalonolide A with 100% yield when reacted with dimethyldioxirane.23 This one-step epoxidation reaction was also applied to taccalonolide B to produce taccalonolide AJ, which resulted in a dramatic 743-fold increase in potency.23 Biochemical experiments with the potent taccalonolides AF and AJ demonstrated, for the first time, a direct interaction with tubulin that promoted microtubule polymerization.23
The identification of the potent taccalonolides AF and AJ facilitated binding studies to identify the interaction of the taccalonolides with microtubules. Although the taccalonolides are similar to paclitaxel and laulimalide in their ability to enhance the extent of microtubule polymerization from purified tubulin, they differ from these microtubule stabilizers in several regards. The taccalonolides are less efficient than either paclitaxel or laulimalide in their ability to initiate microtubule polymerization, as indicated by a prolonged, 5 - 9 min lag period that occurs before the initiation of tubulin polymerization.23 This is in contrast to the almost immediate polymerization observed by the addition of other microtubule stabilizers and suggests that the taccalonolides are less effective at nucleating microtubules.19 However, once microtubules are formed in the presence of taccalonolide AF or AJ, they exhibit an unprecedented level of cold and mechanical stability as compared to paclitaxel or laulimalide.19 The microtubule structures formed in the presence of each of these drugs look essentially identical, as visualized by electron microscopy,19 indicating that the taccalonolides are likely imparting increased stability to a fairly normal microtubule structure in a manner that is distinct from the other major classes of microtubule stabilizers.
6. Identification of the taccalonolide binding site on tubulin
Initial binding studies demonstrated that once bound, the taccalonolides AF and AJ could not be extracted from microtubules under a variety of denaturing conditions, suggesting that the taccalonolides covalently bind to microtubules.19 This was confirmed by mass spectrometry studies with taccalonolide AJ that identified an increase in the mass of the 212-230 peptide of β-tubulin by the exact mass of the drug. Although technical limitations have so far precluded mapping the exact residue(s) of taccalonolide binding, it is important to note that this peptide contains the N226 residue in the luminal taxane binding pocket and the T218 residue in the pore by which taxane-site binding agents gain access to the microtubule lumen, which have both been shown to covalently bind to the microtubule stabilizer cyclostreptin.19, 24 Another microtubule stabilizer that binds covalently to N226 and H227 in the taxane pocket but not to the pore site is zampanolide.25, 26 The taccalonolides were found to be synergistic with either paclitaxel or laulimalide, further suggesting they interact with microtubules in a distinct manner. Interestingly, taccalonolide AJ was not displaced from microtubules by either paclitaxel or laulimalide, although the covalent binding of AJ to microtubules was sufficient to decrease paclitaxel, but not laulimalide, binding. The ability of the taccalonolides to displace paclitaxel is consistent with their binding at or near the luminal and/or pore taxane binding site(s) on microtubules.
In addition to identifying the covalent attachment of taccalonolide AJ to a peptide of β-tubulin, hydrogen/deuterium exchange mass spectrometry was used to map the allosteric effects of taccalonolide binding on microtubules.19 As expected from its biochemical and cellular effects on microtubule polymerization, taccalonolide AJ imparted increased lateral inter-protofilament stability. However, the mechanism of this stabilization differed markedly from classical taxane-site binding agents in that it largely involved stabilization of adjacent α-tubulin protofilaments and did not involve structuring of the M-loop of β-tubulin or the H7 helix at the intradimer interface (Figure 2).19, 26-29 Further studies are needed to clearly define the residues on β-tubulin to which the taccalonolides bind covalently.
Figure 2.
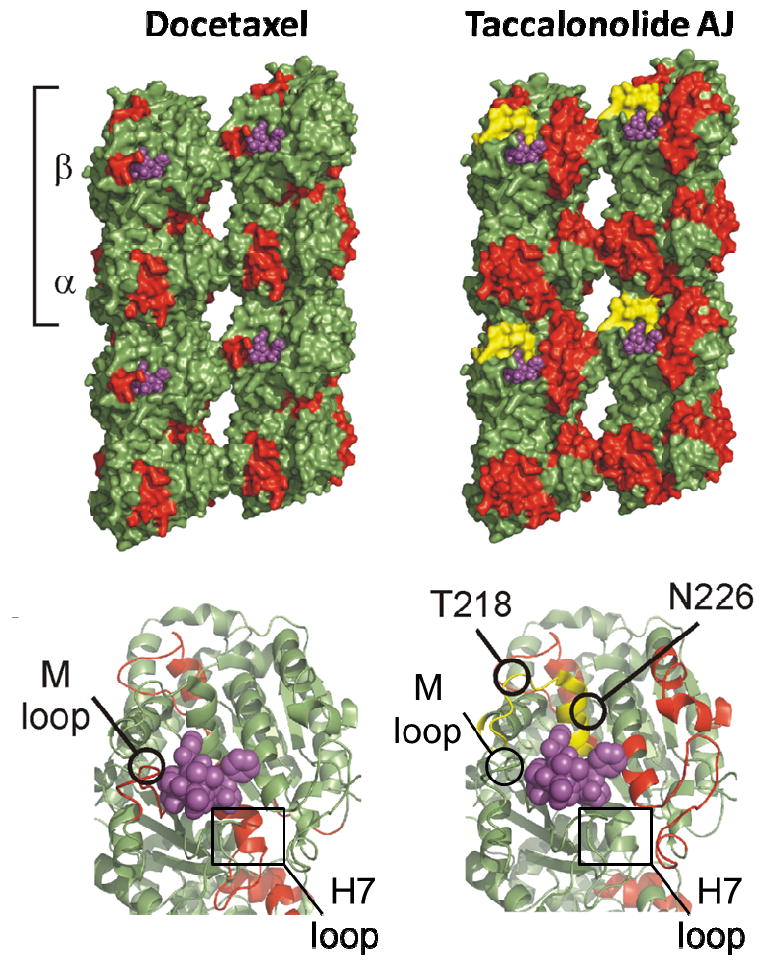
The allosteric effects on microtubule stability elicited by the binding of docetaxel (left) or taccalonolide AJ (right) as determined by hydrogen/deuterium exchange mass spectrometry are depicted in red. Note in the lower panels that the H7 and M-loop, which are affected by docetaxel binding, are not affected by taccalonolide AJ. The covalent binding of taccalonolide AJ to microtubules was mapped to the 212-220 peptide on β-tubulin (yellow), which contains the T218 and N226 residues that covalently bind cyclostreptin. This is distinct, but near to where docetaxel (purple) binds.
The profound microtubule stabilization caused by covalent binding of the taccalonolides to tubulin explains many of their previously observed effects, including their high cellular persistence,18 ability to avoid efflux by ATP-dependent transporters12, 15 and their ability to overcome drug resistance mediated by mutations in the taxane binding site.12
7. In vivo antitumor effects of taccalonolides AF and AJ
The taccalonolides are the first class of covalently bound microtubule stabilizers whose antitumor activities have been extensively explored.15, 16, 19 As previously mentioned, their excellent in vivo potency is likely a direct result of their ability to covalently bind microtubules. The antitumor effects of the taccalonolides AF and AJ were recently evaluated in the MDA-MB-231 breast cancer xenograft model.19 The administration of 2 mg/kg AF twice a week in 5% EtOH : 95% PBS (final concentration) caused significant antitumor effects that were comparable to 10 mg/kg paclitaxel in 10% Cremophor : 90% PBS. A higher, 2.5 mg/kg, dose of AF for 2 days (1 and 5) caused total tumor regression, but was associated with unacceptable delayed toxicity that defined this dose as the LD20. Four doses of 2 mg/kg taccalonolide AJ caused weight loss, but there was no indication of any antitumor effects. Further studies using a variety of doses and schedules of AJ showed slight antitumor effects, but they were associated with unacceptable toxicities. Chemical stability measurements of these taccalonolides in PBS showed that while taccalonolide AJ was quite stable for over 20 h, taccalonolide AF had a t1/2 of 9 h in PBS and was rapidly converted into AJ. These studies suggested the possibility that AF might also convert to AJ in vivo and that this conversion may play a role in the narrow therapeutic window observed for AF.19
Although excellent antitumor effects were obtained with the taccalonolides A, E, N and AF, they correlate with weight loss, indicating a narrow therapeutic window. However, differences have been noted among the taccalonolides with regard to the degree of toxicity that is associated with therapeutic doses, suggesting the feasibility of identifying a taccalonolide with an acceptable therapeutic window for clinical development.19 For instance, it is intriguing to hypothesize that the toxicity observed with AF may be diminished by minimizing its conversion to AJ.
8. Optimization of the C15 hydrolysis reaction
The extensive binding and in vivo studies conducted with the C22-C23 epoxidated form of taccalonolide B, taccalonolide AJ, led to efforts to optimize the hydrolysis conditions to generate taccalonolide B from A. Conditions were identified that provided an 80% yield with a 1:1:1 ratio of taccalonolide A: MeOH: 0.05 M NaHCO3 for 20 h.30 Similarly, taccalonolide N was produced from E under the same hydrolysis conditions. In addition to taccalonolides B and N, five new taccalonolides, AO, AK, AL, AM and AN, were obtained as products of these reactions.30 These hydrolysis reactions confirmed that the taccalonolides are very unstable in alkaline solutions, consistent with a previous report.16 Biological evaluations indicated that none of the new hydrolysis products were potent, with all having IC50 values greater than 1 μM and the taccalonolides AO and AK having no activity up to 50 μM. The inactive taccalonolides AO and AK are the C22-C23 keto-enol tautomeriztion products of taccalonolides A and E, respectively, resulting from the opening and reclosing of the lactone ring between C15 and C26 to form a δ-lactone ring at the bottom of the molecule under basic hydrolysis conditions. These results additionally demonstrated that the C1 acetoxy group of taccalonolide E can be hydrolyzed to form AN, opening up the possibility of introducing different functional groups at C1, which we have shown enhances potency30 (and is discussed in detail below). Interestingly, this hydrolysis was not observed with taccalonolide A, suggesting that an acetoxy group at C11 interferes with this reaction.
9. Isolation of new natural taccalonolides
In addition to taccalonolide AF a number of other rare taccalonolides have been isolated from three Tacca species, Tacca chantrieri, Tacca integrifolia and Tacca plantaginea, using bioassay-guided fractionation. In 2011, three new taccalonolides designated Z, AA and AB, and two known taccalonolides R and T, were isolated from Tacca chantrieri and Tacca integrifolia (Figure 1).16 The taccalonolides Z, AA and AB are similar in structure to taccalonolide A, but each contains a hydroxy group at C5. They have a wide range of potencies from low micromolar AB (1.2 μM) to low nanomolar AA (32 nM) (Table 1). The identification of taccalonolide AA, which has an IC50 of 32 nM, together with taccalonolide AF with an IC50 of 23 nM led to the continued search for rare and potent natural taccalonolides. The isolation and biological evaluations of the taccalonolides T and R, which also contain a C5 hydroxy but are similar to taccalonolide E in that they lack the C11 acetoxy group, provided significant SAR information. Taccalonolide T contains a bulky isovalerate group at C1 and has an IC50 of 340 nM, while taccalonolide R, which contains a C1 acetoxy group, has 39-fold lower potency (Table 1). The fraction from Tacca plantaginea that yielded taccalonolide AF also yielded four additional new taccalonolides in trace quantitities, AC-AE and H2.23
Table 1.
| Compound | IC50 (μM) | Compound | IC50 (μM) |
|---|---|---|---|
| Taccalonolide A | 5.3 ± 0.2 | Taccalonolide T | 0.34 ± 0.02 |
| Taccalonolide B | 3.1 ± 0.2 | Taccalonolide Z | 0.120 ± 0.008 |
| Taccalonolide E | 40 ± 5 | Taccalonolide AA | 0.032 ± 0.002 |
| Taccalonolide N | 8.5 ± 0.4 | Taccalonolide AB | 2.7 ± 0.1 |
| Taccalonolide I | 49 ± 3 | Taccalonolide AC | > 50 |
| Taccalonolide AO | > 50 | Taccalonolide AD | 3.4 ± 0.2 |
| Taccalonolide AK | > 50 | Taccalonolide AE | 5.0 ± 0.2 |
| Taccalonolide AL | 34 ± 8 | Taccalonolide AF | 0.023 ± 0.003 |
| Taccalonolide AM | 2.0 ± 0.1 | Taccalonolide AJ | 0.0042 ± 0.0003 |
| Taccalonolide AN | 1.5 ± 0.1 | Taccalonolide H2 | 0.73 ± 0.02 |
| Taccalonolide R | 13 ± 1 |
10. Structure activity relationships of the taccalonolides
The vast array of natural and semi-synthetic taccalonolide analogues characterized in our laboratory has allowed for rigorous structure activity relationship (SAR) analyses that will greatly facilitate modeling of the interactions between the taccalonolides and microtubules and possibly aid in the identification of an optimal clinical candidate. The current SAR findings clearly demonstrate that there are complex relationships among multiple sites on the taccalonolide backbone, however, the following conclusions can been drawn with regard to the key moieties of the taccalonolide structure that are critical for optimal efficacy and potency of this class of compounds.
10.1. E-ring constituents at C20-C23 play a key role in taccalonolide potency
As discussed previously, analogues such as AO and AK, which have significant structural rearrangements in the C20-23 region, show no antiproliferative or microtubule stabilizing activities at concentrations up to 50 μM.30 A similar lack of potency was observed for taccalonolide AC, which contains an α-hydroperoxyl group at C20 (Figure 1), as opposed to the majority of other taccalonolides which contain an α-methyl group at this site, indicating that this additional α-hydroperoxyl group is not optimal for bioactivity. In contrast, the epoxidation of the C22-C23 double bond was shown to dramatically increase taccalonolide potency as evidenced by the generation of the potent taccalonolides AF and AJ from their parent taccalonolides A and B, respectively (Table 1). Ongoing studies demonstrate that this C22-23 epoxidation increases the potency of other natural and semisynthetic taccalonolides. The substantial changes in potency afforded by small modifications to this region of the taccalonolide backbone strongly suggest that this area is critical for optimal drug binding to microtubules.
10.2. A large, bulky group at C1 provides a significant increase in potency
The first indication that C1 modifications can dramatically impact potency came from the finding that replacing the C1 acetoxy in R with an isovalerate group in T resulted in a 39-fold increase in potency.16 This was confirmed with the 17-fold increase in potency observed by the replacement of the acetoxy group at C1 in taccalonolide AL with an isovalerate group in taccalonolide AM.30 Interestingly, a decrease in bulk at C1 offered by the hydrolysis of the acetoxy group in AL to generate AN in combination with removal of the C5 hydroxy group also offers a similar increase in potency. More highly potent new taccalonolides with isovalerate or other bulky modifications at C1 can likely be isolated or produced semi-synthetically as evidenced by the selective hydrolysis of C1 in the absence of a C11 acetoxy group. These data highlight the importance of the C1 position in facilitating taccalonolide activity and the opportunity to improve potency by hydrolyzing this acetoxyl group with the potential to add steric bulk at this site.
10.3. There are complex indications with regard to the importance of the C5 hydroxy group to potency
Upon comparison of the taccalonolides N and AL, the C5-hydroxy group is the only structural difference, which leads to a 4-fold decrease in potency for taccalonolide AL. By contrast, a significant 44-fold increase in potency was observed by the presence of a C5-hydroxy group in taccalonolide Z as compared to taccalonolide A. Moreover, the presence of the C5-hydroxy group did not have a major effect on the potencies of taccalonolides B and AB. Therefore, the presence of the C5-hydroxy group in itself does not have a predictable effect on potency although this modification can be important when combined with other modifications. For instance, compounds lacking the C5-hydroxy group show increased potency when the C15-acetoxy group is hydrolyzed, such as when A or E are hydrolyzed to generate B and N, respectively. However, this same hydrolysis gives rise to decreased potency in molecules containing the C5-hydroxy group as evidenced by a comparison of Z to AB. These data suggest that multiple interactions across the taccalonolide molecule can interplay in complex ways to affect potency.
10.4. The C5-8 substituents on the lower portion of the B ring can affect taccalonolide potency
One example of the importance of the C5-8 region of the taccalonolide backbone is evidenced by the two natural taccalonolides A and I. The only difference between taccalonolides A and I is a switch between the C6 ketone and the C7-hydroxy. This difference resulted in a 9.25-fold decrease in potency for I, indicating this arrangement is not optimal for biological activity. However, when these same substituents are present in combination with a double bond at C5-C6, as in taccalonolide AD, the potency is slightly increased as compared to A, suggesting that this double bond plays a role in maintaining activity. Additionally, when a second hydroxy group is present at the C7 of taccalonolide A to give the rare geminal diol group on taccalonolide AE, the potency was also unchanged, further indicating that there is some degree of flexibility for modifications in this region. The 7-fold increase in potency of taccalonolide H2 afforded by a C7-C8 double bond compared to taccalonolide A demonstrates that a conjugated B ring with a ketone at C6 position and a double bond at C7-C8 may be optimal for potency in this region.
11. Conclusion
The taccalonolides are a novel class of microtubule stabilizing agents with the ability to circumvent clinically relevant forms of taxane resistance both in vitro and in vivo. The ability of the taccalonolides to covalently bind β-tubulin contributes toward their high cellular persistence and affords in vivo administration at low concentrations in aqueous vehicles. Detailed SAR data from a large number of natural and semisynthesized taccalonolides provides guidance for further modifications that will facilitate more detailed modeling of the taccalonolide binding site and may lead to the identification of a clinical candidate.
Acknowledgments
This work was supported by NCI CA121138 (SLM) and NIDCR DE 14318 (JL). We would like to thank Dr. David Schriemer for assistance with creation of the taccalonolide binding figure and Cristina Rohena and Andrew Robles for their critical reading of the manuscript.
Biographies
Jing Li was born in Neimong Gu, China. She obtained her B.S. degree in Pharmacy from the Beijing University of Chinese Medicine and her Ph.D. from the School of Pharmacy at the University of Mississippi, MS. She is currently a postdoctoral fellow in the Department of Pharmacology at the University of Texas Health Science Center in San Antonio, TX working in the laboratory of Dr. Susan Mooberry. Jing’s research interest is focused on the identification of new anticancer drug leads from natural sources.

Susan L. Mooberry was born in Wilmington Delaware and received her B.S. degree in Biology from St Lawrence University in Canton, NY and her Ph.D. from the Medical University of South Carolina in Charleston, SC. She conducted postdoctoral studies at the Eastern Virginia Medical School and at the University of Hawaii where she then joined the faculty at the Cancer Research Center of Hawaii. She is currently a Professor at the University of Texas Health Science Center at San Antonio in the Department of Pharmacology with a cross appointment in the Department of Medicine. Dr. Mooberry is the co-leader of the Experimental and Developmental Therapeutics program of the Cancer Therapy & Research Center. Her laboratory is focused on drug discovery from natural sources and the identification of the mechanism of action of diverse microtubule targeting agents.

April L. Risinger was born in San Antonio, Texas. She obtained her Bachelors of Science degree in Biochemistry from Texas A&M University in College Station, Texas and her PhD in Cellular Biology from the Massachusetts Institute of Technology in Cambridge, MA. She is currently an Instructor in the Department of Pharmacology at the University of Texas Health Science Center in San Antonio, TX working in the laboratory of Dr. Susan Mooberry. April’s research interests include the in vitro and in vivo characterization of natural compounds with potential efficacy against cancer.

Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Scheuer PJ, Swanholm CE, Madamba LA, Hudgins WR. Lloydia. 1963;26:133. [Google Scholar]
- 2.Chen Z, Wang B, Chen M. Tetrahedron Lett. 1987;28:1673. [Google Scholar]
- 3.Chen Z, Wang B, Shen J. Phytochemistry. 1988;27:2999. [Google Scholar]
- 4.Chen ZL, Shen JH, Gao YS, Wichtl M. Planta Med. 1997;63:40. doi: 10.1055/s-2006-957600. [DOI] [PubMed] [Google Scholar]
- 5.Shen J, Chen Z, Gao Y. Chin J Chem. 1991;9:92. [Google Scholar]
- 6.Shen J, Chen Z, Gao Y. Phytochemistry. 1996;42:891. [Google Scholar]
- 7.Yang J-Y, Zhao R-H, Chen C-X, Ni W, Teng F, Hao X-J, Liu H-Y. Helv Chim Acta. 2008;91:1077. [Google Scholar]
- 8.Muehlbauer A, Gehling M, Velten R, Andersch W, Erdelen C, Harder A, Marczok P, Nauen R, Turberg A, Tran VS, Adam G, Liu J. 2001040256A1. WO. 2001:113.
- 9.Muehlbauer A, Seip S, Nowak A, Tran VS. Helv Chim Acta. 2003;86:2065. [Google Scholar]
- 10.Huang Y, Liu J-K, Muhlbauer A, Henkel T. Helv Chim Acta. 2002;85:2553. [Google Scholar]
- 11.Huang Y, Muehlbauer A, Henkel T, Liu JK. Chin Chem Lett. 2003;14:68. [Google Scholar]
- 12.Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC, Jr, Hemscheidt TK, Mooberry SL. Cancer Res. 2003;63:3211. [PubMed] [Google Scholar]
- 13.Fojo T, Menefee M. Ann Oncol. 2007;18(Suppl 5):v3. doi: 10.1093/annonc/mdm172. [DOI] [PubMed] [Google Scholar]
- 14.Horwitz SB, Cohen D, Rao S, Ringel I, Shen HJ, Yang CP. J Natl Cancer Inst Monogr. 1993:55. [PubMed] [Google Scholar]
- 15.Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD, Mooberry SL. Cancer Res. 2008;68:8881. doi: 10.1158/0008-5472.CAN-08-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng J, Risinger AL, Fest GA, Jackson EM, Helms G, Polin LA, Mooberry SL. J Med Chem. 2011;54:6117. doi: 10.1021/jm200757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buey RM, Barasoain I, Jackson E, Meyer A, Giannakakou P, Paterson I, Mooberry S, Andreu JM, Diaz JF. Chem Biol. 2005;12:1269. doi: 10.1016/j.chembiol.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Risinger AL, Mooberry SL. Cell Cycle. 2011;10:2162. doi: 10.4161/cc.10.13.16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Risinger AL, Li J, Bennett MJ, Rohena CC, Peng J, Schriemer DC, Mooberry SL. Cancer Res. 2013;73:6780. doi: 10.1158/0008-5472.CAN-13-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Nat Rev Clin Oncol. 2011;8:244. doi: 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- 21.Sackett DL, Fojo T. Cell Cycle. 2011;10:3233. doi: 10.4161/cc.10.19.17126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rohena CC, Peng J, Johnson TA, Crews P, Mooberry SL. Biochem Pharmacol (Amsterdam, Neth) 2013;85:1104. doi: 10.1016/j.bcp.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Risinger AL, Peng J, Chen Z, Hu L, Mooberry SL. J Am Chem Soc. 2011;133:19064. doi: 10.1021/ja209045k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buey RM, Calvo E, Barasoain I, Pineda O, Edler MC, Matesanz R, Cerezo G, Vanderwal CD, Day BW, Sorensen EJ, Lopez JA, Andreu JM, Hamel E, Diaz JF. Nat Chem Biol. 2007;3:117. doi: 10.1038/nchembio853. [DOI] [PubMed] [Google Scholar]
- 25.Field JJ, Pera B, Calvo E, Canales A, Zurwerra D, Trigili C, Rodriguez-Salarichs J, Matesanz R, Kanakkanthara A, Wakefield SJ, Singh AJ, Jimenez-Barbero J, Northcote P, Miller JH, Lopez JA, Hamel E, Barasoain I, Altmann KH, Diaz JF. Chem Biol. 2012;19:686. doi: 10.1016/j.chembiol.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO. Science. 2013;339:587. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 27.Huzil JT, Chik JK, Slysz GW, Freedman H, Tuszynski J, Taylor RE, Sackett DL, Schriemer DC. J Mol Biol. 2008;378:1016. doi: 10.1016/j.jmb.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khrapunovich-Baine M, Menon V, Yang CP, Northcote PT, Miller JH, Angeletti RH, Fiser A, Horwitz SB, Xiao H. J Biol Chem. 2011;286:11765. doi: 10.1074/jbc.M110.162214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao H, Verdier-Pinard P, Fernandez-Fuentes N, Burd B, Angeletti R, Fiser A, Horwitz SB, Orr GA. Proc Natl Acad Sci U S A. 2006;103:10166. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Peng J, Risinger AL, Mooberry SL. J Nat Prod. 2013;76:1369. doi: 10.1021/np400435t. [DOI] [PMC free article] [PubMed] [Google Scholar]