

Abstract
Drug administration to avoid unpleasant drug withdrawal symptoms has been hypothesized to be a crucial factor that leads to compulsive drug-taking behavior. However, the neural relationship between the aversive motivational state produced by drug withdrawal and the development of the drug-dependent state still remains elusive. It has been observed that chronic exposure to drugs of abuse increases brain-derived neurotrophic factor (BDNF) levels in ventral tegmental area (VTA) neurons. In particular, BDNF expression is dramatically increased during drug withdrawal, which would suggest a direct connection between the aversive state of withdrawal and BDNF-induced neuronal plasticity. Using lentivirus-mediated gene transfer to locally knock down the expression of the BDNF receptor tropomyosin-receptor-kinase type B in rats and mice, we observed that chronic opiate administration activates BDNF-related neuronal plasticity in the VTA that is necessary for both the establishment of an opiate-dependent state and aversive withdrawal motivation. Our findings highlight the importance of a bivalent, plastic mechanism that drives the negative reinforcement underlying addiction.
Keywords: BDNF, dependent state, drug addiction, opiates, TrkB, withdrawal
Introduction
The initial acute rewarding effect of a drug of abuse activates adaptive neural mechanisms that counteract the departure from a state of equilibrium, producing an aversive motivational state when the drug has been cleared from the organism (Koob and Le Moal, 2001; Vargas-Perez et al., 2007, 2009a). This withdrawal from drugs of abuse produces a negative affective state including symptoms of dysphoria, depression, irritability, and anxiety (Koob and Le Moal, 2001), as well as increases in reward thresholds and decreased tolerance to aversive stimuli (Zacharko and Anisman, 1991; Koob and Le Moal, 2005). Thus, drug intake stimulates a negative response that becomes sensitized during repeated drug withdrawal and even persists into prolonged abstinence, contributing to drug-taking behavior (Koob and Le Moal, 2005). Avoiding the unpleasantness of the withdrawal state is hypothesized to be an important factor that triggers compulsive drug intake, potentially leading to drug addiction (Koob and Bloom, 1988; Koob et al., 1989, 1992; Wise, 1996; Koob and Le Moal, 1997, 2001; Vargas-Perez et al., 2007, 2009a).
The transition to a drug-dependent state is accompanied by a series of plastic changes in brain reward circuits (Crabbe, 2002; Robinson and Berridge, 2003; Kreek et al., 2005), which underlie continuing negative reinforcement driving and maintaining addiction (Nader et al., 1997; Koob and Le Moal, 2005). For instance, the transition from an opiate-naive to an opiate-dependent state is associated with a change, from an inhibitory to an excitatory response, of the GABAA receptors located on GABA neurons in the ventral tegmental area (VTA; Laviolette et al., 2004). This change promotes a shift from a dopamine-independent motivational system, which involves the brainstem tegmental pedunculopontine nucleus (TPP; Bechara et al., 1995), to a dopamine-dependent opiate motivational system (Bechara et al., 1995). Recent evidence demonstrated that an increase in brain-derived neurotrophic factor (BDNF) levels is sufficient to switch the response of GABAA receptors on VTA GABAergic neurons from inhibitory to excitatory, which in turn is sufficient to induce a transition to a drug-dependent motivational state (Vargas-Perez et al., 2009b).
Chronic exposure to drugs of abuse increases BDNF levels in VTA neurons (Bolaños and Nestler, 2004). In particular, BDNF expression is significantly increased during drug withdrawal (Vargas-Perez et al., 2009b), which suggests a direct connection between the aversive state of withdrawal and BDNF-induced neuronal plasticity. Thus, we hypothesize that blocking BDNF signaling in the VTA should block both the aversive motivational state produced by drug withdrawal and the development of the drug-dependent state. To test this hypothesis we used lentivirus-mediated gene transfer to locally knock down the expression of BDNF receptor tropomyosin-receptor-kinase type B (TrkB). We observed that chronic opiate administration activates BDNF-related neuronal plasticity in the VTA that is necessary for both the establishment of an opiate-dependent state and aversive withdrawal motivation, highlighting the importance of a bivalent, plastic mechanism that drives the negative reinforcement underlying addiction.
Materials and Methods
Animals and surgery
All experimental protocols were approved by and conformed to the Institutional and Governmental Animal Care Committee guidelines, in accordance with principles of animal care (http://www.ccac.ca/).
For behavioral tests in which rats were used, subjects were Wistar strain males (Charles River) weighing 350–500 g during experimental training. Rats were housed individually in Plexiglas cages in a room maintained at 22°C and lit from 7:00 A.M. to 7:00 P.M. Rats were given food and water ad libitum throughout the experiment.
In the case of behavioral experiments using mice (VTA BDNF infusions), subjects were wild-type male C57BL/6 weighing 25–30 g. Mice were given food and water ad libitum and maintained at 22°C in Plexiglas cages on a 12 h light/dark cycle.
Male GAD-GFP knock-in mice (>50 d) were used for the iontophoretic recording of VTA GABAergic neurons. Mice were housed in groups of four in Plexiglas mouse cages in a room at a temperature of 22°C with lights on from 7:00 A.M. to 7:00 P.M. No animals were used in more than one experiment.
For VTA infusions, animals were anesthetized with inhaled isoflurane (3%) and placed in a stereotaxic device. The procedure for rats involved bilateral implantations of 22 gauge stainless steel guide cannulae (Plastics One). The cannulae were implanted 2 mm dorsal to the VTA at a 10° angle using the following coordinates relative to bregma: AP: −5.6 mm, ML: ±2.3 mm, and DV: −7.8 mm from the dural surface (Paxinos and Watson, 1986). For TPP temporary lesion 22 gauge stainless steel guide cannulae (Plastics One) were bilaterally implanted 2 mm dorsal to the TPP at a 10° angle using the following coordinates relative to bregma: AP: −7.6 mm, ML: ±3.1 mm, and DV: −6.6 mm from the dural surface (Paxinos and Watson, 1986). At least 2 weeks were allowed for postsurgical recovery preceding behavioral training.
Three stereotaxic surgical procedures were used for the mouse experiments in Figure 5. The first procedure (implemented in Fig. 5a,b) involved the use of 33 gauge needles to bilaterally infuse 0.025 μg or 0.25 μg BDNF in the VTA coordinates previously used in our mouse work (AP: −3.0 mm, ML: ±0.5 mm, DV: −4.2 from bregma). In the case of the 0.025 μg infusions, 0.05 μl was infused over the span of 1 min after which the needle was left stationary for another minute. In the case of the 0.25 μg infusions, 0.5 μl was infused over the span of 5 min after which the needle was left stationary for another minute. The second procedure (implemented in Fig. 5c) involved the infusion of 0.25 μg BDNF in the VTA coordinates used by Koo et al. (2012) (at a 7° angle, AP: −3.2 mm, ML: ±1.0 mm, DV: −4.6 mm from bregma). Mice were left to recover for 7 d before they were used in behavioral experiments.
Figure 5.

Effects of high and low doses of BDNF on 5 and 15 mg/kg morphine-conditioned place preferences in mice. a, Infusions of 0.025 μg BDNF (same dose used in rats by Vargas-Perez et al. (2009b)) or 0.25 μg BDNF (dose used by Koo et al. (2012)) in mouse VTA did not abolish 5 mg/kg morphine place preferences, but resulted in a switch to a DA-dependent motivational state wherein α-flupenthixol blocks the conditioned place preferences. b, Infusions of 0.25 μg BDNF in mouse VTA did not abolish 15 mg/kg morphine-conditioned place preferences, even when the duration of conditioning sessions was extended to 45 min to match the conditions used by Koo et al. (2009). c, Infusions of 0.25 μg BDNF in mouse VTA did not abolish 15 mg/kg morphine preferences even after using the same conditioned place preference paradigm and VTA coordinates used by Koo et al. (2012); *p values <0.05.
Induction of opiate dependence
To induce opiate dependence in rats, animals received 0.5 mg/kg subcutaneous injections of heroin daily for 5 d (in line with the heroin pre-exposure protocol; Laviolette et al., 2004). Animals were conditioned 21 h after their last heroin injection. During conditioning, this dose of heroin was administered as a maintenance dose 3.25 h after the termination of training.
GAD-GFP knock-in mice were made opiate dependent by the implantation of subcutaneous osmotic mini pumps (Model 2001; Alzet Osmotic Pumps) containing morphine solution (70 mg/ml; Dockstader et al., 2001). Pumps were surgically implanted under isoflurane (2%) anesthesia and aseptic conditions. The pumps delivered morphine (1.0 μl/h) for 5–7 d and then experiments were performed 12–16 h following surgical removal of the pumps under isoflurane (2%) anesthesia. In naive animals mini pumps that contained saline solution were implanted as a control. The aversive effects of withdrawal induced by this regimen are similar qualitatively to those observed after the heroin pre-exposure protocol used in rats (Dockstader et al., 2001).
Drugs and microinjection procedure
The drugs used in these experiments were morphine sulfate (Almat Pharmachem), diacetylmorphine hydrochloride (heroin; Almat Pharmachem), muscimol hydrobromide (Sigma), lithium chloride (LiCl; Sigma), α-flupenthixol (Sigma), or lidocaine hydrochloride (Sigma), and were dissolved in physiological saline (pH adjusted to 7.4). Recombinant human BDNF (Sigma) was dissolved in PBS, pH 7.4).
In the rat experiments, bilateral VTA or TPP microinjections (0.5 μl volume per infusion) were performed over 1 min. Injectors were left in place for a further 1 min to ensure adequate diffusion from the injector tip.
Viral vectors
To test for the necessity of BDNF signaling, we locally knocked down TrkB gene expression in vivo, via siRNA-expressing lentiviral vectors (LVs). Through bilateral stereotaxic surgery, we injected 0.5 μl of siRNA-expressing LV (LV-siRNA1, LV-siRNA2, and LV-siRNA3, targeting three separate regions of the mRNA; Bahi et al., 2008) against TrkB, or the green fluorescent protein (GFP)-tagged lentiviral vector pTK433 (LV-GFP; Bahi et al., 2008) as a control, into the VTA of adult rats. It has been observed that this coinfection mix of the three silencers (LV-siRNAs) achieves >93% silencing of TrkB mRNA (Bahi et al., 2008). The three targets were designed according to the rat TrkB mRNA sequence. The TrkB sequences were selected based on Hannon's design criterion: first target, bp 5–27; second target, bp 2438–2460; third target, bp 1500–1522 (Bahi et al., 2008). LV-siRNAs were mixed with LV-GFP to localize injection sites in experimental animals, and the LV-GFP was used alone in the control animals. LVs were injected 1–2 weeks before electrophysiological experiments or behavioral testing.
Histology
At the end of experiments, animals were deeply anesthetized with sodium pentobarbital (35 mg/kg, i.p.) and were perfused intracardially with 200 ml of PBS followed by 400 ml of 4% paraformaldehyde. Brains were removed rapidly and stored for 12 h in a 35% sucrose/PBS solution. Brains then were flash frozen at −80°C, sliced in a freezing microtome at −20°C into 30-μm-thick sections, and mounted on gelatin-coated slides. Brain sections were stained with cresyl violet and subsequently examined by light microscopy.
Immunostaining for TrkB expression in VTA GABAergic neurons
Rats were anesthetized with sodium pentobarbital (35 mg/kg, i.p.) and perfused intracardially with 200 ml of PBS followed by 400 ml of 4% paraformaldehyde. Brains were rapidly removed and stored for 12 h in a 35% sucrose/PBS solution. Brains were then flash frozen at −80°C, sliced in a freezing microtome at −20°C into 20-μm-thick sections, and mounted on gelatin-coated slides. Brain sections were blocked with 10% normal goat serum in PBS for 1 h at room temperature. TrkB (mouse monoclonal antibody, 1:50; BD Transduction Laboratories) and MAP-2 (chicken polyclonal, 1:10,000; Abcam) antibodies were incubated at 4°C overnight. Slides were then washed three times with PBS and incubated with Alexa-conjugated secondary antibodies at a concentration of 1:400 for 1 h at room temperature. All antibody dilutions were made in 10% NGS containing PBS. Slides were washed, dried, and mounted and imaging was performed using an Olympus FV1000 confocal microscope.
To accurately assess the percentage of GFP cells that coexpress the TrkB receptor after LV infection of siRNAs or GFP, coronal sections (approximately −5.6 mm from bregma) containing the VTA (Paxinos and Watson, 1986) from the brain of three animals per condition were immunohistochemically labeled for GFP and TrkB. To measure cell size, ImageJ software (NIH) was used to measure the area of cell bodies of 46 neurons in each condition. A comparison of GFP TrkB-positive VTA neurons between LV-GFP and LV-siRNAs-infected animals was made using a Student's t test, with a minimum of 100 GFP-positive neurons counted per animal.
Iontophoretic recording of VTA GABAergic neurons
Preparation of tissue slices for electrophysiological recordings.
For brain extraction, mice were anesthetized with isoflurane (5%) and intraperitoneal injection of ketamine (60 mg/kg) and decapitated. The brain was quickly removed from the cranium and glued onto a cutting stage. Slicing was performed using a sapphire blade (Electron Microscopy Sciences) on a vibratome tissue slicer in an ice-cold cutting solution containing the following (in mm): 220 sucrose, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 12 MgSO4, 10 glucose, 0.2 CaCl2, and 0.4 ketamine perfused with 95% O2 and 5% CO2. Horizontal slices (210 μm thick) containing the VTA were cut and transferred into an incubation chamber containing artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 2 KCl, 1.25 NaH2PO4, 26 NaHCO3, 12 glucose, and 1.2 MgSO4 2 CaCl2 perfused with 95% O2 and 5% CO2 at 32°C for at least 30 min. Following incubation, slices were transferred to a recording chamber with continuous flow of ACSF maintained at 36°C throughout the experiment.
Characterization of VTA GABA neurons in vitro.
In GAD-GFP knock-in mice (Tamamaki et al., 2003), GABA neurons were studied in horizontal brain slices with the aid of fluorescence microscopy. The VTA was visualized by first locating the substantia nigra reticulata (SNr) in the horizontal slice preparation under low power (4×) with fluorescence illumination. The SNr has a characteristic glow under low magnification with GFP fluorescence optics, likely due to dense GABA terminal innervation. Substantia nigra compacta was then identified medial to SNr. GABA neurons in the VTA were studied by visualizing GAD+ neurons in an area medial to the glowing SNr, posterior to the fasciculus retroflexus and mammillothalamic tract, anterior to the decussation of the superior cerebellar peduncle, and dorsal to the interpeduncular nucleus (Steffensen et al., 2011; Ting-A-Kee et al., 2013). Neurons in the VTA of GAD-GFP mice that did not fluoresce but exhibited a noncation-specific inward rectifying current (Ih) in combination with relatively low input resistance and regular, slow spike activity were assumed to be DA neurons (Steffensen et al., 2011; Ting-A-Kee et al., 2013).
Cell-attached, voltage-clamp recording of spike activity in brain slices.
Electrodes were pulled from borosilicate glass capillaries and then filled with 150 mm NaCl (3–5 MΩ). Positive pressure was applied to the electrode when approaching the neuron. By applying suction to the electrode, a seal (10 MΩ–1 GΩ) was created between the cell membrane and the recording pipette. Spontaneous GABA firing activity was recorded in voltage-clamp mode with a Molecular Devices Multiclamp 700B amplifier and sampled at 10 kHz using an Axon 1440A digitizer, and collected and analyzed using pClamp10 software. Neurons were voltage clamped at 0 mV throughout the experiment. First, a stable baseline recording of firing activity was obtained for 5–10 min. Two methods were used to assess the effects of muscimol on VTA GABA neuron firing rate. In some slices, only one dose of muscimol (Sigma-Aldrich; 0.01–1 μm) was studied on one cell in each slice. In other slices, a wash was performed between superfusions of different concentrations of muscimol on the same cell (5–10 min at each dose with 10 min between doses). Regardless, no more than one cell was studied per slice and no more than three slices were studied per animal. One-way repeated-measures ANOVA and Tukey's post hoc test were performed to compare LV-GFP-naive, LV-siRNAs-naive, LV-GFP-dependent, and LV-siRNAs-dependent groups.
Rat place preference test
The place-conditioning apparatus and conditioning procedures were identical to previous studies (Vargas-Perez et al., 2009a). Conditioning took place in one of two distinct environments (41 × 41 × 38 cm), which differed in color, texture, and smell. One environment was white, with a wire mesh floor. The other environment was black, with a smooth Plexiglas floor that was wiped down with a 12% acetic acid solution before each conditioning session. These conditioning environments are motivationally balanced such that animals show no initial preference for either environment before conditioning (Laviolette et al., 2004). During testing, each rat was placed at the neutral gray zone (41 × 10 cm) that separated the two compartments and was allowed to explore both environments freely for a period of 10 min. Testing was performed drug free, at least 2 d after the final conditioning session.
For experiments that examined the direct effect of a drug, rats were conditioned with a fully counterbalanced place-conditioning procedure described previously as procedure B (Vargas-Perez et al., 2009a). In this procedure, rats were exposed to both conditioning environments in a fully counterbalanced order. All experimental groups received four drug-environment and four saline-environment conditioning sessions for 40 min over 8 consecutive days. Two days later, rats were tested drug free and the time spent in each environment was recorded over a 10 min test period.
Under this place-conditioning procedure we assessed the rewarding effects of morphine (10 mg/kg), the aversive effects of LiCl (5 mg/kg), and the aversive effects of naloxone-precipitated withdrawal (5 mg/kg) in LV-GFP against LV-siRNAs-infected, opiate-dependent rats. The rewarding effects of morphine and naloxone were tested, in naive and dependent animals. Morphine was administrated after the blockade of the dopaminergic system with the neuroleptic α-flupenthixol (0.8 mg/kg, i.p,)–administered 2.5 h before conditioning. Additionally, in a new group of animals, we test the rewarding effects of morphine after the temporary inactivation of the TPP. Bilateral TPP temporary inactivation was performed 2 min before conditioning by microinfusing 5 μl of lidocaine (4%; Vargas-Perez et al., 2009a). Sham temporary-inactivation animals received bilateral injections of PBS.
To assess the effect of spontaneous withdrawal from morphine in dependent rats, we used a modified place-conditioning procedure for which conditioning took place in only one compartment (Procedure W or withdrawal-paired) of the place-conditioning apparatus (Vargas-Perez et al., 2009a). Briefly, each dependent rat, by means of the heroin pre-exposure protocol, intraperitoneally received 3 mg/kg of morphine. Approximately 16 h after the morphine injection (Bechara et al., 1995; Vargas-Perez et al., 2007, 2009a), each rat was injected with saline vehicle and then exposed immediately to a distinct conditioning environment for 40 min. This procedure was repeated four times over 8 d. As a result, one of the two compartments was paired with the absence of morphine and the other was an unfamiliar, neutral environment. The time spent in each environment was recorded over a 10 min test period. Times spent in each environment were scored separately for each animal.
Mouse place preference test
The place conditioning apparatus (Med-Associates SOF-700RA-25) used consisted of a black compartment with metal rod floor, a white compartment with a wire mesh floor, and a gray compartment in the middle that contained a partition on both sides. Two unbiased conditioned place preference paradigms were used. The first was composed of eight, 15 min long conditioning sessions conducted over the span of 8 d (i.e., one session per day). The compartments to which the morphine (5 mg/kg) and saline injections were paired were counterbalanced. On the ninth day, the partitions between the compartments were removed and mice were placed in the gray zone and allowed to roam freely for 10 min. The second conditioning paradigm (Koo et al., 2012) was composed of six or eight, 45 min long conditioning sessions conducted over the span of 3 d or 4 d, respectively (i.e., two sessions per day). Saline conditioning was always performed in the morning, while morphine (15 mg/kg) conditioning was performed in the afternoon. On the day following the last day of conditioning, animals were placed in the gray zone and allowed to roam freely between the compartments for 20 min.
For place-conditioning experiments, statistical analysis was performed using a two-way or three-way ANOVA. Post hoc Student–Newman–Keuls tests or Student's t tests were performed where appropriate.
Opiate-withdrawal somatic signs, ultrasonic distress vocalizations, and locomotor sensitization
Rats were observed for somatic signs of opiate withdrawal 16 h after heroin injections compared with saline-treated rats. Typical abstinence signs in rats included body and head shakes, cheek tremors, eye blinks, ptosis, foot and genital licks, scratches, writhes, and gasps (Grieder et al., 2010). Each rat's percentage abstinence scores were its number of observed abstinence signs divided by the average of abstinence signs observed in saline-treated rats and multiplied by 100. One-way repeated-measures ANOVA and post hoc Fisher's least-significant difference tests were performed to compare the differences in the number of percentage abstinence scores among groups.
Ultrasonic distress vocalizations in the 20–28 kHz range were recorded for 10 min following the administration of the opioid antagonist naloxone in naive and chronic opiate-treated LV-siRNAs rats and chronic opiate-treated LV-GFP rats. Animals were brought to an empty room and placed inside a gray chamber (41 × 41 × 38 cm), where ultrasonic vocalizations (s) were recorded and transformed into an audible signal with the aid of Mini Bat Detectors (Ultra Sound Advice). The signal was sent to a computer, where it was digitized for analysis. A Student's t test was performed to compare the difference in the number of ultrasonic distress vocalizations emitted by the two groups.
The test of locomotor activation produced by repeated opiate administration was performed in a gray chamber (41 × 41 × 38 cm) during daylight. A camera positioned above the chamber recorded distance traveled (meters) using EthoVision XT (version 5) software (Noldus Information Technology). Locomotor activity was monitored at least 2 weeks after surgery. The first day, rats were placed drug free in the activity chamber for 10 min after the subcutaneous injection of saline solution. On subsequent days, rats were placed in the activity chamber for 10 min immediately after the subcutaneous injection of 0.5 mg/kg of heroin. Rat locomotion was recorded on drug-free day (day 0) and on days 1 and 4 after heroin administrations. One-way repeated-measures ANOVA and post hoc Student–Newman–Keuls tests were performed to compare the effects of heroin on locomotion in LV-GFP- against LV-siRNAs-infected rats. The experimental time line of LV infections and intra-VTA BDNF infusion experiments are described in Table 1 and Table 2, respectively.
Table 1.
Description of experimental time line for LV infections experiments
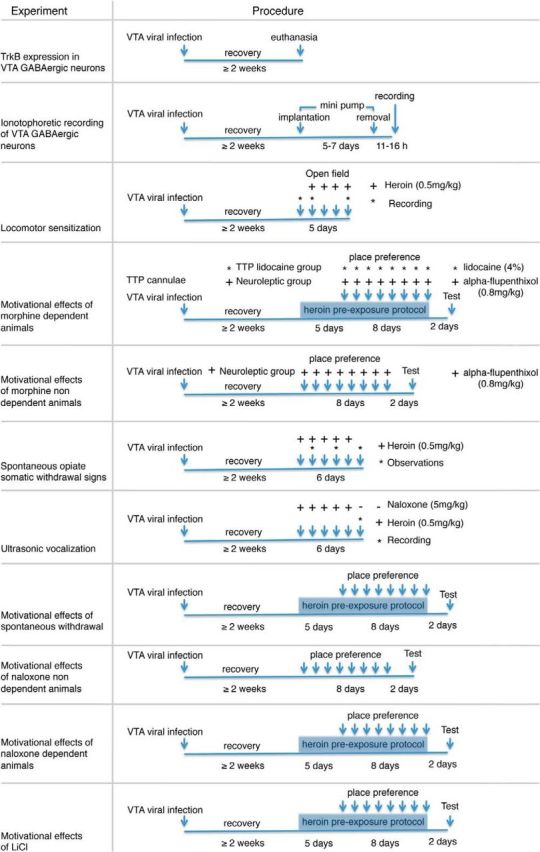
Table 2.
Description of experimental time line for intra-VTA BDNF infusions experiments

Results
We found that rats coinjected with LV-siRNAs against TrkB showed a selective loss of TrkB expression in the VTA as determined by immunohistochemistry (Fig. 1). In slices taken from the VTA (Fig. 1c), confocal microscopy revealed a >80% reduction (t(5) = 2.77, p = 0.003) in the detection of TrkB receptor-expressing cells after injection of GFP-labeled LV-siRNAs compared with LV-GFP control (Fig. 1a,d). Furthermore, as described previously (Bolaños and Nestler, 2004), an overall trend, but not significant, decrease in the soma size (micrometers) of VTA LV-siRNAs-positive neurons with respect to the VTA LV-GFP control was detected (mean ± SEM: LV-siRNAs, 55.94 ± 3.74; LV-GFP, 50.16 ± 7.39; t(91) = 1.08, p = 0.57; Fig. 1b).
Figure 1.

Photomicrographs of rat coronal sections identifying lentiviral infections in the VTA. a, Double-labeled cell with GFP (green) and TrkB (red). Nuclei are in blue, in GFP lentiviral vector pTK433 (LV-GFP)-infected animals (left) and siRNAs expressing lentiviral-infected (LV-siRNAs) and LV-GFP animals (right). Yellow denotes the overlapping of LV-GFP and TrkB labeling, which is very prominent is control animals (LV-GFP) and almost absent in LV-siRNAs-infected animals (arrow). b, Identification of LV-GFP (left) and LV-siRNAs (right) infections in VTA neurons labeled with the neuronal marker MAP2 (cyan). An overall decrease in sizes of VTA LV-siRNAs-positive neurons with respect to the VTA LV-GFP control can be observed (compare arrowed neurons on the left and right). c, A schematic of the anatomical region from which the section displayed in a and b was taken. d, Inhibition of TrkB expression in the VTA mediated by LV-siRNAs (n = 6) as opposed to control LV-GFP (n = 6; *p < 0.05). Data are numbers of TrkB-positive cells, expressed as a percentage of the LV-GFP control ± SEM.
The TrkB receptor mediates the adaptive response of VTA GABA neurons after chronic morphine administration
The shift from inhibitory to excitatory GABAA receptor signaling induced by chronic opiate administration and withdrawal was blocked in LV-siRNAs-treated animals, but not in LV-GFP control animals. The average firing rate of VTA GABA neurons recorded in LV-GFP Naive mice in the horizontal slice preparation ex vivo was 12.9 ± 1.9 Hz (n = 14), in LV-siRNAs Naive mice was 9.0 ± 2.5 Hz (n = 6), in LV-GFP Dependent mice was 22.3 ± 4.9 Hz (n = 9), and in LV-siRNAs Dependent mice was 17.2 ± 3.5 Hz (n = 9). Although the mean baseline firing rate of LV-GFP Dependent mice in the slice preparation was greater than LV-GFP Naive mice, as we have previously demonstrated in morphine-dependent mice in vivo (Vargas-Perez, 2009b), it was not significantly greater (p > 0.05), perhaps because of the pronounced variability in baseline rate across cells in the slice preparation. However, muscimol (0.01–1.0 mm) briskly and markedly inhibited VTA GABA neuron firing rate in LV-GFP Naive mice (Fig. 2a) and LV-siRNAs Naive mice (Fig. 2b), slightly enhanced firing rate in LV-GFP Dependent mice (Fig. 2c), and inhibited VTA GABA neuron firing rate in LV-siRNAs Dependent mice (Fig. 2d), regardless of baseline firing rate. In these examples, cells with similar firing rates (∼10 Hz) were chosen for comparison. Figure 2e summarizes the effects of muscimol (0.01–1.0 mm) across all groups of mice. ANOVA revealed no overall difference between groups at 0.01 mm muscimol (p = 0.055, F(1,26) = 2.93), but there were significant overall differences between groups at 0.1 mm muscimol (F(1,30) = 13.62, p = 0.0001) and 1.0 mm muscimol (F(1,25) = 6.76, p = 0.002). Tukey's post hoc test revealed a significant difference between LV-GFP Naive versus LV-GFP Dependent mice at the 0.1 mm (p = 0.0001) and 1.0 mm (p = 0.003) muscimol levels and LV-GFP Dependent versus LV-siRNAs Dependent mice at the 0.1 mm (p = 0.0001) and 1.0 mm (p = 0.002) muscimol levels. There was no significant difference between LV-GFP Naive versus LV-siRNAs Naive mice or between LV-siRNAs Naive versus LV-siRNAs Dependent mice at any dose level (p > 0.05).
Figure 2.

Blockade of TrkB receptors prevents the switch in GABAA receptor function in VTA GABA neurons. VTA GABA neurons in GAD GFP mice were visualized with fluorescent optics and recorded in cell-attached mode under voltage-clamp conditions. Insets i and ii in a–d are representative 5 s traces of VTA GABA neuron spiking before and during 0.1 mm muscimol application taken at the places indicated on each respective rate meter. Representative cells from each group of mice were chosen based on similarity in baseline firing rate (∼10 Hz). Rate meters from each group are shown in response to 0.1 mm muscimol. Although most cells recovered fully from muscimol, wash data are not shown, as the kinetics of recovery from muscimol varied considerably. All axes in a–d are normalized to facilitate comparisons between groups. a, This rate meter shows a representative VTA GABA neuron recorded in an LV-GFP Naive mouse. Superfusion of muscimol markedly inhibited the firing rate of this VTA GABA neuron. b, Similarly, muscimol markedly inhibited the firing rate of a representative VTA GABA neuron in an LV-siRNAs Naive mouse. c, However, muscimol slightly enhanced the firing rate of a representative VTA GABA neuron in an LV-GFP Dependent mouse. d, Similar to LV-GFP Naive and LV-siRNAs mice, muscimol inhibited the firing rate of a representative VTA GABA neuron in an LV-siRNA Dependent mouse. e, Comparison of muscimol (0.01–1.0 mm) effects on the firing rate of all VTA GABA neurons recorded in the four groups of mice following 16 h morphine withdrawal. Muscimol significantly reduced VTA GABA neuron firing rate in LV-GFP Naive, LV-siRNAs Naive, and LV-siRNAs Dependent mice, but not in LV-GFP Dependent mice (*p < 0.05).
BDNF signaling in the VTA is necessary for the switch to a drug-dependent state motivational system
Blocking TrkB function in the VTA was sufficient to reduce the increased locomotor activation produced by repeated opiate administration observed in LV-GFP (p = 0.04, n = 7); however there was still an effect of heroin on locomotion activation in the LV-siRNAs (n = 7) group compared with LV-siRNAs (n = 8) naive animals (p = 0.035). No significant differences were found between LV-siRNAs and LV-GFP (n = 8)-infected opiate naive groups (effect of heroin treatment: F(3,89) = 3.7, p = 0.024; LV treatment: F(1,89) = 3.4, p = 0.039; effects of days: F(2,89) = 3.89, p = 0.024; Fig. 3a). Additionally, blocking TrkB function in the VTA was sufficient to keep the animals in a motivationally opiate naive-like state, even after chronic opiate exposure (Fig. 3b,c). Using conditioned place preference procedures in rats infected with LV-siRNAs or LV-GFP in the VTA, we observed that BDNF in the VTA is necessary for the switching mechanism from a nondopamine, opiate-naive motivational reward system to a dopaminergic, opiate-dependent motivational system. In contrast to control LV-GFP (n = 8 per group)-infected animals, where antagonism of the dopaminergic system with the neuroleptic α-flupenthixol (0.8 mg/kg) blocked the rewarding effects of systemic morphine in drug-dependent rats (p = 0.12), similarly treated rats infected with LV-siRNAs (n = 8 per group) showed conditioned place preferences that were not blocked by α-flupenthixol (p = 0.001; F(1,63) = 4.0, p = 0.001, interaction of morphine, neuroleptic, and LV treatment; Fig. 3b). However, as is the case in opiate-naive animals (Vargas-Perez et al., 2009a), the rewarding properties of acute morphine administration were blocked by TPP inactivation in LV-siRNAs (n = 8) rats chronically opiate treated and in withdrawal (p = 0.73). TPP inactivation during conditioning did not affect the morphine place preferences in LV-GFP (n = 8)-infected animals with similar chronic opiate treatments (p = 0.0001). PBS Sham TPP inactivations, in chronic opiate-treated and withdrawn animals, did not affect the expression of morphine place preferences in LV-GFP (n = 8) and LV-siRNAs (n = 8; p = 0.0001; F(1,63) = 8.0, p = 0.006, interaction of morphine, TPP inactivation and LV treatment; Fig. 3c). It is possible that intra-VTA LV-siRNAs infections modify the ability of opiates to produce conditioned place preferences. However, neither LV-siRNAs nor LV-GFP infections had any effect on the sizes of the conditioned place preferences produced by morphine in naive animals pretreated with α-flupenthixol (p = 0.001) or in the control group (p = 0.001; F(1,57) = 1.96, p = 0.168, interaction of drug and LV treatment; LV-siRNAs neuroleptic, n = 7; LV-siRNAs control, n = 8; LV-GFP neuroleptic, n = 7; LV-GFP neuroleptic n = 7; Fig. 3d).
Figure 3.

Opiate motivational effects in opiate-dependent animals after blocking the function of BDNF in the VTA. a, Depletion of TrkB expression in the VTA reduces but does not block locomotor sensitization induced by chronic heroin administration. Animals infected with LV-GFP (n = 7) show an increase in locomotion after repeated heroin administrations, compared with animals infected with LV-siRNAs (n = 7; *p < 0.05). However, there is an effect of heroin in locomotion activation in LV-siRNAs groups compared with naive LV-siRNAs (n = 8) and LV-GFP (n = 8) animals (*p < 0.05). b, Blockade of the dopaminergic system with neuroleptics (α-flupenthixol, 0.8 mg/kg) fails to block the rewarding effects of morphine (10 mg/kg) in dependent rats after intra-VTA LV-siRNAs infection (n = 8 per group; *p < 0.05), opposite to LV-GFP-infected animals (n = 8 per group), where the pretreatment with neuroleptics blocks any morphine place preference. c, Reversible lidocaine (4%) TPP lesions (n = 8), but no Sham lesions (n = 8), block the rewarding properties of morphine administration in drug-dependent animals after intra-VTA LV-siRNAs infection, but the same inactivation of the TPP (n = 8), as occurs in Sham lesions (n = 8), does not affect morphine place preference in LV-GFP animals (*p < 0.05). d, LV-siRNAs (n = 8), compared with LV-GFP (n = 7), infections do not affect the size of the conditioned place preference produced by acute morphine administration in drug-nondependent rats (*p < 0.05). Data represent means ± SEM the absolute times spent during testing in the previously saline and previously morphine-paired compartments.
BDNF signaling in the VTA is necessary for the aversive motivation of opiate withdrawal in dependent animals, but not for the expression of somatic withdrawal signs
Chronic opiate exposure produced a daily increase in the expression of spontaneous somatic withdrawal signs in both LV-siRNAs (n = 8) and LV-GFP (n = 8) after 16 h of heroin administration (F(2,23) = 5.6, p = 0.0001), compared with nonopiate-treated control rats (n = 8; p = 0.00001; Fig. 4a). However, the aversive state associated with opiate withdrawal was blocked in LV-siRNAs (n = 8) rats compared with the control LV-GFP (n = 8) animals. This aversive state was inferred by monitoring low-frequency s, which are associated with negative events (Hamdani and White, 2011). A decreased expression of vocalizations in the 22 kHz range was observed following the administration of the opioid antagonist naloxone in LV-siRNAs rats as compared with LV-GFP in naive (p = 0.01) and chronic opiate-treated (p = 0.001) rats (F(1,27) = 0.48, p = 0.001, effect of LV treatment; Fig. 4b).
Figure 4.

Motivational effects of withdrawal in opiate-dependent animals after blocking BDNF function in the VTA. a, Knocking down TrkB in the VTA with LV-siRNAs did not affect spontaneous opiate somatic withdrawal signs. Both LV-siRNAs (n = 8) and LV-GFP (n = 8) animals show an increase in percentage abstinence scores over time after 16 h heroin administration with respect to saline control animals (n = 8; *p < 0.05). b, Opiate naive (n = 7) and dependent (n = 7) LV-siRNAs-treated animals display a reduction in low-frequency (22 kHz) s compared with opiate naive (n = 7) and dependent (n = 7) LV-GFP-treated animals (*p < 0.05) during naloxone-precipitated chronic opiate withdrawal during the course of a 10 min test session. c, Knocking down TrkB in the VTA with LV-siRNAs blocks the place aversion for 16 h of abstinence from morphine (3 mg/kg) in dependent rats (n = 8). In contrast, control opiate-dependent animals infected with LV-GFP (n = 8) show conditioned place aversions (*p < 0.05). d, Naloxone-conditioned (5 mg/kg) place aversions were blocked in LV-siRNAs-treated (n = 8) dependent animals, but not in LV-GFP-treated (n = 8) animals (*p < 0.05). e, Both LV-siRNAs (n = 8) and LV-GFP (n = 8)-infected animals display a conditioning place aversion to LiCl (5 mg/kg; *p < 0.05). Data represent the means ± SEM the absolute times spent in the previously saline and previously drug paired compartments.
We observed that knocking down TrkB expression with LV-siRNAs (n = 8) reduced the expression of the aversive motivational effects of withdrawal in chronic opiate-treated animals, in contrast to LV-GFP (n = 8) animals, which show conditioned place aversions to an environment paired with 16 h of opiate withdrawal (F(1,31) = 5.20, p = 0.03, interaction of LV treatment and withdrawal; Fig. 4c). In addition, the aversive motivational effects of the opioid antagonist naloxone were blocked in both naive (n = 8) and chronic (n = 8) opiate-treated rats infected with LV-siRNAs in the VTA, but not in LV-GFP naive (p = 0.001 n = 8) and chronic opiate-treated rats (p = 0.001, n = 8; F(1,63) = 16.4, p = 0.001, interaction of LV treatment and naloxone; Fig. 4d).
LV-siRNAs infection in the VTA did not block the aversive effects of all drug stimuli, as LV-siRNAs (n = 8) and LV-GFP (n = 8)-treated rats expressed similar conditioned place aversions produced by intraperitoneal LiCl administration (F(1,31) = 1.22, p = 0.27, interaction of LiCl and LV treatment; Fig. 4e). These results demonstrate the importance of BDNF-related neuronal plasticity in the VTA for mediating the negative motivational state associated with opiate withdrawal.
BDNF-TrkB signaling in the VTA is sufficient for inducing opiate dependence in mice
Recent work has suggested that VTA BDNF may block morphine place preferences (Koo et al., 2012), in contrast to our data showing that VTA BDNF preserves morphine place preferences but switches the brain substrates underlying the morphine place preferences (Vargas-Perez et al., 2009b). Given that this more recent work used a mouse model, a different conditioned place preference paradigm, larger BDNF and morphine doses, and different VTA coordinates from our mouse and rat work, we performed a series of experiments to investigate whether BDNF-TrkB signaling played a dual role with regards to opiate reward. First, we tested the possibility that high doses of BDNF (0.25 μg/mouse; Koo et al., 2012) in mice give rise to different effects from doses equivalent to what we had previously used in our rat work (0.025 μg/mouse; Vargas-Perez et al., 2009a). We found that, similar to controls (n = 5), mice receiving the smaller dose of BDNF (n = 13) and those treated with the higher dose showed similar morphine place preferences (n = 9, F(2,24) = 0.507, p = 0.608 interaction of morphine and treatment) and those preferences were blocked by α-flupenthixol in both BDNF groups (n = 5 for the 0.025 μg BDNF group and n = 4; for the 0.25 μg BDNF group; F(1,15) = 5.317, p = 0.03, interaction of α-flupenthixol and BDNF treatment; Fig. 5a). Second, we investigated whether the use of high doses of morphine in conjunction with a longer conditioning time (Koo et al., 2012) results in a BDNF-dependent suppression of morphine place preferences perhaps due to the prolonged duration revealing some aversive properties of the morphine. Once again, BDNF had no suppressive effects on morphine place preferences, such that controls showed an equivalent preference as compared with BDNF-treated animals (controls: n = 7; BDNF group: n = 9; F(1,14) = 0.232, p = 0.637, interaction of morphine and treatment; Fig. 5b). Third, we used the same VTA coordinates used by Koo et al. (2012) and found that, even after accounting for all the major differences in methodology, we could not replicate their effect; controls and BDNF-treated animals spent similar amounts of time in the morphine-paired compartment (n = 7 and n = 9, respectively; F(1,14) = 1.037, p = 0.325, interaction of morphine and treatment; Fig. 5c). Therefore, VTA BDNF did not attenuate morphine place preferences; instead, it changed the neural substrates mediating the preferences.
Discussion
Selectively knocking down TrkB receptor expression in the VTA blocks the shift from inhibitory to excitatory GABAA receptor signaling induced by chronic opiate administration and withdrawal, keeping the animals in a motivational opiate naive-like state. We observed that muscimol robustly inhibited the firing rate of VTA GABA neurons at low concentrations (≤1.0 mm) in all LV-GFP naive mice, LV-siRNAs naive mice, and LV-siRNAs-dependent mice, but not in LV-GFP-dependent mice, indicating that the adaptive response of VTA GABA neurons to chronic morphine treatment is due to activation of TrkB receptors. Consequently, BDNF signaling is necessary for the shift in GABAA receptors on GABAergic VTA neurons, from inhibitory to excitatory. This neuronal change would cause the sensitization of inhibitory GABAergic signaling over dopaminergic neurons in the VTA, altering the activity of the mesolimbic dopaminergic system (White, 1996). Converging lines of evidence suggest that a change in the function of the mesolimbic dopaminergic system is implicated in the aversive motivational response to drug withdrawal (Grieder et al., 2010; George et al., 2012). Accordingly, we observed that blocking BDNF signaling prevented the expression of conditioned (i.e., conditioned place aversion) and unconditioned (i.e., vocalizations) aversive motivational states related to opiate withdrawal. However, these manipulations did not cause a general loss of the ability to detect aversive events or the expression of unconditioned withdrawal, as LV-siRNAs-infected animals were able to learn conditioned place aversion caused by LiCl and the expression of withdrawal somatic sign were not affected. Furthermore, the intra-VTA infection with LV-siRNAs by itself did not cause any significant effect on behavior as shown by the size of the conditioned place preference produced by morphine in naive animals.
In naive rats, activation of GABAA receptors located on VTA GABAergic neurons results in an inhibitory conductance mediated by Cl− influx. In naive rats, morphine activation of mu-opiate receptors on presynaptic GABAergic terminals in the VTA–which form inhibitory synapses on VTA GABAergic neurons–inhibit them by decreasing the level of GABA release onto their GABAA receptors. This disinhibition of GABAergic neuron activity inhibits the mesolimbic dopaminergic pathway and produces reward through a nondopaminergic, TPP-mediated pathway (Vargas-Perez et al., 2009b).
When rats are opiate dependent and in withdrawal, GABAA receptors on VTA GABA neurons switch their signaling properties from inhibitory to excitatory (Vargas-Perez et al., 2009b). It has been observed that a single bilateral intra-VTA BDNF infusion (0.25 μg/0.5 μl each, but not at doses 50× larger and 10× the volume; Koo et al., 2012) promotes this switching mechanism. BDNF may reduce the levels of the potassium chloride cotransporter KCC2, thereby increasing the intracellular chloride concentration. GABAA receptor activation then would result in anions flooding out of the neuron (Ting-A-Kee et al., 2013). Alternatively, BDNF infusions may elevate intracellular carbonic anhydrase enzyme levels, thereby encouraging HCO3− efflux in response to GABAA receptor activation (Ting-A-Kee et al., 2013). These possible changes would make the neuron's membrane potential more positive, or depolarized, relative to the resting membrane potential underlying both the drug-dependent state and the aversion to withdrawal. In opiate-dependent rats, opioid inhibition of GABA release from afferents to VTA GABAergic neurons results in less activation of this now excitatory GABAergic input. This limits the inhibitory GABAergic input to the VTA dopamine neurons, thereby increasing the activity of the mesolimbic dopaminergic system. Thus, blockade of BDNF signaling, via the knockdown of BDNF TrkB receptors (using LV-siRNAs), does not block morphine-conditioned place preference (Koo et al., 2012), but prevents the switch from a TPP-dependent reward system to a dopamine-dependent motivational system and also prevents the expression of a withdrawal aversive motivational state.
In addition to this specific effect of BDNF on VTA GABA neuron TrkB receptors, other BDNF-related plastic changes also might occur. For example, it has been observed that BDNF in high doses can also bind p75 receptors, inducing cell death. Thus, higher doses and larger volumes of external BDNF (that cause blockade of the rewarding effects of morphine administration; Koo et al., 2012) could be due to nonspecific neuroplastic changes on VTA neurons that are unrelated to TrkB signaling. We hypothesized that the nonspecific effects of external infusion of BDNF, at high doses, on the motivational properties of morphine would be independent of TrkB signaling, but found that regardless of the dose, BDNF always had the same effect; it induced an opiate-dependent state wherein dopamine signaling was necessary for morphine-conditioned place preferences, and did not suppress morphine preferences as previously indicated (Koo et al., 2012). This effect was very robust considering that the dose of morphine, length of conditioning, and specific VTA coordinates did not reveal a different role for BDNF as suggested by Koo et al. (2012). It is important to note that, unlike Koo et al. (2012), our work does not directly address the role of BDNF-TrkB signaling in the dopamine neurons of the VTA. However, if it is in fact the case that BDNF-mediated neuroplastic changes at the level of VTA dopamine neurons negatively modulate morphine reward, it may be the case that TrkB signaling following exogenous application of BDNF in the VTA or throughout the brain results in more efficacious signaling at the level of the GABA neurons. This would explain our BDNF effects and would not contradict the dopamine-specific manipulations undertaken by Koo et al. (2012) (e.g., the ablation of TrkB in dopaminergic cells of the VTA resulting in increased morphine place preferences). Whether VTA dopamine neurons express less TrkB receptors than the GABA population is unclear, but such a difference might help explain the discrepancy.
The upregulation of BDNF expression has been associated with the administration of several drugs of abuse (Kauer and Malenka, 2007). In particular, it has been observed that BDNF expression is dramatically increased in the mesolimbic system during prolonged drug withdrawal (Grimm et al., 2003; Kauer and Malenka, 2007; Thomas et al., 2008; Vargas-Perez et al., 2009b) and chronic stress (Berton et al., 2006). Increased levels of BDNF in the VTA are related to the incubation of cocaine craving, cue-induced cocaine seeking (Thomas et al., 2008), and the switch to an opiate-dependent state (Vargas-Perez et al., 2009b). The present results suggest that BDNF signaling is directly related to the expression and consolidation of the aversive motivation of drug withdrawal that lead to a drug-dependent state. The precise VTA BDNF-related neural adaptations that lead to this outcome are unclear. High levels of BDNF have been suggested to induce and sustain early and late long-term potentiation (Minichiello, 2009), regulating both rapid and long-lasting modifications of circuit activity (Kauer and Malenka, 2007; Minichiello, 2009). Due to the critical role that BDNF is thought to have in synaptic plasticity during learning and memory (Kauer and Malenka, 2007; Minichiello, 2009), these findings align with the hypothesis that processes comparable to associative learning are essential for the integration and development of drug withdrawal motivation in the VTA (Berton et al., 2006; Thomas et al., 2008). Our results are consistent with the conjecture that the withdrawal from administration of drugs of abuse elicits an aversive stress response (Koob and Le Moal, 2005; Kauer and Malenka, 2007; Thomas et al., 2008; George et al., 2012) that is responsible for the neuronal plastic changes associated to the development and progression of drug addiction.
Thus, drug-taking behavior to alleviate a chronic drug-generated aversive state (e.g., withdrawal) activates adaptive neuronal changes (dependent on BDNF) that produce a state of dependency and, in turn, set up aversive motivational mechanisms in response to drug withdrawal. This negative reinforcement generates a feedforward mechanism that progressively leads to an allostatic state involving physiological and behavioral changes that drive and maintain addiction. Therefore, the manipulation of VTA TrkB signaling pathway is a promising target for novel addiction treatments.
Footnotes
This work was funded by the Canadian Institutes of Health Research (CIHR), CIHR Training Grant in Population Intervention for Chronic Disease Prevention: A Pan-Canadian Program (53893), the Swiss National Foundation grants 3100-059350 and 3100AO-100686 (J.L.-D.), and grant AA020919 to S.S.
The authors have no competing financial interests.
References
- Bahi A, Boyer F, Chandrasekar V, Dreyer JL. Role of accumbens BDNF and TrkB in cocaine-induced psychomotor sensitization, conditioned-place preference, and reinstatement in rats. Psychopharmacology. 2008;199:169–182. doi: 10.1007/s00213-008-1164-1. [DOI] [PubMed] [Google Scholar]
- Bechara A, Nader K, van der Kooy D. Neurobiology of withdrawal motivation: evidence for two separate aversive effects produced in morphine-naive versus morphine-dependent rats by both naloxone and spontaneous withdrawal. Behav Neurosci. 1995;109:91–105. doi: 10.1037/0735-7044.109.1.91. [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bolaños CA, Nestler EJ. Neurotrophic mechanisms in drug addiction. Neuromolecular Med. 2004;5:69–83. doi: 10.1385/NMM:5:1:069. [DOI] [PubMed] [Google Scholar]
- Crabbe JC. Genetic contributions to addiction. Annu Rev Psychol. 2002;53:435–462. doi: 10.1146/annurev.psych.53.100901.135142. [DOI] [PubMed] [Google Scholar]
- Dockstader CL, Rubinstein M, Grandy DK, Low MJ, van der Kooy D. The D2 receptor is critical in mediating opiate motivation only in opiate-dependent and withdrawn mice. Eur J Neurosci. 2001;13:995–1001. doi: 10.1046/j.1460-9568.2001.01455.x. [DOI] [PubMed] [Google Scholar]
- George O, Le Moal M, Koob GF. Allostasis and addiction: role of the dopamine and corticotropin-releasing factor systems. Physiol Behav. 2012;106:58–64. doi: 10.1016/j.physbeh.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieder TE, Sellings LH, Vargas-Perez H, Ting-A-Kee R, Siu EC, Tyndale RF, van der Kooy D. Dopaminergic signaling mediates the motivational response underlying the opponent process to chronic but not acute nicotine. Neuropsychopharmacology. 2010;35:943–954. doi: 10.1038/npp.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–747. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdani S, White NM. Ultrasonic vocalization ratios reflect the influence of motivational state and amygdala lesions on different types of taste avoidance learning. Behav Brain Res. 2011;217:88–98. doi: 10.1016/j.bbr.2010.09.026. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Koo JW, Mazei-Robison MS, Chaudhury D, Juarez B, LaPlant Q, Ferguson D, Feng J, Sun H, Scobie KN, Damez-Werno D, Crumiller M, Ohnishi YN, Ohnishi YH, Mouzon E, Dietz DM, Lobo MK, Neve RL, Russo SJ, Han MH, Nestler EJ. BDNF is a negative modulator of morphine action. Science. 2012;338:124–128. doi: 10.1126/science.1222265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Koob GF, Stinus L, Le Moal M, Bloom FE. Opponent process theory of motivation: neurobiological evidence from studies of opiate dependence. Neurosci Biobehav Rev. 1989;13:135–140. doi: 10.1016/S0149-7634(89)80022-3. [DOI] [PubMed] [Google Scholar]
- Koob GF, Maldonado R, Stinus L. Neural substrates of opiate withdrawal. Trends Neurosci. 1992;15:186–191. doi: 10.1016/0166-2236(92)90171-4. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Nielsen DA, Butelman ER, LaForge KS. Genetic influences on impulsivity, risk taking, stress responsivity and vulnerability to drug abuse and addiction. Nat Neurosci. 2005;8:1450–1457. doi: 10.1038/nn1583. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, Gallegos RA, Henriksen SJ, van der Kooy D. Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat Neurosci. 2004;7:160–169. doi: 10.1038/nn1182. [DOI] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Nader K, Bechara A, van der Kooy D. Neurobiological constraints on behavioral models of motivation. Annu Rev Psychol. 1997;48:85–114. doi: 10.1146/annurev.psych.48.1.85. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Orlando, FL: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Addiction. Annu Rev Psychol. 2003;54:25–53. doi: 10.1146/annurev.psych.54.101601.145237. [DOI] [PubMed] [Google Scholar]
- Steffensen SC, Bradley KD, Hansen DM, Wilcox JD, Wilcox RS, Allison DW, Merrill CB, Edwards JG. The role of connexin-36 gap junctions in alcohol intoxication and consumption. Synapse. 2011;65:695–707. doi: 10.1002/syn.20885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting-A-Kee R, Vargas-Perez H, Mabey JK, Shin SI, Steffensen SC, van der Kooy D. Ventral tegmental area GABA neurons and opiate motivation. Psychopharmacology. 2013;227:697–709. doi: 10.1007/s00213-013-3002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Perez H, Ting-A-Kee RA, Heinmiller A, Sturgess JE, van der Kooy D. A test of the opponent-process theory of motivation using lesions that selectively block morphine reward. Eur J Neurosci. 2007;25:3713–3718. doi: 10.1111/j.1460-9568.2007.05599.x. [DOI] [PubMed] [Google Scholar]
- Vargas-Perez H, Ting-A-Kee R, van der Kooy D. Different neural systems mediate morphine reward and its spontaneous withdrawal aversion. Eur J Neurosci. 2009a;29:2029–2034. doi: 10.1111/j.1460-9568.2009.06749.x. [DOI] [PubMed] [Google Scholar]
- Vargas-Perez H, Ting-A-Kee R, Walton CH, Hansen DM, Razavi R, Clarke L, Bufalino MR, Allison DW, Steffensen SC, van der Kooy D. Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science. 2009b;324:1732–1734. doi: 10.1126/science.1168501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FJ. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–251. doi: 10.1016/S0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- Zacharko RM, Anisman H. Stressor-induced anhedonia in the mesocorticolimbic system. Neurosci Biobehav Rev. 1991;15:391–405. doi: 10.1016/S0149-7634(05)80032-6. [DOI] [PubMed] [Google Scholar]