

Abstract
Glucose and GLP-1 stimulate not only insulin secretion, but also the post-transcriptional induction of insulin granule biogenesis. This process involves the nucleocytoplasmic translocation of the RNA binding protein PTBP1. Binding of PTBP1 to the 3′-UTRs of mRNAs for insulin and other cargoes of beta cell granules increases their stability. Here we show that glucose enhances also the binding of PTBP1 to the 5′-UTRs of these transcripts, which display IRES activity, and their translation exclusively in a cap-independent fashion. Accordingly, glucose-induced biosynthesis of granule cargoes was unaffected by pharmacological, genetic or Coxsackievirus-mediated inhibition of cap-dependent translation. Infection with Coxsackieviruses, which also depend on PTBP1 for their own cap-independent translation, reduced instead granule stores and insulin release. These findings provide insight into the mechanism for glucose-induction of insulin granule production and on how Coxsackieviruses, which have been implicated in the pathogenesis of type 1 diabetes, can foster beta cell failure.
Keywords: Beta cells, Diabetes, Insulin, Polypyrimidine tract-binding protein, Secretory granules, Translation, Virus
Abbreviations: CV, Coxsackievirus; eIF4E-V5, eIF4E tagged at its C-terminus with a V5-epitope; ER, endoplasmic reticulum; EV, Enterovirus; F, Faulkner; FL, firefly luciferase; IRES, internal ribosomal entry site; ITAF, IRES-trans-acting factor; mTORC1, mammalian Target Of Rapamycin Complex 1; MCA, MIN6 cell adapted; PABP, poly(A)-binding protein; PC, prohormone convertase; PTBP1, polypyrimidine tract-binding protein 1; S6K1, p70S6 Kinase 1; T1D, type 1 diabetes; UTR, untranslated region
1. Introduction
Hyperglycemia and incretins prompt pancreatic beta cells to produce and release insulin. Rapid induction of insulin granule biogenesis to replenish the hormone stores is of physiological relevance given the preferential release of newly synthetized insulin [1–3]. This process is not transcriptionally regulated, but depends on post-transcriptional mechanisms involving polypyrimidine tract-binding protein 1 (PTBP1) [4,5]. PTBP1 binds to polypyrimidine-rich sequences [6] of single-stranded RNAs and has been implicated in alternative splicing [6,7] and polyadenylation of pre-mRNAs [8] as well as stability [4,5] and translation initiation [9] of mRNAs.
Stimulation of insulinoma and primary beta cells with glucose and GLP-1 induces PTBP1 translocation from the nucleus to the cytosol [5,10,11]. Binding of cytosolic PTBP1 to the 3′-untranslated regions (UTRs) of mRNAs for insulin and other insulin granule proteins enhances their stability and translation, and thereby granule biogenesis, while silencing of PTBP1 in insulinoma cells inhibits glucose- and incretin-stimulated insulin secretion [4,5,10,12], Notably, the impaired ability of diabetic islets isolated from partially pancreatectomized subjects to rapidly upregulate total insulin levels in response to glucose-stimulation correlates with increased nuclear retention of PTBP1 [11]. Furthermore, PTBP1 has been identified as a novel risk gene for type 2 diabetes associated with reduced insulin secretion [13]. Hence, the study of PTBP1 function in beta cells is relevant for type 2 diabetes.
PTBP1 is a key internal ribosomal entry site (IRES)-trans-acting factor (ITAF) for cap-independent translation of various positive single-stranded RNA viruses [7,14–16], including human Enteroviruses (EVs). In the case of cap-dependent translation interaction of eIF4G/A complex with the cap-binding protein eIF4E results in the recruitment of the 40S ribosomal subunit to the very 5′-terminus of m7GpppN-capped mRNAs. Cap-independent translation involves instead the recruitment of the 40S ribosomal subunit to cis-acting IRES located within the 5′-UTR of RNAs, in closer proximity to the start codon for translation [15,17,18]. EVs inhibit translation of the host cell while exploiting its translational machinery for cap-independent translation of their uncapped RNA genome [19,20]. Intriguingly, EVs such as Coxsackieviruses are regarded among the environmental factors that may trigger or accentuate loss of immune self-tolerance towards beta cells in type 1 diabetes (T1D) [21–24].
In this study we sought to investigate whether glucose-dependent, PTBP1-regulated translation of mRNAs for insulin granule proteins is cap-independent and whether this process is affected upon infection of rodent insulinoma INS-1 and MIN6 cells as well as mouse pancreatic islets with Coxsackieviruses.
2. Material and methods
2.1. Material
The following commercial antibodies were employed: mouse monoclonal antibodies anti-PTBP1, anti-V5 (Invitrogen), anti-m3G/m7G-cap (Synaptic System), anti-γ-tubulin and anti-insulin (for western blotting) (Sigma), rabbit polyclonal antibodies anti-eIF4E, anti-ph-eIF4E (Ser-209), anti-4E-BP, anti-eIF4G, anti-AKT, anti-ph-AKT (Ser-473), anti-p70S6K, anti-ph-p70S6K (Thr-389) and anti-PABP (Cell Signaling Technology), anti-CgA (Abcam), anti-PC1/3 and anti-PC2 (GeneTex), guinea pig antibody anti-insulin (for immunocytochemistry) (Abcam), goat anti-mouse, anti-rabbit and anti-guinea pig IgGs conjugated with Alexa 488 or Alexa 568 (Molecular Probes), goat anti-mouse and anti-rabbit IgGs conjugated to horseradish peroxidase (Bio-Rad). The mouse anti-ICA512 mAb has been previously described in Ref. [25]. The following reagents were from commercial sources: Rapamycin and LY294002 (Cell Signaling Technology), the eIF4E/eIF4G interaction inhibitor (Calbiochem), m7GpppG cap-analog (New England Biolabs), m7GTP-Sepharose (GE Healthcare) and Dynabeads M270 streptavidin (Invitrogen), reagents for luciferase assay (Promega), 35S-methionine (Hartmann-Analytics), pro-/insulin ELISA (Mercodia) and insulin RIA (Millipore).
2.2. Islet isolation and cell culture
Pancreatic islets were isolated from C57BL/6JRj mice by collagenase digestion and density gradient centrifugation as described previously in Ref. [26]. Mouse insulinoma MIN6 and rat insulinoma INS-1 cells were kind gifts from Jun-ichi Miyazaki (Osaka, Japan) and Claes Wollheim (Geneve, Switzerland), respectively, and were cultured as described in Refs. [5,27]. Cells were kept in resting medium (15 mM HEPES, pH 7.4, 5 mM KCl, 120 mM NaCl, 24 mM NaHCO3, 1 mM MgCl2, 2 mM CaCl2, 3.3 mM glucose, 1 mg/ml ovalbumin) for 1 h before stimulation for 2 h by addition of glucose as indicated. All inhibitors were added at the indicated concentrations to the resting and stimulating media. Two hours after stimulation with glucose cells or islets were harvested. De novo protein synthesis was validated by metabolic labeling of MIN6 cells with 100 μCi 35S-methionine/35 mm well for 2 h. After 5 washes the cells were extracted in lysis buffer containing 1% Triton X-100. After normalization of the protein concentrations the immunoprecipitations was carried out following standard protocols [28]. Biosynthesis of insulin was investigated as described in Ref. [29].
2.3. Cloning
The 5′-UTRs of rat PC1/3, PC2, insulin1 and 2 were amplified by RT-PCR from INS-1 cell total RNA and cloned into pGL3-B (Promega). The 5′-UTR of rat ICA512 mRNA was obtained by 5′-RACE based on the public sequence NM_053881. Mutation of polypyrimidine tracts in rat ICA512 and PC2 mRNA 5′-UTRs was carried out with the QuikChange Site-Directed Mutagenesis Kit (Stratagene). For overexpression of eIF4E the coding sequence was cloned into pcDNA3.1 (Invitrogen) using the Directional cloning kit.
2.4. Transfection
DNA was transiently transfected into MIN6 or INS-1 cells using an AMAXA electroporator, as described in Ref. [5]. siRNAs were transfected into MIN6 cells with Dharmafect 4 solution (Thermo Scientific). Longer RNA molecules as well as the m7GpppG cap-analog were transfected using Lipofectamin 2000 (Invitrogen).
2.5. Virus infection
CVB5 F or MCA were propagated in GMK cells as described in Ref. [30]. For infection, 5 × 105 MIN6 cells were incubated with 2.25 × 105 TCID50 CVB5 F or MCA/5 × 105 cells as described in Ref. [30].
2.6. Protein analysis
Cells and purified islets were extracted in lysis buffer [20 mM TRIS/HCl, pH 8.0, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% protease inhibitor cocktail (Sigma) and 1% phosphatase inhibitor cocktail (Calbiochem)]. Protein concentration in the detergent soluble material was measured by BCA assay (Pierce). Cell extracts were separated by SDS-PAGE and immunoblotted as described in Ref. [5]. Chemiluminescence was performed using the Supersignal West Pico Substrate (Pierce) and detected with a LAS 3000 Bioimaging System (Fuji). Total protein synthesis was validated by metabolic labeling of insulinoma cells with 100 μCi 35S-methionine/35 mm well for 2 h. After 5 washes the cells were extracted and the protein precipitated with 10% TCA.
2.7. Immunocytochemistry
MIN6 cells were grown on cover slides, fixed after treatments with 3% paraformaldehyde and permeabilized with 0.2% saponin. Immunostaining, image acquisition and processing were performed as described in Ref. [5].
2.8. RNA interference
Short interfering double-stranded RNA (siRNA) oligonucleotides for mouse eIF4E (NCBI accession number NM_007917) were synthesized with the Silencer siRNA Construction Kit (Ambion) using the following primers: eIF4E sense primer 1, 5′-aaccttcgattgatctctaagccctgtctc; eIF4E antisense primer 1, 5′-aacttagagatcaatcgaaggcctgtctc; eIF4E sense primer 2, 5′-aaggtgataagatagcaatatcctgtctc; eIF4E antisense primer 2, 5′-aaatattgctatcttatcacccctgtctc; eIF4E sense primer 3, 5′-aagtctcattcgcctttgtctcctgtctc; eIF4E antisense primer 3, 5′-aaagacaaaggcgaatgagaccctgtctc. Control scrambled siRNA oligonucleotides were previously described in Ref. [5]. siRNA oligos for mouse PTBP1 (NCBI accession number NM_008956) and the corresponding control siRNA were obtained from RIBOXX.
2.9. Real-time PCR
Total RNA from INS-1 cells was prepared with the RNeasy Kit (QIAGEN). 1 μg total RNA was reverse transcribed with SuperScript II reverse transcriptase (Invitrogen) and oligo d(T) primer. mRNA levels were measured by quantitative real-time PCR with the qPCR GoTaq Kit (Promega) and a MX4000 Thermocycler (Stratagene). Normalization of real-time PCR data was performed by parallel amplification of rat β-actin mRNA. The used primers have been previously described in Ref. [10].
2.10. Luciferase assays
INS-1 cells were co-transfected with firefly and renilla luciferase constructs. The firefly luciferase activity was measured 4 days after transfection and normalized versus that of renilla luciferase. Luciferase activity in MIN6 cells 1 day after transfection of reporter RNA was measured as for INS-1 cells.
2.11. In vitro RNA binding assay
The mRNA 5′-UTRs of ICA512, PC1/3, PC2, insulin1 and 2 as well as CVB5 MCA RNA were biotinylated by T7 in vitro transcription (Biozym). Dynabeads M270 streptavidin were loaded with 50 pmol biotinylated RNA, washed twice and then incubated with INS-1 and MIN6 cell extracts for 2 h. The beads were washed 3 times and the bound PTBP1 was then analyzed by western blotting.
2.12. Cap-binding assay
eIF4E and associated proteins were isolated according to Ref. [31] using extracts from MIN6 cells incubated with 25 μl of m7GTP-Sepharose for 30 min. After 3 washes the beads were resolved in 50 μl 1 × SDS loading buffer and bound proteins were analyzed by western blotting.
2.13. Insulin analysis
Total insulin content in INS-1 cells and in the corresponding medium was measured with the Sensitive Rat Insulin RIA Kit (Millipore). For all experiments with MIN6 cells and mouse islets the Rat/Mouse proinsulin and Rat insulin ELISA (Mercodia) were used.
2.14. Statistics
Statistical analyses were done by using ANOVA test (STAT PLUS:mac LE2009). Data were shown as means ± SD. Significance from at least 3 independent experiments was indicated in the figures as follows: *p < 0.05, **p < 0.01 and ***p < 0.001.
3. Results
3.1. The 5′-UTRs of mRNAs for granule proteins bind PTBP1 and display IRES activity
Previous studies indicated that PTBP1 is critical for post-transcriptional up-regulation of proinsulin and other granule protein biosynthesis shortly after glucose stimulation [4,5]. Accordingly, glucose-stimulation of rat insulinoma INS-1 cells for 2 h led to a significant increase in the amounts of insulin1/2 and the secretory granule markers ICA512/IA-2, PC1/3 and PC2 mRNAs co-immunoprecipitated with PTBP1 (Figure 1A and B and Supplementary Table 1). Conversely, comparable amounts of carboxypeptidase H/E (CPH) mRNA, which is not translated in a glucose-regulated fashion and lacks putative binding sites for PTBP1 [5], were co-immunoprecipitated with PTBP1 from extracts of resting and stimulated cells.
Figure 1.
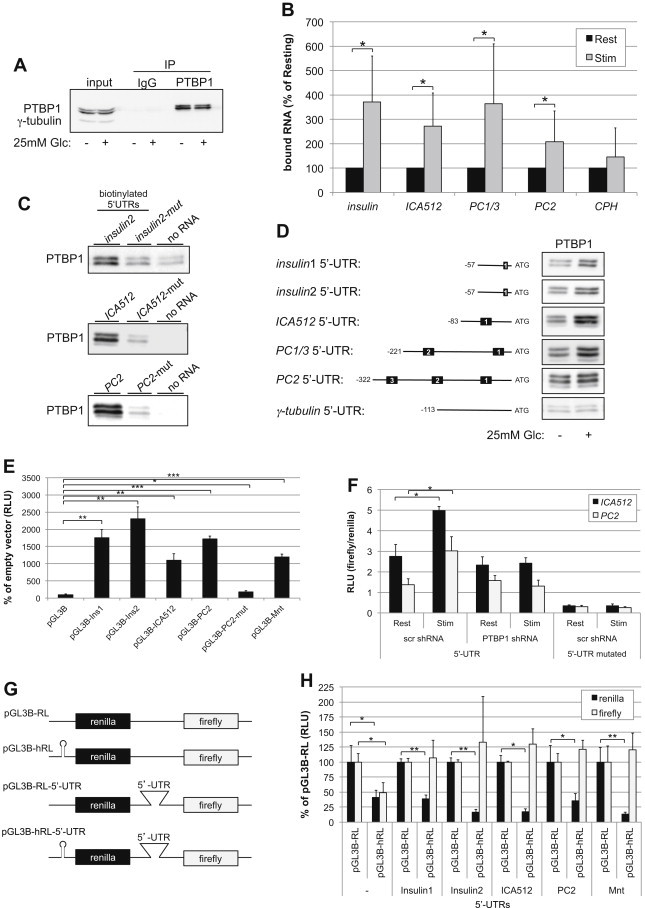
PTBP1 binds to the 5′-UTRs of mRNAs encoding SG proteins. (A) Immunoblot for PTBP1 immunoprecipitated from extracts of resting or glucose-stimulated INS-1 cells. (B) Levels of mRNAs encoding SG proteins co-immunoprecipitated with PTBP1 were assessed by qPCR (n = 6). (C) The polypyrimidine tracts in the biotinylated 5′-UTRs of insulin2, ICA512 and PC2 mRNAs were mutated to verify with in vitro RNA binding assays the specific binding of PTBP1. (D) Left: Schemes of the mRNA 5′-UTRs of insulin1 and 2, ICA512, PC1/3, PC2, and γ-tubulin used for in vitro RNA binding assays. Polypyrimidine tracts are shown as black boxes. Right: The amount of PTBP1 recovered with the corresponding construct from extracts of INS-1 cells stimulated or not with 25 mM glucose was detected by immunoblotting. (E) Dual luciferase assays with the mRNA 5′-UTRs of ICA512, PC1/3, PC2, insulin1 and 2 and Mnt inserted into pGL3-B. In pGL3-B-PC2-mut the consensus for PTBP1 binding was mutated. The activity ratio between firefly luciferase and the co-transfected renilla luciferase in the empty pGL3-B was set as 100%. (F) Dual luciferase assays in INS-1 cells depleted of PTBP1 with shRNA or treated for control with a scrambled (scr) shRNA. (G) Schemes of the pGL3-B bicistronic vectors. (H) Luciferase activity in INS-1 cell extracts. The values of the constructs lacking the hairpin (pGL3-B-RL and pGL3-B-RL-5′-UTR) were set as 100%. All luciferase assays were independently repeated 3 times in triplicates. Significance in (B) (E) (F) and (H) was determined by ANOVA test (*p < 0.05, **p < 0.01, ***p < 0.001).
The mRNA 5′-UTRs of rat insulin1 and 2 contain a single polypyrimidine stretch conforming the “core” consensus (UUGUUCC) for PTBP1-binding [32]. The mRNA 5′-UTRs of rat ICA512, prohormone convertase 1/3 (PC1/3) and 2 (PC2) (Figure 1D and Figure S1A), include instead one or more “extended” consensus (CYYYYCYYYYYG) for PTBP1-binding [4]. In the case of the rat ICA512 mRNA the presence of this site was identified by 5′-RACE (Figure S1C). The interaction of PTBP1 with the mRNA 5′-UTRs of these granule markers, but not of γ-tubulin, which lacks a predicted PTBP1-binding site, was verified by in vitro RNA binding assays (Figure 1D). Disruption of the single PTBP1-binding motif in the 5′-UTRs of insulin2 and ICA512 mRNAs or of the corresponding sites 2 and 3 in rat PC2 mRNA reduced the recovery of PTBP1 (Figure 1C). Significantly more PTBP1 was recovered with the mRNA 5′-UTRs of insulin1, ICA512 and PC2, but not with those of insulin2, PC1/3 and γ-tubulin, when this assay was performed using extracts from glucose-stimulated INS-1 cells as the source for PTBP1 (Figure 1D and Figure S1D). Similar results were obtained by performing this assay with the 5′-UTRs of the corresponding mouse transcripts and extracts of mouse insulinoma MIN6 cells as the source for PTBP1 (Figure S1A). The specific binding of PTBP1 to the mRNA 5′-UTRs of mouse insulin2, ICA512 and PC2 was corroborated by competitive displacement with the corresponding non-biotinylated mRNA 5′-UTRs (Figure S1B).
In addition to RNA splicing and stability, PTBP1 promotes cap-independent translation by binding to IRES [15,33]. Inclusion of the 5′-UTRs of rat insulin1 and 2, ICA512 or PC2 mRNAs in firefly luciferase (FL) reporter constructs increased the expression/activity of FL in transfected INS-1 cells relative to the empty control vector (Figure 1E). This increase was comparable with that observed upon inclusion in the FL reporter vector of the mRNA 5′-UTR of Mnt, which has been reported to be translated in a cap-independent fashion [34]. Glucose-stimulation of INS-1 cells augmented the FL activity when the reporter vector included the 5′-UTRs of ICA512 or PC2 mRNAs (Figure 1F). Such increments, however, were specifically abolished upon knockdown of PTBP1 by 70% with a silencing hairpin (Figure 1F, Figure S1E and F). The glucose-dependent increase of FL activity was also not detected upon mutation of the PTBP1-binding sites (Figure 1F).
The presence of an IRES within a transcript can be investigated using bicistronic reporter vectors including the 5′-UTR of interest between the coding sequences for renilla luciferase (RL) and FL. We used this strategy to assess whether the mRNA 5′-UTRs of insulin1 and 2, ICA512 and PC2 display IRES activity (Figure 1G). Consistent with previous data (Figure 1E), their inclusion upstream of FL increased the expression of the latter in transfected INS-1 cells relative to the basic bicistronic vector (data not shown). To exclude that expression of FL reflected a read-through translation downstream of RL rather than the activity of an IRES introduced between the coding sequences for RL and FL, we transfected vectors in which the first AUG for RL was preceded by a hairpin that blocks ribosomal scanning (Figure 1G). The presence of this hairpin reduced indeed the expression of RL in all cases, while that of FL in the presence of insulin1 and 2, ICA512, PC2 or Mnt mRNA 5′-UTRs was unchanged (Figure 1H). Another concern regarding the use of bicistronic vectors to test for IRES in eukaryotic transcripts is the potential inclusion of cryptic promoter elements within the intervening 5′-UTRs [35]. To verify this possibility we quantified the levels of FL and RL transcripts upon silencing of the latter by RNA interference. Reduction of RL mRNA levels by 80%, as measured by qPCR, did not increase the FL/RL mRNA ratio, thus excluding the transcription of a capped monocistronic mRNA for FL alone (Figure S1E). Taken together, these data imply that the 5′-UTRs of mRNAs for several granule proteins contain IRES and bind to the ITAF PTBP1, which is critical for glucose-induced translation of these transcripts.
3.2. Glucose-stimulated up-regulation of granule precursor biosynthesis is Rapamycin- and LY294002-insensitive
Next, we employed MIN6 cells, another surrogate model of beta cells, to investigate whether glucose enhances the biosynthesis of insulin granule proteins by cap-dependent translation or by eliciting their IRES-mediated, cap-independent translation. As IRES-containing transcripts may escape repression of global translation in condition of endoplasmic reticulum (ER) stress [36] we first verified that in our experimental conditions glucose-stimulation did not elicit an unfolded protein response. In MIN6 cells stimulated with 25 mM glucose for 2 h the ER stress markers phospho-eIF2α and phospho-IRE1α were reduced compared with cells kept at rest without glucose or exposed to the ER stress inducer thapsigargin, while the levels of BIP were unaffected (Figure S2A). Unlike glucose starvation or thapsigargin treatment, glucose-stimulation also did not induce the splicing of Xbp1 (Figure S2B).
Mammalian target of Rapamycin complex 1 (mTORC1) play a key role in promoting cap-dependent translation downstream of nutrient and insulin receptor/AKT signalings. Specifically, mTORC1 phosphorylates the regulators of translation p70S6 Kinase 1 (S6K1) and eIF4E-BP/4E-BP. As reported in Refs. [37,38], glucose-stimulation of MIN6 cells increased the phosphorylation of AKT and S6K1 (Figure S3A), the recovery of eIF4E with eIF4G (Figure S3B) and global protein biosynthesis (Figure 2A). Conversely, inhibition of mTORC1 with Rapamycin reduced S6K1 phosphorylation (Figure S3A), the recovery of the eIF4E-eIF4G complex (Figure S3B) and total protein biosynthesis (Figure 2A). Similarly, the PI3K/AKT inhibitor LY294002 reduced the levels of phosphoAKT and phosphoS6K1 (Figure S3A) and total protein biosynthesis (Figure 2A). Remarkably, however, neither Rapamycin nor LY294002 prevented glucose-stimulation from rapidly enhancing the levels of proinsulin (Figure 2B). Both drugs also did not inhibit the glucose-induced biosynthesis of proICA512 and proPC1/3, as assessed either with conventional 35S-metabolic labeling followed by immunoprecipitation and autoradiography (Figure 2C) or immunoblotting of the cell extracts (Figure 2D). Analysis by immunoblotting revealed that also granule precursor proteins proChromogranin A (proCgA), and proPC2 (Figure S3C and D) were also up-regulated by glucose-stimulation in a Rapamycin- and LY294002-insensitive fashion, although the increase of proPC2, unlike that of the other granule markers, was not always statistically significant (Figure S3D). Similarly, glucose-induced up-regulation of proICA512 and proCgA expression in INS-1 cells was insensitive to either Rapamycin or LY294002 treatments (Figure S3E). The observed increments of granule precursor proteins, as revealed by immunoblotting, resulted clearly from de novo biosynthesis, as verified by inhibition of translation with cycloheximide (Figure S3F and G). Importantly, also in mouse islets the glucose-stimulated up-regulation of insulin (Figure 2E) and several other granule cargoes (Figure 2F and G) was insensitive to Rapamycin or LY294002, despite their ability to inhibit phosphorylation of S6K1, and also of AKT in the specific case of LY294002 (Figure 2F). Taken together, these results support the possibility that glucose-stimulation enhances the biosynthesis of granule cargoes in a cap-independent fashion.
Figure 2.
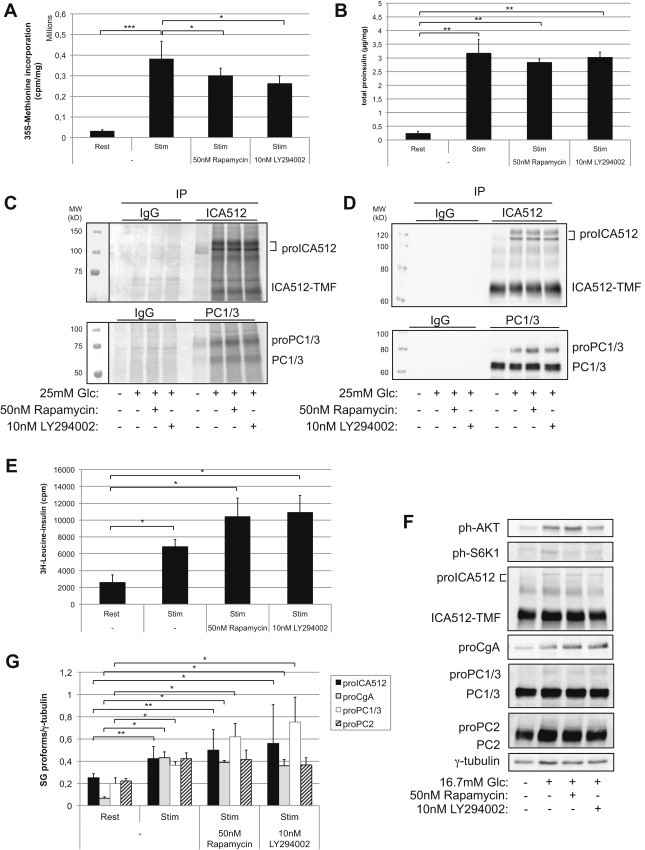
Inhibition of mTOR does not affect glucose-stimulated translation of SG proteins. MIN6 cells were incubated with Rapamycin or LY294002 for 1 h before being glucose-stimulated. (A) Total protein biosynthesis as measured by 35S-methionine incorporation (n = 3). (B) Total proinsulin as measured by ELISA (n = 3). (C) Autoradioagraphy and (D) corresponding immunoblots of pulse-chase labeled ICA512 and PC1/3. (E) Pulse-chase labeling of insulin in isolated mouse islets (n = 3) (F) Immunoblots and quantification (G) for SG proteins and γ-tubulin on extracts of mouse isolated islets treated with Rapamycin and LY294002 before glucose-stimulation (n = 3).
3.3. Glucose-stimulated up-regulation of granule precursor biosynthesis is insensitive to pharmacological inhibition of eIF4E activity
To investigate further this hypothesis MIN6 cells were treated with an eIF4E/eIF4EG inhibitor, which reduced the glucose-stimulated binding of eIF4G to eIF4E (Figure S4A). In eIF4E/eIF4EG inhibitor treated cells glucose-stimulated global protein translation was blunted relative to untreated MIN6 (Figure 3A) and INS-1 (not shown) cells. However, treatment with the eIF4E/eIF4G inhibitor did not impair glucose-induced biosynthesis of proICA512 and proPC1/3 as assessed again with either pulse-chase labeling (Figure 3B) or immunoblotting of the same cell extracts (Figure 3C). A similar behavior was observed for proCgA and proPC2 (Figure S4B and C). The persistent elevated expression of these granule precursor proteins upon treatment with the eIF4E/eIF4G did not result from inhibition of their processing along the secretory pathway. Indeed, treatment with furin inhibitor 1, which inhibits protein convertases, could enhance proICA512 and proPC1/3 levels above those found in cells treated with eIF4E/eIF4G inhibitor alone (Figure 3B and C). As in MIN6 cells, treatment of mouse islets with eIF4E/eIF4G inhibitor did not downregulate the glucose-enhanced biosynthesis of insulin (Figure 3D) and other granule cargoes (Figure 3E and F).
Figure 3.
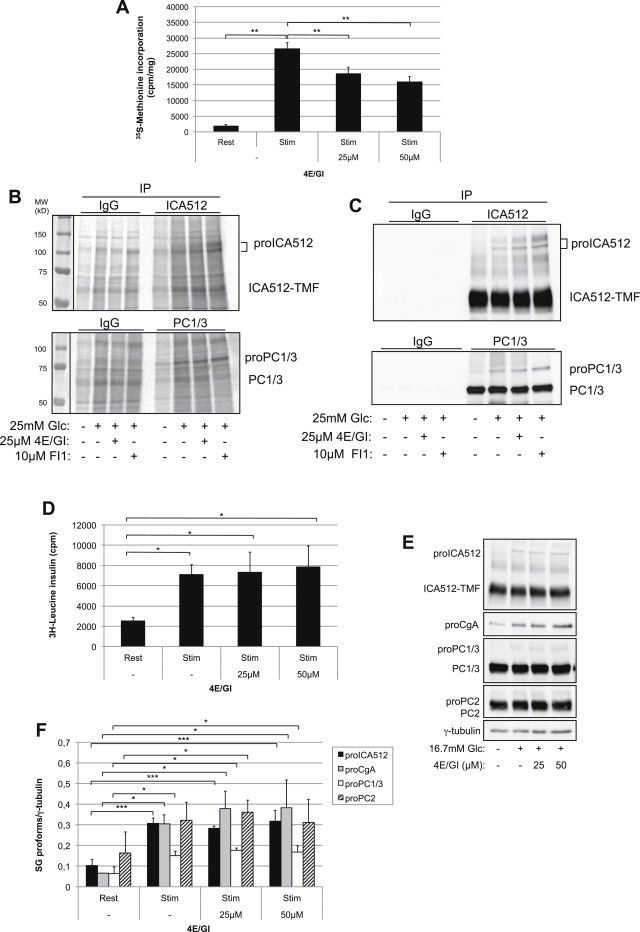
Inhibition of eIF4E does not affect glucose-stimulated translation of SG proteins. (A) Rate of total protein biosynthesis as measured by 35S-methionine incorporation after treatment of MIN6 cells with the eIF4E/4G inhibitor (n = 3). (B) Autoradioagraphy and (C) corresponding immunoblots of pulse-chase labeled ICA512 and PC1/3 on extracts of MIN6 cells treated with the eIF4E/4G inhibitor or furin inhibitor 1 before glucose-stimulation. (D) Pulse-chase labeling of insulin in isolated mouse islets (n = 3). (E) Immunoblots for SG proteins and γ-tubulin on extracts of mouse isolated islets pretreated with the eIF4E/4G inhibitor prior to glucose-stimulation. (F) Quantification of SG precursor proteins as detected in (E) (n = 3).
As an additional pharmacological method to verify the cap-independent translation of insulin granule cargoes, both MIN6 and INS-1 cells were transfected with m7GpppG, which competes with the 7-methyl-guanosine triphosphate cap-structure at the 5′-end of mRNAs for the binding to eIF4E. Transfection of this cap-analog was also unable to prevent up-regulation of granule precursor proteins in response to glucose in both MIN6 (Figure S4D and S4E) and INS-1 cells (Figure S4F), despite the ability to inhibit enhancement of global protein synthesis (Figure S4G).
3.4. Glucose-stimulated up-regulation of granule precursor biosynthesis is insensitive to genetic modification of eIF4E expression
Next, the ability of glucose to rapidly stimulate cap-independent translation of insulin granule proteins was investigated genetically by knockdown of eIF4E in MIN6 (Figure 4A) or INS-1 (Figure S5A–C) cells. Glucose-stimulation enhanced the phosphorylation of eIF4E (Figure 4A), thereby increasing its affinity for the mRNA 5′-cap. As reported in Ref. [39], we verified that knockdown of eIF4E reduced cell replication, as measured by counting BrdU+ MIN6 and INS-1 cells (Figure S5D and E). Silencing of eIF4E lowered also glucose-stimulated translation in both cell lines (Figure 4B and Figure S5F). However, eIF4E-depletion did not prevent glucose-stimulation from significantly upregulating the levels of total proinsulin and other granule markers in MIN6 cells (Figure 4C–E). Similarly, proICA512 and proCgA were induced in glucose-stimulated, eIF4E-depleted INS-1 cells (Figure S5G).
Figure 4.
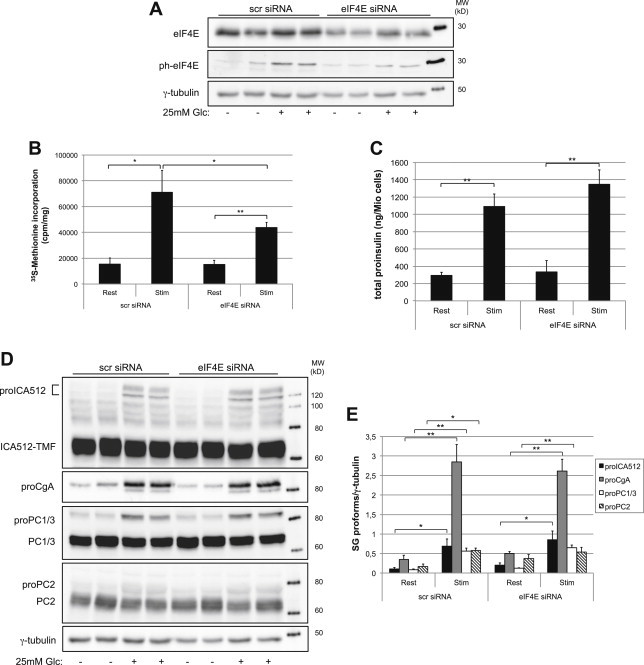
Depletion of eIF4E does not reduce glucose-stimulated translation of SG proteins. MIN6 cells were analyzed 4 days after treatment with scrambled (scr) or eIF4E siRNA oligos. (A) Immunoblottings for eIF4E, phospho-eIF4E and γ-tubulin in MIN6 cells stimulated or not with glucose. (B) Total protein biosynthesis upon eIF4E knockdown as measured by 35S-methionine incorporation (n = 3). (C) Total proinsulin values after eIF4E knockdown as measured by ELISA (n = 3). (D) Immunoblots for SG proteins and γ-tubulin. (E) Quantification of SG precursor proteins as detected in D (n = 4).
Similarly, we analyzed glucose-induction of granule markers in MIN6 cells overexpressing eIF4E tagged at its C-terminus with a V5-epitope (eIF4E-V5). This tag did not prevent eIF4E-V5 from binding to the cap-structure and dissociating from eIF4E-BP in a glucose-regulated fashion (Figure 5A). While overexpression of eIF4E-V5 increased cell replication (Figure S5H and I) and global protein translation (Figure 5B), it did not enhance glucose-induced biosynthesis of proinsulin (Figure 5C) and other granule precursor proteins (Figure 5D and E) relative to control cells. Hence, both pharmacological and genetic treatments consistently indicate that glucose-stimulation rapidly enhances the biogenesis of insulin granules by promoting the translation of their secretory cargoes in a cap-independent fashion.
Figure 5.
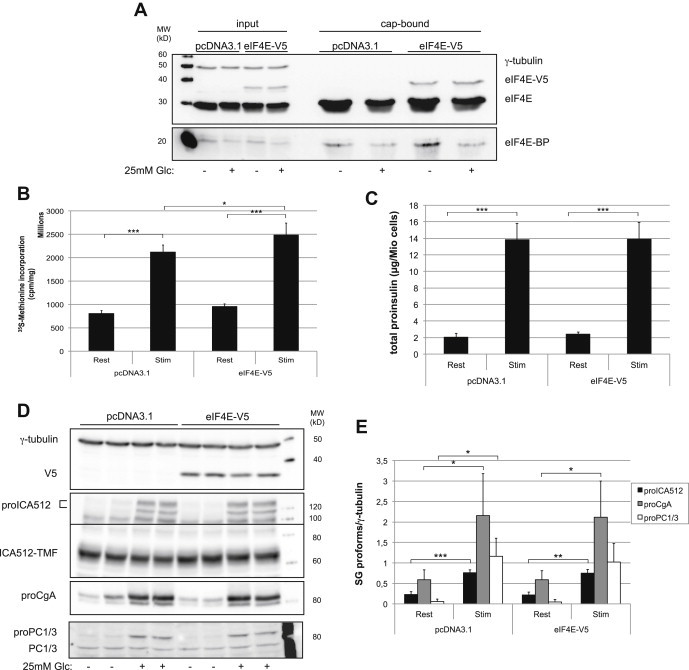
Overexpression of eIF4E does not alter glucose-stimulated translation of SG proteins. MIN6 cells were analyzed 4 days after transient transfection with V5-tagged eIF4E in pcDNA3.1 or the pcDNA3.1 vector alone. (A) Immunoblotting for eIF4E-V5, eIF4E and eIF4E-BP and γ-tubulin recovered by cap-binding assay from extracts of MIN6 cells stimulated or not with glucose. (B) Total protein biosynthesis as measured by 35S-methionine incorporation (n = 3). (C) Total proinsulin values as measured by ELISA (n = 3). (D) Immunoblots for SG proteins, V5 and γ-tubulin. (E) Quantification of the SG precursor proteins as detected in (D) (n = 4).
3.5. Glucose-stimulated up-regulation of granule precursor biosynthesis is insensitive to inhibition of cap-dependent translation by Coxsackieviruses
Coxsackieviruses (CVs) belong to the family Picornaviridae and have been often being implicated as potential triggering or precipitating agents for autoimmunity in type 1 diabetes. As their genome is a positive-sense single-stranded RNA lacking the 5′-cap, CVs strictly depend on cap-independent translation for replication and potently inhibit cap-dependent translation in the host cells. Interestingly, PTBP1 has been shown to acts as ITAF for cap-independent translation of several Picornaviruses [40]. To test if and how CVs interferes with glucose-stimulated granule biogenesis, we infected MIN6 cells with either Coxsackievirus B5 Faulkner (CVB5 F) or MIN6 cell adapted (CVB5 MCA). The latter serotype was isolated from human subjects and selected for its ability to infect mouse insulinoma cells [30]. To verify first the modalities of MIN6 cell infection with CVB5 F and MCA we monitored the expression of their capsid protein VP1 (Figure 6B and Figure S6B). Increased labeling of infected MIN 6 cell cultures with ethidium homodimer III suggested that both serotypes induced necrosis (Figure 6A), but not apoptosis, as the cell surface staining with Annexin V was negative (Figure 6A). Unlike Staurosporin, which induces apoptosis, CVB5 F and MCA-infected MIN6 cells were also negative for TUNEL (Figure S6A), and for the cleavage of Caspase 3 and PARP (Figure S6B).
Figure 6.
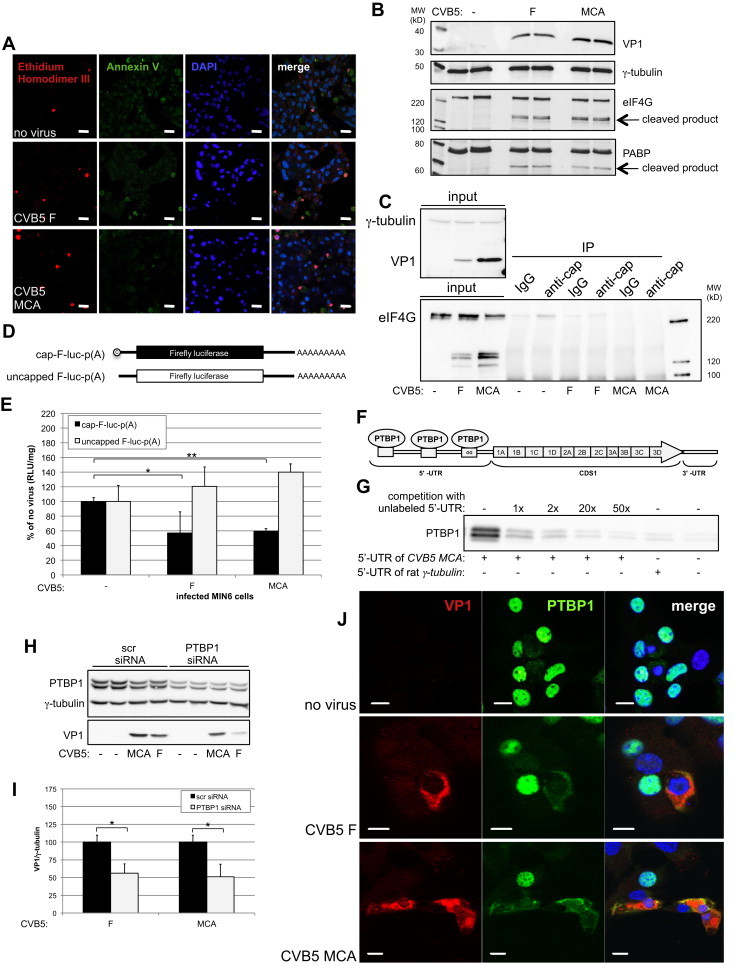
Infection of MIN6 cells with CVB5 inhibits glucose-stimulated cap-dependent translation. MIN6 cells were infected with different CVB5 strains and analyzed 4 days thereafter. (A) Stainings of MIN6 cells with FITC-annexin V and ethidium homodimer III. Scale bars: 20 μm. (B) Immunoblots for eIF4G, PABP, γ-tubulin and VP1. (C) Top left panel: levels of eIF4G, VP1 and γ-tubulin in the cell extracts used as input for immunoprecipations. Bottom panel: immunoblots for eIF4G co-immunoprecipitated with mRNA using an anti-cap antibody. (D) Scheme of in vitro transcribed capped and uncapped reporter luciferase RNAs transfected in CVB5-infected MIN6 cells. (E) Luciferase activity in CVB5-infected cells 1 day after transfection of the RNA constructs shown in (D) (n = 3). (F) Diagram of the CVB5 genome. In the third polypyrimidine tract there is a mismatch of 2 Gs relative to the canonic sequence for PTBP1-binding. (G) Immunoblot for PTBP1 recovered from MIN6 cell extracts following in vitro RNA binding assays with the biotinylated RNA 5′-UTR of CVB5 MCA. Specificity of PTBP1-binding was verified by competition with the corresponding unlabeled RNA 5′-UTR. (H) Immunoblots for VP1, PTBP1 and γ-tubulin in MIN6 cells transfected with scrambled (scr) or PTBP1 siRNAs and infected 2 days later with CVB5 strains. (I) Quantification of VP1 as detected in (H) (n = 3). (J) Confocal microscopy for VP1 (red) and PTBP1 (green) in CVB5-infected MIN6 cells. Nuclei were counterstained with DAPI (blue). Scale bars: 20 μm.
As described for HeLa cells infected with other Picornaviruses [19], CVB5 F or MCA infection of MIN6 cells led instead to the cleavage of eIF4G (Figure 6B and C) and poly(A)-binding protein (PABP) (Figure 6B). Proteolysis of eIF4G prevents its interaction with eIF4E, whereas cleaved PABP cannot bind to eIF4G, thereby precluding the assembly of the eIF4E/eIF4G/PABP complex required for cap-dependent translation. Accordingly, the recovery of eIF4G with the cap-structure was abolished in CVB5 F- and MCA-infected MIN6 cells (Figure 6C). To verify the impact of CVB5 F and MCA on translation, MIN6 cells were then transiently transfected with in vitro transcribed RNAs for FL including or lacking the 5′-cap (Figure 6D). In infected cells cap-dependent and -independent translation of these reporter constructs were either reduced or increased, respectively (Figure 6E), consistent with the expectation that CVs inhibit the former, but not the latter.
The 5′-UTRs of CVB5 F and MCA contain 3 putative PTBP1-binding sites, with the one most proximal to the initiation codon being the least conformed to the consensus for PTBP1-binding (Figure 6F). Using total protein extracts of MIN6 cells for in vitro RNA binding assay, we confirmed the interaction of PTBP1 with the biotinylated 5′-UTR of CVB5 MCA (Figure 6G). Moreover, in PTBP1-depleted MIN6 cells the expression of CVB5 F and MCA, as measured based on VP1 levels, was impaired (Figure 6H and I). In non-infected MIN6 cells, PTBP1 is mostly restricted to the nucleus (Figure 6J). However, in MIN6 cells infected with either CVB5 F- or MCA-PTBP1 was detected throughout the cytoplasm, while its nuclear pool was drastically depleted. Sequestration of PTBP1 in the cytosol is conceivably instrumental to support the high demand for cap-independent translation of the virus genome.
Having shown that CVB5 F and MCA are powerful inhibitors of cap-dependent translation in insulinoma cells, while requiring PTBP1 for their own translation, we investigated if they affected glucose-induced translation of granule proteins. Both viruses impaired glucose-stimulated translation in MIN6 cells (Figure 7A), but not that of proinsulin (Figure 7B), proICA512, proCgA, proPC1/3 and proPC2 (Figure 7D and E). Similar observations were made in isolated mouse islets infected with CVB5 MCA (Figure 7F, G and H). Remarkably, however, infection of MIN6 cells correlated with depletion of the converted, mature granule cargoes insulin (Figure 7C and Figure S6C), ICA512-TMF and PC2 (Figure 7D). As infected cells released less insulin compared with control cells (Figure S6E) the depletion of these granule markers could not result from granule exocytosis. Depletion of mature granule markers was also not secondary to inhibition of their conversion, since in CVB5-infected cells, unlike in cells treated with furin inhibitor 1, the levels of the corresponding proproteins were not increased (Figure S6D). Notably, the levels of insulin, ICA512-TMF and PC2 were also reduced in resting CVB5-infected MIN6 cells relative to resting, non-infected islets (Figure 7C and D). Hence, we conclude that CVB5 infection of beta cells, which blocks cap-dependent translation, does not inhibit glucose-stimulated biosynthesis of granule precursor proteins, but reduces nevertheless insulin stores, conceivably by targeting granule proteins for destruction prior or after their conversion along the secretory pathway.
Figure 7.
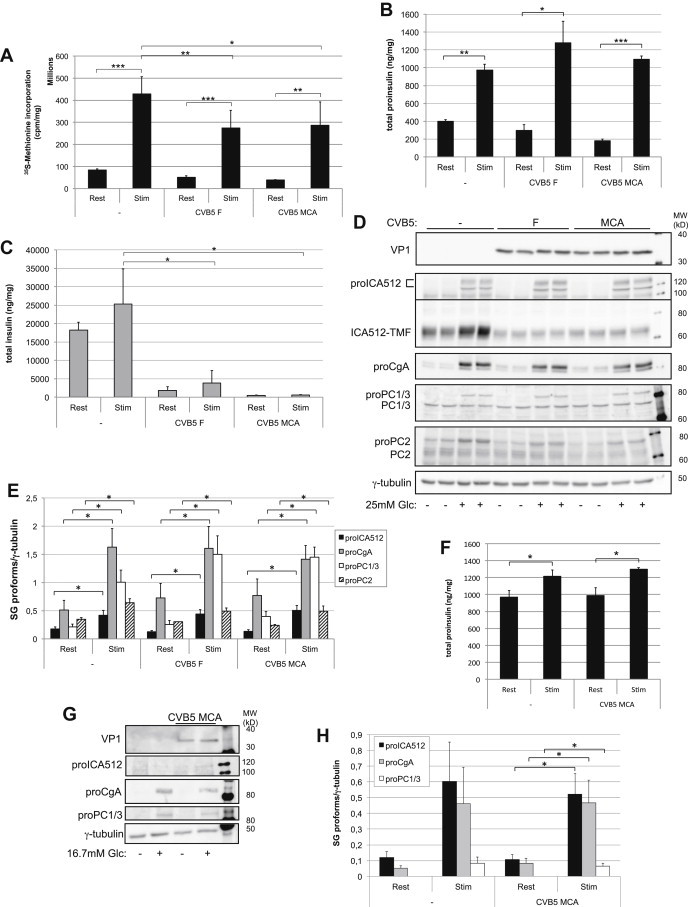
CVB5 infection does not prevent glucose-stimulated translation of SG proteins. MIN6 cells were glucose-stimulated 4 days after CVB5 infection. (A) Total protein biosynthesis as measured by 35S-methionine incorporation (n = 3). (B, C) Total proinsulin (B) and insulin (C) as measured by ELISA (n = 3). (D) Immunoblots for VP1, SG proteins and γ-tubulin. (E) Quantification of SG precursor proteins as detected in (D) (n = 4). (F, G) Total proinsulin and insulin levels as measured by ELISA (F) and immunoblots for VP1, SG proteins and γ-tubulin in mouse isolated islets stimulated with glucose 3 days after infection with CVB5 MCA. (H) Quantification of SG precursor proteins as detected in (G) (n = 3).
4. Discussion
In this manuscript we provide compelling evidence that rodent insulinoma MIN6 and INS-1 cells as well as isolated mouse pancreatic islets exploit cap-independent translation for rapid up-regulation of insulin granule biosynthesis in response to glucose-stimulation. Mechanistic studies in insulinoma cells further indicate that this dedicated pathway is PTBP1-dependent. The mRNAs of insulin1 and 2 and several other granule cargoes, including ICA512, CgA, PC1/3 and PC2, were shown to contain PTBP1-binding sites in their 5′-UTR and to rely on the activity of PTBP1 as an ITAF for their translation. These findings, therefore, extend those of a previous study in which evidence for cap-independent translation was provided for human insulin mRNA alone [29] in conditions of pharmacologically induced nitrosative stress. At variance with that study, we could show that glucose stimulation increases the binding of PTBP1 to the 5′-UTRs of mRNAs for granule components in vitro, and in the case of insulin and ICA512, also in cells. Unlike previously suggested, we also did not find evidence pointing to insulin biosynthesis being mainly cap-dependent in the absence of stress. On the contrary, all data independently obtained upon pharmacological inhibition of cap-dependent translation with Rapamycin, LY294003, eIF4E/4G inhibitor or m7GpppG, or alteration of eIF4E expression point to cap-dependent translation being irrelevant for the biosynthesis of the investigated granule proteins. Our findings could also provide a mechanistic explanation for the inability of Rapamycin to inhibit proinsulin biosynthesis not only in islets from Akita mice, but also in unstressed islets from control mice [41]. A correlate of these findings is that the rate of insulin granule biosynthesis is mainly susceptible to changes in glycemia rather than on pathways regulating global protein biosynthesis, such as insulin receptor signaling. Accordingly, pharmacological depolarization of beta cells with agents such as high potassium effectively triggers insulin release, but unlike glucose, does not induce biosynthesis of granule proteins.
Translation of eukaryotic transcripts is typically cap-dependent. Cap-independent translation is commonly regarded to be an exception exploited only in the case of unusually long 5′-UTRs (>500 bps), which may interfere with efficient initiation due to their length and secondary structures, and for cell protection in conditions of stress and global inhibition of protein synthesis, such as hypoxia, amino acid starvation and apoptosis [35]. The first case is unlikely to account for our findings, as the 5′-UTRs of insulin and the other granule cargo mRNAs investigated here are short, ranging between 60 and 322 bps. On the other hand, hyperglycemia per se could be considered as a stress condition for beta cells, the relief of which depends on increased insulin output. Thus, also in this case the tenet of cap-independent translation being a protective mechanism would hold true. At variance with our findings, Lipson and coworkers reported that short-term glucose-stimulation of isolated mouse islets and INS-1 cells induced phosphorylation of IRE1α [44]. Intriguingly, however, that IRE1α phosphorylation rather than been a sign of ER stress, was instrumental for the selective up-regulation of insulin biosynthesis and secretion. Hence, despite the apparent discrepancy on glucose-induced IRE1α phosphorylation, possibly due to experimental conditions, both studies reached the conclusion that transient glucose-stimulation does not elicit a conventional ER-stress response.
Being pivotal for translation of insulin, by far the most abundant protein produced by beta cells, it is not surprising that PTBP1 is also exploited by EVs such as CVBs for their own propagation in these cells. This convergence may actually account, at least in part, for their beta cell tropism. Strikingly, however, hijacking of PTBP1 by CVB did not competitively reduce the glucose-stimulated biosynthesis of granule precursor proteins. Most likely this was due to the massive nucleocytoplasmic redistribution of PTBP1 in infected cells. Even more strikingly, we found that in CVB5-infected cells the levels of mature granule proteins, and thus of the insulin stores, were dramatically decreased, although their translation was unaffected. As infected cells released much less insulin, this reduction cannot be explained with a paroxysmal exocytic activity. Impaired conversion after CVB infection is also unlikely, as the levels of granule proproteins in infected cells were not increased, as instead was the case in infected cells treated with a prohormone convertase inhibitor. Therefore the only suitable explanation is that virus infection targets either precursor or mature granule proteins to intracellular degradation along the secretory pathway. It would be interesting to investigate whether in beta cells infected with presumed diabetogenic EVs this phenomenon, concomitantly with decreased translation of housekeeping proteins and increased MHC I expression [42,43], favors the antigenic presentation of granule-derived peptides at the cell surface and thus accounts for the preferential loss of tolerance toward granule antigens in type 1 diabetes [45].
5. Conclusion
Regulation of insulin translation is key for understanding the physiology of pancreatic beta cells and their failure in diabetes. This study demonstrates that beta cells selectively exploit cap-independent translation to rapidly upregulate the biosynthesis of insulin and other insulin granule cargoes and illustrates how this process may be influenced by diabetogenic viruses. Evidence that rapamycin, which impairs proliferation of transplanted beta cells [46], does not block glucose-stimulated insulin production can be of clinical interest, since this immunosuppressive drug is commonly administered to type 1 diabetic patients undergoing islet transplantation. Furthermore our results suggest that antiviral therapies targeting cap-independent translation may have a detrimental impact on insulin granule biosynthesis.
Funding
This work was supported by the EU-FP7 consortium DIAPREPP (EU-FP7 Health-2007-2.4.3-1) (M.S. and M.R.) and by a grant from the German Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD e.V.) (FKZ 82DZD00101) (M.S.).
Contribution statement
Conception and design of the study: K.-P.K., M.R., M.S.; acquisition and analysis of the data: K.-P.K; S.N.-S., A.P., H.S., M.B., C.W., A.S., C.M., A. F.; writing of the article: K.-P.K, S.N.-S., M.S. All authors approved the final version of the article.
Acknowledgments
We thank Claes Wollheim, Jun-ichi and Susumu Seino for the gift of INS-1 and MIN6 cells, Martina Lachnit for technical assistance, Maria Grazia Magro for critical reading of the manuscript, all members of the Solimena lab and Ezio Bonifacio for discussion and advices, Katja Pfriem for outstanding administrative assistance.
Conflict of interest
The authors do not have any conflict of interest with the results and conclusion of this study.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
PTBP1 binding to the 5′-UTRs of mouse and rat mRNAs encoding SG proteins is glucose-stimulated. PTBP1 binding to mRNA 5′-UTRs of mouse mRNAs encoding SG proteins is glucose-stimulated (A) Left panel: schemes of the 5′-UTRs of mouse insulin2, ICA512, PC2 and γ-tubulin mRNAs which were synthesized by in vitro transcription and used for in vitro RNA binding assays. Polypyrimidine tracts are shown as black boxes. Right panel: immunoblottings for PTBP1 recovered with the corresponding mRNA 5′-UTRs from extracts of MIN6 cells stimulated or not with glucose. (B) To verify specificity, the binding of PTBP1 was competed with increasing amounts of the corresponding non-biotinylated 5′-UTR. (C) Alignment of the extended sequence of rat ICA512 mRNA 5′-UTR obtained by 5′-RACE based on sequences for rat ICA512 available in public databases. The alignment was obtained using the software ClustralW at <http://www.ch.EMBnet.org>. The red box indicates the putative PTBP1-binding motif. The translation start is indicated in bold and italic. (D) Quantification of immunoblots for PTBP1 recovered with in vitro RNA binding assays as shown in Figure 1D (n = 6). (E) Immunoblotting for PTBP1 and γ-tubulin and quantification (F) using INS-1 extracts 4 days after transfection with indicated shRNA vectors. (G) The ratios between firefly and renilla luciferase mRNAs in INS-1 cells transfected with different pGL3-B-RL vectors were assessed by qPCR 3 days after knockdown of renilla luciferase (n = 3). β-actin mRNA levels were used for normalization.
: Rapamycin and LY294002 treatment inhibits glucose-dependent phosphorylation of AKT and S6K1 but not glucose-stimulated translation of SG proteins in insulinoma cell lines. MIN6 cells were incubated with Rapamycin or LY294002 for 1 h before being glucose-stimulated. (A) Immunoblots for AKT, phospho-AKT, p70S6 kinase, phospho-p70S6 kinase and γ-tubulin. (B) Immunoblots for eIF4E and eIF4G recovered with m7GTP-sepharose beads (cap-binding assay) from extracts of MIN6 cells treated as indicated. (C, D) Immunoblots for SG proteins and γ-tubulin (C) and quantification of the SG preforms (D) of the respective signals (n = 4) in MIN6 cells treated as indicated. (E) Immunoblots for proICA512, proCgA and γ-tubulin on extracts of INS-1 cells treated with Rapamycin or LY294002 for 1 h before glucose-stimulation. (F) Immunoblots for SG proteins and γ-tubulin in MIN6 cells treated with 10 μg/ml cycloheximide for 1 h before glucose-stimulation. (G) Quantification of SG precursor proteins as detected in (A) (n = 3).
: CVB5 infection does not induce apoptosis of MIN6 cells but degranulation. MIN6 cells were analyzed for apoptosis 4 days after infection with CVB5 MCA. Alternatively, as a positive control for apoptosis, cells were treated with the inducer of apoptosis Staurosporin. (A) Immunostainings for VP1 (red) and for apoptosis by TUNEL assay (green). Nuclei were counterstained with DAPI (blue). Scale bars: 20 μm. (B) Detection of Caspase 3 and PARP cleavage as biomarkers of apoptosis by immunoblotting. (C) Confocal microscopy for VP1 (green) and insulin (red) immunostainings in MIN6 cells 4 days after infection with CVB5 MCA. Nuclei were counterstained with DAPI (blue). Right panel: magnification of the inset area on the left. Scale bars: left panels 20 μm; right panel: 10 μm. (D) Accumulation of SG precursor proteins in CVB5 infected MIN6 cells treated with furin inhibitor 1. Immunoblottings for VP1, γ-tubulin and SGs markers ICA512, proCgA, PC1/3, PC2 in CVB5-infected or control MIN6 cells treated with furin inhibitor 1 for 1 h before stimulation with glucose for 2 h. (E) Insulin amounts in media of MIN6 cells stimulated with glucose for 2 h (n = 3).
Figure S2.
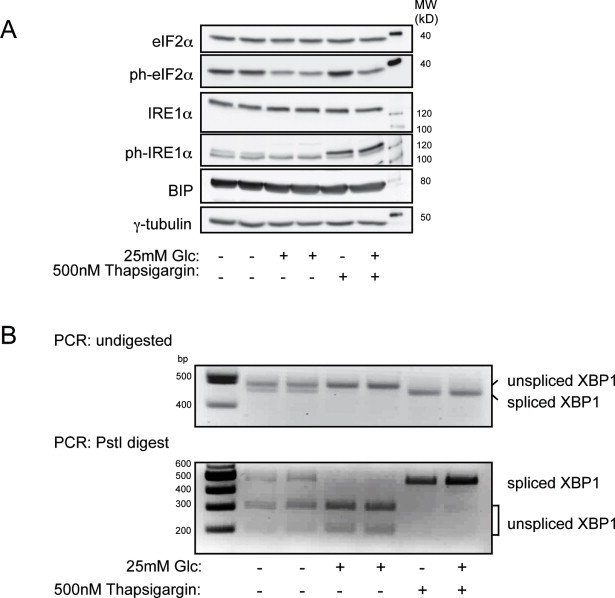
Glucose-stimulation of MIN6 cells for 2 h does not evoke ER stress. (A) Immunoblots for ER stress markers and γ-tubulin in MIN6 cells glucose-stimulated for 2 h. For control, ER stress was induced with 500 nM Thapsigargin. (B) RT-PCR for detection of Xbp1 spliced variants. The unspliced Xbp1 mRNA (479bp) contains a PstI restriction site. PstI restriction digestion generates fragments of 287 bp and 192 bp.
Figure S4.
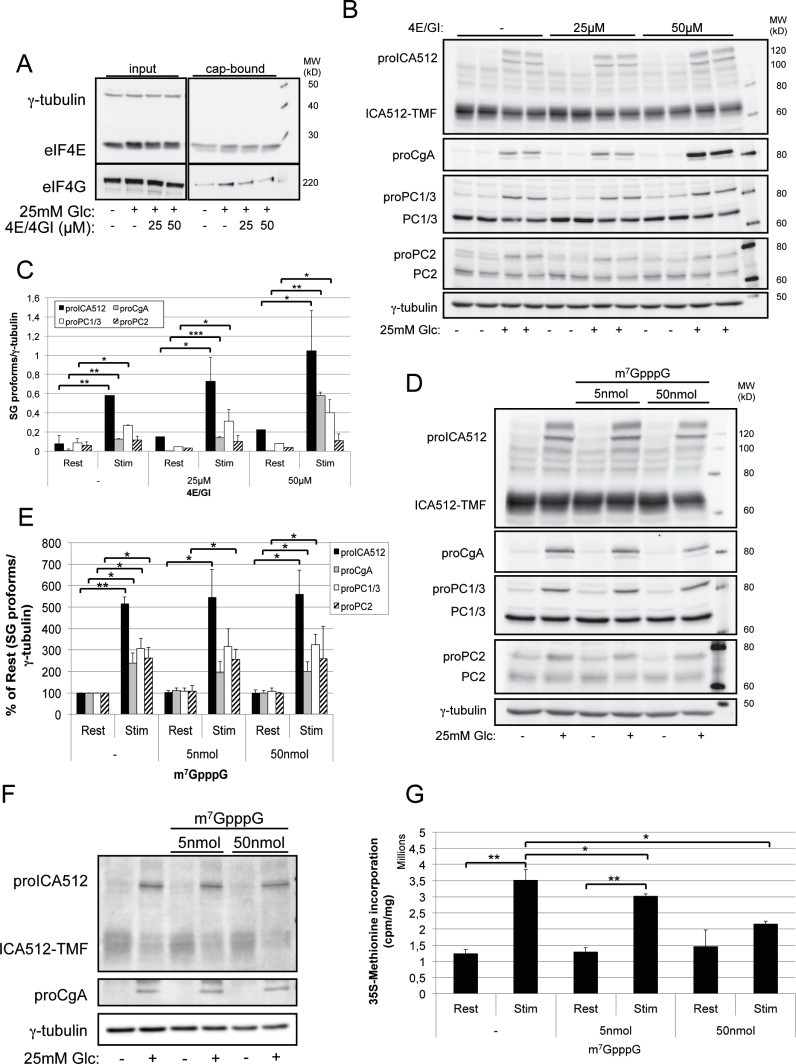
: Inhibition of the eIF4E/eIF4G binding does not affect glucose-stimulated translation of SG proteins in insulinoma cell lines. (A) Immunoblots for eIF4E, eIF4G and γ-tubulin recovered in cap-binding assays with m7GTP-sepharose beads from extracts of MIN6 cells treated or not with the eIF4E/4G inhibitor for 1 h prior to glucose-stimulation. (B) Immunoblots for SG proteins and γ-tubulin in MIN6 cells pretreated with the eIF4E/4G inhibitor prior to glucose-stimulation. (C) Quantification of SG precursor proteins as detected in (B). (D) Immunoblots for SG proteins and γ-tubulin in MIN6 cells transfected with the m7GpppG cap-analog prior to glucose-stimulation. (E) Quantification of SG precursor proteins as detected in (D) (n = 4). (F) Immunoblots for ICA512, proCgA and γ-tubulin in glucose-stimulated, m7GpppG-transfected INS-1 cells. (G) Glucose-stimulated total protein biosynthesis as measured by 35S-methionine incorporation in m7GpppG-transfected INS-1 (n = 3).
Figure S5.
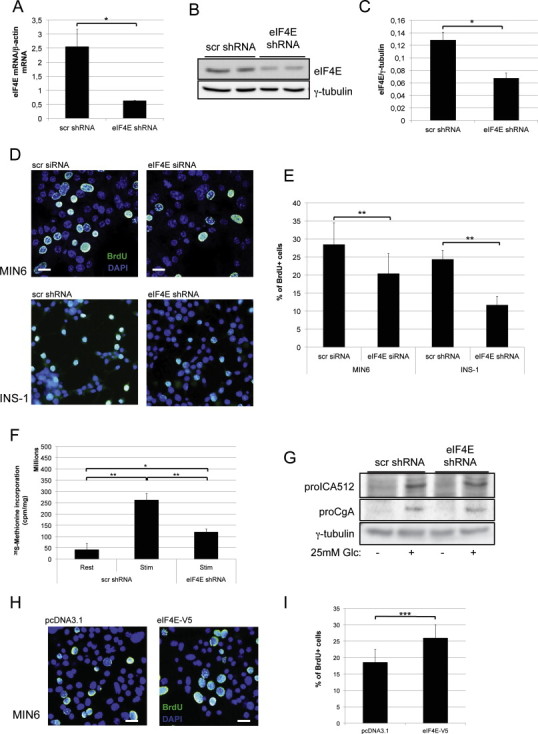
: Modifications of eIF4E expression do not reduce glucose-stimulated translation of SG proteins in insulinoma cell lines. INS-1 cells were analyzed 4 days after transfection with scrambled (scr) shRNA or eIF4E shRNA. (A) Quantification of eIF4E mRNA levels by qPCR (n = 5). (B) Immunoblots for eIF4E and γ-tubulin. (C) Quantification of the immunoblots for eIF4E as shown in (B) (n = 5). (D) Confocal microscopy of MIN6 cells and INS-1 cells incubated with BrdU (green) for 1 h. Nuclei were counterstained with DAPI (blue). (E) Quantification of BrdU+ MIN6 and INS-1 cells as detected in (D) (n = 4). (F) Total protein biosynthesis in INS-1 cells as measured by 35S-methionine incorporation (n = 3). (G) Immunoblots for proICA512, proCgA and γ-tubulin in INS-1 cells transfected with scrambled or eIF4E shRNAs prior to glucose-stimulation. (H) MIN6 cells were analyzed 4 days after transfection of V5-tagged eIF4E in pcDNA3.1 or of the pcDNA3.1 vector alone. Confocal microscopy of MIN6 cells incubated with BrdU (green). Nuclei were counterstained with DAPI (blue). Scale bars: 20 μm. (I) Quantification of BrdU+ cells (n = 4).
References
- 1.Schatz H., Nierle C., Pfeiffer E.F. (Pro-) insulin biosynthesis and release of newly synthesized (pro-) insulin from isolated islets of rat pancreas in the presence of amino acids and sulphonylureas. European Journal of Clinical Investigation. 1975;5(6):477–485. doi: 10.1111/j.1365-2362.1975.tb00480.x. [DOI] [PubMed] [Google Scholar]
- 2.Halban P.A. Differential rates of release of newly synthesized and of stored insulin from pancreatic islets. Endocrinology. 1982;110(4):1183–1188. doi: 10.1210/endo-110-4-1183. [DOI] [PubMed] [Google Scholar]
- 3.Ivanova A., Kalaidzidis Y., Dirkx R., Sarov M., Gerlach M., Schroth-Diez B. Age-dependent labeling and imaging of insulin secretory granules. Diabetes. 2013;62(11):3687–3696. doi: 10.2337/db12-1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tillmar L., Carlsson C., Welsh N. Control of insulin mRNA stability in rat pancreatic islets. Regulatory role of a 3′-untranslated region pyrimidine-rich sequence. Journal of Biological Chemistry. 2002;277(2):1099–1106. doi: 10.1074/jbc.M108340200. [DOI] [PubMed] [Google Scholar]
- 5.Knoch K.-P., Bergert H., Borgonovo B., Saeger H.-D., Altkrüger A., Verkade P. Polypyrimidine tract-binding protein promotes insulin secretory granule biogenesis. Nature Cell Biology. 2004;6(3):207–214. doi: 10.1038/ncb1099. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Blanco M.A., Jamison S.F., Sharp P.A. Identification and purification of a 62,000-dalton protein that binds specifically to the polypyrimidine tract of introns. Genes & Development. 1989;3(12):1874–1886. doi: 10.1101/gad.3.12a.1874. [DOI] [PubMed] [Google Scholar]
- 7.Kafasla P., Mickleburgh I., Llorian M., Coelho M., Gooding C., Cherny D. Defining the roles and interactions of PTB. Biochemical Society Transactions. 2012;40(4):815–820. doi: 10.1042/BST20120044. [DOI] [PubMed] [Google Scholar]
- 8.Lou H., Helfman D.M., Gagel R.F., Berget S.M. Polypyrimidine tract-binding protein positively regulates inclusion of an alternative 3′-terminal exon. Molecular and Cellular Biology. 1999;19(1):78–85. doi: 10.1128/mcb.19.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jang S.K., Wimmer E. Cap-independent translation of encephalomyocarditis virus RNA: structural elements of the internal ribosomal entry site and involvement of a cellular 57-kD RNA-binding protein. Genes & Development. 1990;4(9):1560–1572. doi: 10.1101/gad.4.9.1560. [DOI] [PubMed] [Google Scholar]
- 10.Knoch K.-P., Meisterfeld R., Kersting S., Bergert H., Altkrüger A., Wegbrod C. cAMP-dependent phosphorylation of PTB1 promotes the expression of insulin secretory granule proteins in beta cells. Cell Metabolism. 2006;3(2):123–134. doi: 10.1016/j.cmet.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Ehehalt F., Knoch K.-P., Erdmann K., Krautz C., Jäger M., Steffen A. Impaired insulin turnover in islets from type 2 diabetic patients. Islets. 2010;2(1):30–36. doi: 10.4161/isl.2.1.10098. [DOI] [PubMed] [Google Scholar]
- 12.Fred R.G., Welsh N. The importance of RNA binding proteins in preproinsulin mRNA stability. Molecular and Cellular Endocrinology. 2008;15(2):28–33. doi: 10.1016/j.mce.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Heni M., Ketterer C., Wagner R., Linder K., Böhm A., Herzberg-Schäfer S.A. Polymorphism rs11085226 in the gene encoding polypyrimidine tract-binding protein 1 negatively affects glucose-stimulated insulin secretion. PLOS One. 2012;7(10):e46154. doi: 10.1371/journal.pone.0046154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell S.A., Spriggs K.A., Bushell M., Evans J.R., Stoneley M., Le Quesne J.P. Identification of a motif that mediates polypyrimidine tract-binding protein-dependent internal ribosome entry. Genes & Development. 2005;19(13):1556–1571. doi: 10.1101/gad.339105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoneley M., Willis A.E. Cellular internal ribosome entry segments: structures, trans-acting factors and regulation of gene expression. Oncogene. 2004;23(18):3200–3207. doi: 10.1038/sj.onc.1207551. [DOI] [PubMed] [Google Scholar]
- 16.Kafasla P., Morgner N., Pöyry T.A.A., Curry S., Robinson C.V., Jackson R.J. Polypyrimidine tract binding protein stabilizes the encephalomyocarditis virus IRES structure via binding multiple sites in a unique orientation. Molecular Cell. 2009;34(5):556–568. doi: 10.1016/j.molcel.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 17.Vagner S., Galy B., Pyronnet S. Irresistible IRES. Attracting the translation machinery to internal ribosome entry sites. European Molecular Biology Organization Reports. 2001;2(10):893–898. doi: 10.1093/embo-reports/kve208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts L., Holcik M. RNA structure: new messages in translation, replication and disease. Workshop on the role of RNA structures in the translation of viral and cellular RNAs. European Molecular Biology Organization Reports. 2009;10(5):449–453. doi: 10.1038/embor.2009.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belsham G.J., Sonenberg N. Picornavirus RNA translation: roles for cellular proteins. Trends in Microbiology. 2000;8(7):330–335. doi: 10.1016/s0966-842x(00)01788-1. [DOI] [PubMed] [Google Scholar]
- 20.Schneider R.J., Mohr I. Translation initiation and viral tricks. Trends in Biochemical Sciences. 2003;28(3):130–136. doi: 10.1016/S0968-0004(03)00029-X. [DOI] [PubMed] [Google Scholar]
- 21.Yeung W.C., Rawlinson W.D., Craig M.E. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ. 2011;342:d35. doi: 10.1136/bmj.d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grieco F.A., Sebastiani G., Spagnuolo I., Patti A., Dotta F. Immunology in the clinic review series; focus on type 1 diabetes and viruses: how viral infections modulate beta cell function. Clinical and Experimental Immunology. 2012;168(1):24–29. doi: 10.1111/j.1365-2249.2011.04556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dotta F., Censini S., van Halteren A.G.S., Marselli L., Masini M., Dionisi S. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(12):5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roivainen M., Klingel K. Virus infections and type 1 diabetes risk. Current Diabetes Reports. 2010;10:350–356. doi: 10.1007/s11892-010-0139-x. [DOI] [PubMed] [Google Scholar]
- 25.Mziaut H., Trajkovski M., Kersting S., Ehninger A., Altkrüger A., Lemaitre R.P. Synergy of glucose and growth hormone signalling in islet cells through ICA512 and STAT5. Nature Cell Biology. 2006;8(5):435–445. doi: 10.1038/ncb1395. [DOI] [PubMed] [Google Scholar]
- 26.Gotoh M., Maki T., Kiyoizumi T., Satomi S., Monaco A.P. An improved method for isolation of mouse pancreatic islets. Transplantation. 1985;40:437–438. doi: 10.1097/00007890-198510000-00018. [DOI] [PubMed] [Google Scholar]
- 27.Ishihara H., Asano T., Tsukuda K., Katagiri H., Inukai K., Anai M. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia. 1993;36(11):1139–1145. doi: 10.1007/BF00401058. [DOI] [PubMed] [Google Scholar]
- 28.Schubert S., Knoch K.-P., Ouwendijk J., Mohammed S., Bodrov Y., Jäger M. β2-Syntrophin is a Cdk5 substrate that restrains the motility of insulin secretory granules. PLoS One. 2010;5(9):e12929. doi: 10.1371/journal.pone.0012929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fred R.G., Sandberg M., Pelletier J., Welsh N. The human insulin mRNA is partly translated via a cap- and eIF4A-independent mechanism. Biochemical and Biophysical Research Communication. 2011;412(4):693–698. doi: 10.1016/j.bbrc.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 30.Al-Hello H., Ylipaasto P., Smura T., Rieder E., Hovi T., Roivainen M. Amino acids of coxsackie B5 virus are critical for infection of the murine insulinoma cell line, MIN-6. Journal of Medical Virology. 2009;81(2):296–304. doi: 10.1002/jmv.21391. [DOI] [PubMed] [Google Scholar]
- 31.Morley S.J., Pain V.M. Translational regulation during activation of porcine peripheral blood lymphocytes: association and phosphorylation of the alpha and gamma subunits of the initiation factor complex eIF-4F. Biochemical Journal. 1995;312:627–635. doi: 10.1042/bj3120627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez I., Lin C.-H., McAfee J.G., Patton J.G. Mutation of PTB binding sites causes mirsregulation of alternative 3′- splice site selection in vivo. RNA. 1997;3:764–778. [PMC free article] [PubMed] [Google Scholar]
- 33.Clerte C., Hall K.B. The domains of polypyrimidine tract binding protein have distinct RNA structural preferences. Biochemistry. 2009;48(10):2063–2074. doi: 10.1021/bi8016872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graber T.E., Holcik M. Cap-independent regulation of gene expression in apoptosis. Molecular Biosystems. 2007;4(1):825–834. doi: 10.1039/b708867a. [DOI] [PubMed] [Google Scholar]
- 35.Gilbert W.V. Alternative ways to think about cellular internal ribosome entry. Journal of Biological Chemistry. 2010;285(38):29033–29038. doi: 10.1074/jbc.R110.150532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoneley M., Spencer J.F., Wright S.C. An internal ribosome entry segment in the 5′ untranslated region of the mnt gene. Oncogene. 2001;15(7):893–897. doi: 10.1038/sj.onc.1204157. [DOI] [PubMed] [Google Scholar]
- 37.Srinivasan S., Bernal-Mizrachi E., Ohsugi M., Permutt M.A. Glucose promotes pancreatic islet β-cell survival through a PI 3-kinase/Akt-signaling pathway. American Journal of Physiology. Endocrinology and Metabolism. 2002;283(4):784–793. doi: 10.1152/ajpendo.00177.2002. [DOI] [PubMed] [Google Scholar]
- 38.Ohsugi M., Cras-Méneur C., Zhou Y., Bernal-Mizrachi E., Johnson J.D., Luciani D.S. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. Journal of Biological Chemistry. 2005;280(6):4992–5003. doi: 10.1074/jbc.M411727200. [DOI] [PubMed] [Google Scholar]
- 39.Li I., Fan S., Koo J., Yue P., Chen Z., Owonikoko T.K. Elevated expression of eukaryotic translation initiation factor 4E is associated with proliferation, invasion and acquired resistance to erlotinib in lung cancer. Cancer Biology & Therapy. 2012;13(5):272–280. doi: 10.4161/cbt.18923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verma H., Bhattacharyya S., Das S. Polypyrimidine tract-binding protein interacts with coxsackievirus B3 RNA and influences its translation. Journal of General Virology. 2010;91(5):1245–1255. doi: 10.1099/vir.0.018507-0. [DOI] [PubMed] [Google Scholar]
- 41.Bachar-Wikstrom E., Wikstrom J.D., Ariav Y., Tirosh B., Kaiser N., Cerasi E. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes. 2013;62(4):1227–1237. doi: 10.2337/db12-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seewaldt S., Thomas H.E., Ejrnaes M., Christen U., Wolfe T., Rodrigo E. Virus-induced autoimmune diabetes most β-cells die through inflammatory cytokines and not perforin from autoreactive cytotoxic T-lymphocytes. Diabetes. 2000;49(11):1801–1809. doi: 10.2337/diabetes.49.11.1801. [DOI] [PubMed] [Google Scholar]
- 43.Filippi C.M., von Herrath M.G. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: viruses, autoimmunity and immunoregulation. Clinical and Experimental Immunology. 2010;160(1):113–119. doi: 10.1111/j.1365-2249.2010.04128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lipson K.L., Fonseca S.G., Ishigaki S., Nguyen L.X., Foss E., Bortell R. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metabolism. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 45.Solimena M. Vesicular autoantigens of type 1 diabetes. Diabetes Metabolism Reviews. 1998;14(3):227–240. doi: 10.1002/(sici)1099-0895(1998090)14:3<227::aid-dmr215>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 46.Krautz C., Wolk S., Steffen A., Knoch K.-P., Ceklareg U., Thiery J. Effect of immunosuppression on alpha and beta cells renewal in transplanted mouse islets. Diabetologia. 2013;56(7):1596–1604. doi: 10.1007/s00125-013-2895-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PTBP1 binding to the 5′-UTRs of mouse and rat mRNAs encoding SG proteins is glucose-stimulated. PTBP1 binding to mRNA 5′-UTRs of mouse mRNAs encoding SG proteins is glucose-stimulated (A) Left panel: schemes of the 5′-UTRs of mouse insulin2, ICA512, PC2 and γ-tubulin mRNAs which were synthesized by in vitro transcription and used for in vitro RNA binding assays. Polypyrimidine tracts are shown as black boxes. Right panel: immunoblottings for PTBP1 recovered with the corresponding mRNA 5′-UTRs from extracts of MIN6 cells stimulated or not with glucose. (B) To verify specificity, the binding of PTBP1 was competed with increasing amounts of the corresponding non-biotinylated 5′-UTR. (C) Alignment of the extended sequence of rat ICA512 mRNA 5′-UTR obtained by 5′-RACE based on sequences for rat ICA512 available in public databases. The alignment was obtained using the software ClustralW at <http://www.ch.EMBnet.org>. The red box indicates the putative PTBP1-binding motif. The translation start is indicated in bold and italic. (D) Quantification of immunoblots for PTBP1 recovered with in vitro RNA binding assays as shown in Figure 1D (n = 6). (E) Immunoblotting for PTBP1 and γ-tubulin and quantification (F) using INS-1 extracts 4 days after transfection with indicated shRNA vectors. (G) The ratios between firefly and renilla luciferase mRNAs in INS-1 cells transfected with different pGL3-B-RL vectors were assessed by qPCR 3 days after knockdown of renilla luciferase (n = 3). β-actin mRNA levels were used for normalization.
: Rapamycin and LY294002 treatment inhibits glucose-dependent phosphorylation of AKT and S6K1 but not glucose-stimulated translation of SG proteins in insulinoma cell lines. MIN6 cells were incubated with Rapamycin or LY294002 for 1 h before being glucose-stimulated. (A) Immunoblots for AKT, phospho-AKT, p70S6 kinase, phospho-p70S6 kinase and γ-tubulin. (B) Immunoblots for eIF4E and eIF4G recovered with m7GTP-sepharose beads (cap-binding assay) from extracts of MIN6 cells treated as indicated. (C, D) Immunoblots for SG proteins and γ-tubulin (C) and quantification of the SG preforms (D) of the respective signals (n = 4) in MIN6 cells treated as indicated. (E) Immunoblots for proICA512, proCgA and γ-tubulin on extracts of INS-1 cells treated with Rapamycin or LY294002 for 1 h before glucose-stimulation. (F) Immunoblots for SG proteins and γ-tubulin in MIN6 cells treated with 10 μg/ml cycloheximide for 1 h before glucose-stimulation. (G) Quantification of SG precursor proteins as detected in (A) (n = 3).
: CVB5 infection does not induce apoptosis of MIN6 cells but degranulation. MIN6 cells were analyzed for apoptosis 4 days after infection with CVB5 MCA. Alternatively, as a positive control for apoptosis, cells were treated with the inducer of apoptosis Staurosporin. (A) Immunostainings for VP1 (red) and for apoptosis by TUNEL assay (green). Nuclei were counterstained with DAPI (blue). Scale bars: 20 μm. (B) Detection of Caspase 3 and PARP cleavage as biomarkers of apoptosis by immunoblotting. (C) Confocal microscopy for VP1 (green) and insulin (red) immunostainings in MIN6 cells 4 days after infection with CVB5 MCA. Nuclei were counterstained with DAPI (blue). Right panel: magnification of the inset area on the left. Scale bars: left panels 20 μm; right panel: 10 μm. (D) Accumulation of SG precursor proteins in CVB5 infected MIN6 cells treated with furin inhibitor 1. Immunoblottings for VP1, γ-tubulin and SGs markers ICA512, proCgA, PC1/3, PC2 in CVB5-infected or control MIN6 cells treated with furin inhibitor 1 for 1 h before stimulation with glucose for 2 h. (E) Insulin amounts in media of MIN6 cells stimulated with glucose for 2 h (n = 3).