

Abstract
Oxidative stress is a hallmark of Alzheimer’s disease (AD). We propose that rather than causing damage because of the action of free radicals, oxidative stress deranges signaling pathways leading to tau hyperphosphorylation, a hallmark of the disease. Indeed, incubation of neurons in culture with 5 µM beta-amyloid peptide (Aβ) causes an activation of p38 MAPK (p38) that leads to tau hyperphosphorylation. Inhibition of p38 prevents Aβ-induced tau phosphorylation. Aβ-induced effects are prevented when neurons are co-incubated with trolox (the water-soluble analog of vitamin E).
We have confirmed these results in vivo, in APP/PS1 double transgenic mice of AD. We have found that APP/PS1 transgenic mice exhibit a high level of P-p38 in the hippocampus but not in cortex and this is prevented by feeding animals with a diet supplemented with vitamin E.
Our results underpin the role of oxidative stress in the altered cell signaling in AD pathology and suggest that antioxidant prevention may be useful in AD therapeutics.
Keywords: Vitamin E, Antioxidant, Beta-amyloid, P-p38
Introduction
The p38 MAPK is a 38 kD polypeptide with 4 isoforms (α, β, γ, δ) all of which are activated by dual phosphorylation at Thr180 and Tyr182 residues [1]. Activated p38 phosphorylates serine and threonine residues in a great variety of substrates, mostly kinases and transcription factors. In Alzheimer’s disease (AD), increased p38 MAPK activity in human brains was observed more than a decade ago [2]. Post-mortem brains of AD patients revealed that phospho-p38 MAPK immune reactivity occurs at a very early stage of the disease [3,4].
In AD, tau, which is a target of p38, turns into a hyperphosphorylated state by the action of several kinases one of which is p38 [5–7]. The link between p38, tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer’s was shown by Zhu et al. [8]. Studies using transgenic mice exhibiting hyperphosphorylated tau also show positive correlation between phosphorylated (activated) p38 and the level of aggregated tau [9]. We previously established that Aβ produces free radicals by binding to heme which interferes with the respiratory chain and subsequently increases peroxide production in mitochondria [10]. Thus, the aim of our work is to show if p38 links Aβ-induced oxidative stress with tau hyperphosphorylation both in vitro (primary neurons incubated with Aβ) and in vivo (an animal model of AD, double transgenic for amyloid precursor protein/presenilin1). We also checked the possible beneficial effects of vitamin E in the prevention of Aβ-associated damage. Vitamin E is a powerful antioxidant but its beneficial effects in AD are not clear. Our results showed that P-p38 increases after Aβ treatment. We also found that the increased tau hyperphosphorylation caused by Aβ is dependent on p38 as it is prevented by specific inhibition of this kinase. Finally, we showed that incubation with trolox (the soluble vitamin E analog) prevents Aβ-induced p38 activation. Furthermore, supplementation of mice chow with vitamin E for 21 days prevents p38 activation in APP/PS1 mice.
Materials and methods
Primary culture of cortical neurons
Primary cultures of rat cortical neurons were prepared from the cerebral cortex of 14- or 15-day-old rat fetuses. Briefly, the cerebral cortex of fetuses obtained under sterile conditions was dissected and dissociated mechanically, by pipetting 10 times with a 10 mL pipette in DMEM (Gibco Invitrogen Corporation, Barcelona, Spain). The cell suspension was filtered through a nylon mesh with a pore size of 90 μm. Cell suspensions were plated (5 × 104 cells/cm2) on poly-l-lysine-coated dishes. After attachment of the cells (1 h), the plating medium was changed to DMEM containing 10% FBS supplemented with antibiotics (1%) and fungizone (1%). Cultures were grown in a humidified atmosphere of 5% CO2/95% air at 37 °C for three days. Cells were then exposed to cytosine-β-d-arabinofuranoside (5 μM) for 24 h to inhibit proliferation of non-neuronal cells. The purity of neurons and possible contamination by astrocytes were assessed by immunofluorescence using anti-GFAP and anti-MAP-2 antibodies. Under our isolation conditions, 99% of the cells were neurons.
Cell treatment
Four days after seeding, cells were treated with 5 µM soluble Aβ(1-42). Briefly, soluble Aβ(1-42) peptides were dissolved as follows: 1 mg of peptide was dissolved in 100 µl 1% NH4OH and 2100 µl of sterile PBS. (Gibco, Invitrogen Corporation, Barcelona, Spain), and incubated for 24 h at 4 °C. Cells were also treated with 1 mM of Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid), a water-soluble derivative of vitamin E (Sigma-Aldrich, St. Louis, MO).
To study the effect of inhibiting p38 on tau phosphorylation, cells were pre-incubated with 10 µM of SB203580 (Sigma-Aldrich, St. Louis, MO) for 30 min prior to the addition of 5 µM of Aβ(1-42).
Animal model
Nine APPswe, PSEN1dE9-85Dbo/J transgenic mice and nine wild type mice from the same colony aged nine months were used for this study. Mice were maintained individually under a 12:12-h dark-light cycle at 23 ± 1 °C and 60% relative humidity, and were provided with a standard chow diet (PANLAB S.L.) and water ad libitum. Control animals were fed a standard 2014 diet (Harlan laboratories) and the treated group was fed the same diet but enriched with 800 IU/kg of α-tocopheryl acetate. Both groups ate the same amount of food (data not shown).
The animals were anesthetized with inhalatory anesthesia (SEVOrane®) and then sacrificed. Hippocampus and cortex were isolated, freeze-clamped and stored at −80 °C. The tissues were lysed in cold lysis buffer for Western blot determinations (Tris: 76.5 mM; pH: 6.8; SDS: 2%; Glicerol: 10%; supplemented with 2 mM sodium orthovanadate and proteases inhibitor [Sigma-Aldrich]). Homogenates were sonicated on ice for 3 s and incubated for 10 min at 4 °C. Protein concentration in the samples was determined using the Lowry method.
All animal experimental procedures used in this study were approved by the Committee on Ethics in Research of the Faculty of Medicine, University of Valencia, Spain.
Western blotting analysis
Protein extracts from cultured neurons were mixed with equal volumes of sodium dodecyl sulphate (SDS) buffer [0.125 M Tris–HCl, pH 6.8, 2% SDS, 0.5% (v/v) 2-mercaptoethanol, 1% bromophenol blue, and 19% glycerol] and then boiled for 5 min. Proteins were separated by SDS- polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes which were incubated overnight at 4 °C with appropriate primary antibodies: anti-p38 (Cell Signaling), anti-p-p38 (Cell Signaling) and anti-p-tau 231 (Genscript). The protein levels of α-tubulin (Santa Cruz Biotechnology, Inc.) were used as a loading control. Thereafter, membranes were incubated with a secondary antibody for 1 h at room temperature. Specific proteins were visualized by using the enhanced chemiluminescence procedure as specified by the manufacturer (Amersham) and quantified by densitometry using a Bio-Rad scanning densitometer (Bio-Rad, Hercules, CA, USA).
Statistics
Measurements from individual cultures were always performed in triplicate. Results are expressed as mean ± SD. Statistical analysis was performed by the least-significant difference test, which consists of two steps: first an analysis of variance was performed. The null hypothesis was accepted for all numbers of those sets in which F was non-significant, at the level of p > 0.05. Second, the sets of data in which F was significant were examined by the modified t-test using p ≤ 0.05 as the critical limit. We used the Student T-test to compare two means in animal experiments and T-test for paired samples in experiments in neurons in culture. Our analysis of variance shows that samples did not follow a normal distribution and thus, we used the Mann–Whitney test for nonparametric samples.
Results
Vitamin E analogue, trolox, prevents p38 activation in Aβ treated neurons in primary culture
Incubation of neurons in culture with Aβ induced an activation of p38 (Fig. 1). This activation which was determined as the ratio between P-p38/total p38, was prevented by co-incubation with trolox.
Fig. 1.
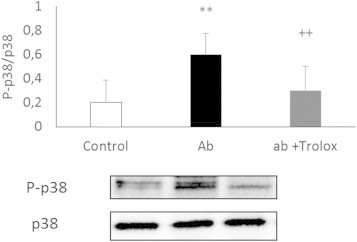
Trolox prevents Aβ dependent p38 activation in neurons in primary culture. Representative Western blots are shown. In all cases, values are means ± SD of 9 experiments. Data were normalized with tubulin values. **p < 0.01 compared to control values; ++p< 0.01 compared to values obtained with Aβ.
Vitamin E supplementation prevents the activation of p38 in AD mice in vivo
To determine if our findings in cells in culture could be confirmed in vivo we used the double transgenic mice APP/PS1. Our results showed that the hippocampus of AD mice expressed high levels of P-p38 compared to WT animals (Fig. 2A) and this was prevented when animals were fed with a diet supplemented with vitamin E (Fig. 2B). This increase did not occur in samples from brain cortex (data not shown).
Fig. 2.
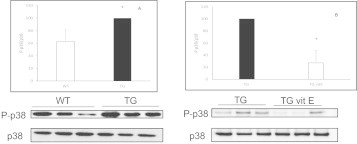
Vitamin E supplementation prevents activation of p38 in AD mice in vivo. Representative Western blots are shown. In all cases, values are means ± SD of 9 mice (A) or 6 mice (B). Data were normalized with tubulin values, *p < 0.05 compared to values found in wild type mice (WT) or in mice fed with standard diet (TG).
p38 activation leads to an increase in P-tau in neurons in primary culture. Prevention by trolox
Using a specific inhibitor of p38 (SB203580), we confirmed that p38 activation, leads to tau hyperphosphorylation. Fig. 3 shows that Aβ-induced tau hyperphosphorylation is p38 dependent as incubation with SB203580 prevents it. When we cultured neurons in the presence of Aβ we observed an increase in tau hyperphosphorylation that was prevented by trolox (see Fig. 4). We have also measured the effect of p38 inhibitor together with trolox and while both independently protect from Aβ toxicity, their effects are not additive, further suggesting that the protective effects of trolox may be mediated by p38.
Fig. 3.
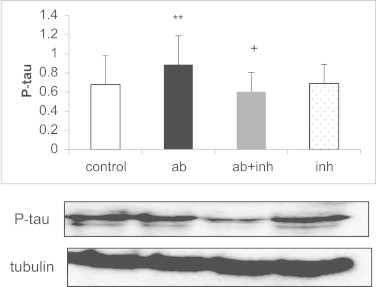
P38 activation leads to an increase in p-tau. Cultured neurons were pre-incubated with a p38 inhibitor (SB203580) for 30 min. Prior to the treatment with 5 µM of Aβ. Results showed that when p38 is inhibited there is a decrease in tau-phosphorylation compared to neurons without p38 inhibitor. Representative Western blots are shown. In all cases, values are means ± SD of 4 experiments. **p < 0.01 compared to control values. +p < 0.05 compared to values obtained with Aβ.
Fig. 4.
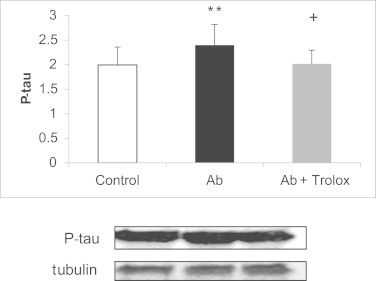
Effect of Aβ peptide and trolox in the expression of p-tau. Cultured neurons treated with 5 µM of Aβ showed significantly higher expression of p-tau (p < 0.05). Trolox reverted the Aβ-induced effects. Representative Western blots are shown. In all cases, values are means ± SD of 9 experiments. **p < 0.05 compared to control values. +p < 0.05 compared to values obtained with Aβ.
P-tau in the double transgenic murine model of AD
The content of P-tau in the hippocampus of AD mice is significantly higher than that of controls (see Fig. 5). This difference does not occur in cortex (data not shown). When we fed animals a diet supplemented with vitamin E we did not find a decrease in P-tau.
Fig. 5.
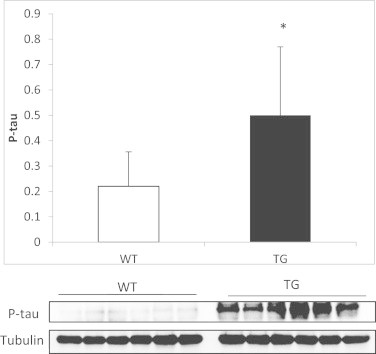
P-tau in AD mice model. P-tau was higher in the hippocampus of AD transgenic mice compared to wild type mice. Representative Western blots are shown. In all cases, values are means ± SD of 9 experiments. *p < 0.05 compared to WT animals.
Discussion
Amyloid β toxicity and tau phosphorylation are related via oxidative stress and p38 activation
Patients that have developed full-blown AD suffer from oxidative stress. This has been shown in post-mortem brain samples. A molecular response to oxidative stress is the phosphorylation, and thus activation, of p38 [11]. To study the possible role of p38 in the relationship between Aβ toxicity and tau phosphorylation, we used fetal rat neurons in culture. We incubated them with Aβ and found a significant increase in p38 and tau phosphorylation. This activation was prevented by co-incubation with trolox, the soluble vitamin E analogue (see Fig. 1). To show that Aβ-induced tau phosphorylation occurred due to the activity of p38, we inhibited it and observed a prevention of tau phosphorylation induced by Aβ (Fig. 3).
In the previous years, the two major pathophysiological mechanisms associated with the development of AD, namely Aβ toxicity and tau hyperphosphorylation were seen as independent factors [11]. Recent evidence, including some from our own laboratory, has shown that this may not be the case. Indeed, it was previously shown that glycogen synthase kinase 3 (GSK3β) can be activated by the presence of Aβ and can itself phosphorylate tau. We further developed the concept that Aβ and tau are mechanistically related and showed that Aβ causes oxidative stress [12–14]. The group of Davies showed that oxidative stress results in increased expression of an adaptive enzyme formally called Adapt-78 and now known as RCAN1 which is an inhibitor of calcineurin [15], the phosphatase that dephosphorylates Tau. We described a role of the oxidative stress-induced upregulation of RCAN1 in AD [16]. We now propose that Aβ causes an increase in oxidative stress that leads to phosphorylation of p38 which is a kinase that uses tau (in its T231 residue) as a substrate. Thus, Aβ toxicity can be mechanistically linked to an increased phosphorylation of tau via p38 as we show here.
We have shown that p38 is involved in linking Aβ and tau, not only in neurons in culture but also in vivo using the double transgenic model of AD (APP/PS1). This is a good model of AD which, for instance shows Aβ plaques and also tau hyperphosphorylation. It is well-known from clinical autopsies that the cellular damage that occurs in AD is not uniform. Some structures like the hippocampus are very heavily damaged whereas others, like the motor, somatic sensory, and primary visual areas are spared [17]. When we measured activation of p38, in hippocampal cells and cortex we found that it is activated in hippocampal cells but not in other cortex areas and that this follows a pattern very similar to that of tau phosphorylation. The similar topology of activation of p38 and phosphorylation of tau is a further indication that p38 may be very important in linking Aβ toxicity with tau hyperphosphorylation.
The role of the antioxidant properties of vitamin E in the treatment of Alzheimer’s disease
The work of Sano et al. [18] gave impetus to the idea of vitamin E as the treatment of AD. Recently, the same group has reported that vitamin E benefits also patients with mild to moderate Alzheimer’s disease by slowing functional decline [19]. However there are a number of studies questioning the efficacy of vitamin E treatment [20,21]. We reported that the high variability of results on the effect of vitamin E in Alzheimer’s is due to differences in the individual response to its antioxidant properties [22]. Vitamin E does not act as an antioxidant equally in all patients. Only in those in whom the vitamin lowered the oxidation of blood glutathione and the peroxidation of plasma lipids did vitamin E cause an improvement in Alzheimer’s disease. We suggest that there are two sub-populations who react differently to treatment with vitamin E: patients for whom vitamin E does act as an antioxidant, react favorably to treatment, especially in terms of preventing functional decline, but on the other hand, other patients for whom vitamin E does not act as an antioxidant, do not respond favorably to treatment with this vitamin. The paradox of vitamin E is that it is efficient for some patients, but not for others [22].
Because of the critical importance of reactive oxygen species in abnormal signaling in AD [10] we tested the effect of p38 signaling in AD and the protection by the vitamin E analogue, trolox. Vitamin E acts not only as an antioxidant but also as modulator of several cell signaling pathways, in fact it can act as a modulator of gene expression. Recently it has been shown a significant increase in mRNA levels of the scavenger receptor CD36 in aortae of cholesterol fed rabbits and shown that vitamin E treatment attenuated increased CD36 mRNA expression [23]. Furthermore, CD36 mediates the upregulation of Aβ toxicity by lipid peroxidation products [24].
On the other hand vitamin E inhibits the activation of p38, but this is mediated by a lowering of oxidative stress [25]. Our results indicate that oxidative stress is therefore central in the pathophysiology of the disease because it links Aβ with tau by various mechanisms, one of them shown in this paper (see schematic summary in Fig. 6) and that seriously preventing it (and not just giving antioxidant vitamins to all patients [22]) may be a good therapeutic approach if applied in the early stages of the disease.
Fig. 6.
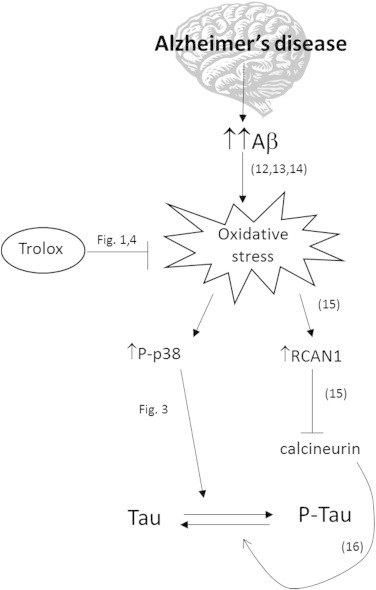
Schematic representation of the role of oxidative stress linking Aβ and tau toxicities. Numbers near the arrows indicate the pertinent reference in the reference list. Figures refer to those in the paper.
Conflicts of interest
The authors declare that no conflict of interest exists.
Acknowledgment
This work was supported by grants SAF2010-19498, from the Spanish Ministry of Education and Science (MEC); ISCIII2006-RED13-027 and ISCIII2012-RED-43-029 from the “Red Tematica de investigacion cooperativa en envejecimiento y fragilidad” (RETICEF); PROMETEO2010/074 from “Conselleria d’Educació, Cultura i Esport de la Generalitat Valenciana”; 35NEURO GentxGent from “Fundació Gent Per Gent de la Comunitat Valenciana”; RS2012-609 Intramural Grant from INCLIVA and EU Funded CM1001 and FRAILOMIC-HEALTH.2012.2.1.1-2. This study has been co-financed by FEDER funds from the European Union. E. G. was beneficiary of a Sara Borrel grant.
References
- 1.Ashwell J.D. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nature Reviews. Immunology. 2006;6:532–540. doi: 10.1038/nri1865. 16799472 [DOI] [PubMed] [Google Scholar]
- 2.Hensley K., Floyd R.A., Zheng N.Y., Nael R., Robinson K.A., Nguyen X., Pye Q.N., Stewart C.A., Geddes J., Markesbery W.R. p38 kinase is activated in the Alzheimer’s disease brain. Journal of Neurochemistry. 1999;72:2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x. 10217284 [DOI] [PubMed] [Google Scholar]
- 3.Pei J.J., Braak E., Braak H., Grundke-Iqbal I., Iqbal K., Winblad B., Cowburn R.F. Localization of active forms of c-Jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. Journal of Alzheimer’s Disease. 2001;3:41–48. doi: 10.3233/jad-2001-3107. 12214071 [DOI] [PubMed] [Google Scholar]
- 4.Sun A., Liu M., Nguyen X.V., Bing G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Experimental Neurology. 2003;183:394–405. doi: 10.1016/s0014-4886(03)00180-8. 14552880 [DOI] [PubMed] [Google Scholar]
- 5.Sheng J.G., Jones R.A., Zhou X.Q., McGinness J.M., Van Eldik L.J., Mrak R.E., Griffin W.S. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: potential significance for Tau protein phosphorylation. Neurochemistry International. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. 11578769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feijoo C., Campbell D.G., Jakes R., Goedert M., Cuenda A. Evidence that phosphorylation of the microtubule-associated protein tau by SAPK4/p38δ at Thr50 promotes microtubule assembly. Journal of Cell Science. 2005;118:397–408. doi: 10.1242/jcs.01655. 15632108 [DOI] [PubMed] [Google Scholar]
- 7.Yoshida H., Goedert M. Sequential phosphorylation of Tau protein by cAMP-dependent protein kinase and SAPK4/p38 delta or JNK2 in the presence of heparin generates the AT100 epitope. Journal of Neurochemistry. 2006;99:154–164. doi: 10.1111/j.1471-4159.2006.04052.x. 16987243 [DOI] [PubMed] [Google Scholar]
- 8.Zhu X.M., Rottkamp C.A., Boux H.P., Takeda A.P., Perry G.P., Smith M.A.P. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2000;59:880–888. doi: 10.1093/jnen/59.10.880. 11079778 [DOI] [PubMed] [Google Scholar]
- 9.Kelleher I., Garwood C., Hanger D.P., Anderton B.H., Noble W. Kinase activities increase during the development of tauopathy in htau mice. Journal of Neurochemistry. 2007;103:2256–2267. doi: 10.1111/j.1471-4159.2007.04930.x. 17908241 [DOI] [PubMed] [Google Scholar]
- 10.Lloret A., Badía M.C., Mora N.J., Ortega A., Pallardó F.V., Alonso M.D., Atamna H., Viña J. Gender and age-dependent differences in the mitochondrial apoptogenic pathway in Alzheimer’s disease. Free Radical Biology & Medicine. 2008;44:2019–2025. doi: 10.1016/j.freeradbiomed.2008.02.017. 18387371 [DOI] [PubMed] [Google Scholar]
- 11.Pearson G., Robinson F., Beers Gibson T., Xu B.E., Karandikar M., Berman K., Cobb M.H. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrine Reviews. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. 11294822 [DOI] [PubMed] [Google Scholar]
- 12.Mudher A., Lovestone S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends in Neurosciences. 2002;25:22–26. doi: 10.1016/s0166-2236(00)02031-2. 11801334 [DOI] [PubMed] [Google Scholar]
- 13.Butterfield D.A., Yatin S.M., Varadarajan S., Koppal T. Amyloid beta-peptide-associated free radical oxidative stress, neurotoxicity, and Alzheimer’s disease. Methods in Enzymology. 1999;309:746–768. doi: 10.1016/s0076-6879(99)09050-3. 10507060 [DOI] [PubMed] [Google Scholar]
- 14.Cardoso S.M., Santana I., Swerdlow R.H., Oliveira C.R. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. Journal of Neurochemistry. 2004;89:1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. 15189344 [DOI] [PubMed] [Google Scholar]
- 15.Ermak G., Harris C.D., Davies K.J.A. The DSCR1 (Adapt78) isoform 1 protein calcipressin 1 inhibits calcineurin and protects against acute calcium-mediated stress damage, including transient oxidative stress. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2002;16:814–824. doi: 10.1096/fj.01-0846com. 12039863 [DOI] [PubMed] [Google Scholar]
- 16.Lloret A., Badia M.C., Giraldo E., Ermak G., Alonso M.D., Pallardó F.V., Davies K.J., Viña J. Amyloid-β toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2011;27:701–709. doi: 10.3233/JAD-2011-110890. 21876249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearson R.C., Esiri M.M., Hiorns R.W., Wilcock G.K., Powell T.P. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:4531–4534. doi: 10.1073/pnas.82.13.4531. 3859874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sano M., Ernesto C., Thomas R.G., Klauber M.R., Schafer K., Grundman M., Woodbury P., Growdon J., Cotman C.W., Pfeiffer E., Schneider L.S., Thal L.J. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s disease Cooperative Study. New England Journal of Medicine. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. 9110909 [DOI] [PubMed] [Google Scholar]
- 19.Dysken M.W., Sano M., Asthana S., Vertrees J.E., Pallaki M., Llorente M., Love S., Schellenberg G.D., McCarten J.R., Malphurs J., Prieto S., Chen P., Loreck D.J., Trapp G., Bakshi R.S., Mintzer J.E., Heidebrink J.L., Vidal-Cardona A., Arroyo L.M., Cruz A.R., Zachariah S., Kowall N.W., Chopra M.P., Craft S., Thielke S., Turvey C.L., Woodman C., Monnell K.A., Gordon K., Tomaska J., Segal Y., Peduzzi P.N., Guarino P.D. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. Journal of the American Medical Association. 2014;311:33–44. doi: 10.1001/jama.2013.282834. 24381967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farina, N., Isaac, M.G., Clark, A.R., Rusted, J., Tabet, N., Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database of Systematic Reviews, 11 (2012) CD002854, CD002854. Pubmed: 23152215 [DOI] [PMC free article] [PubMed]
- 21.Tabet N., Birks J., Evans J.G., Orrel M., Spector A. Vitamin E for Alzheimer’s disease. Cochrane Database of Systematic Reviews. 2000;4 doi: 10.1002/14651858.CD002854. CD002854. [DOI] [PubMed] [Google Scholar]
- 22.Lloret A., Badía M.C., Mora N.J., Pallardó F.V., Alonso M.D., Viña J. Vitamin E paradox in Alzheimer’s disease: it does not prevent loss of cognition and may even be detrimental. Journal of Alzheimer’s Disease. 2009;17:143–149. doi: 10.3233/JAD-2009-1033. 19494439 [DOI] [PubMed] [Google Scholar]
- 23.Bozaykut P., Karademir B., Yazgan B., Sozen E., Siow R.C., Mann G.E., Ozer N.K. Effects of vitamin E on peroxisome proliferator-activated receptor γ and nuclear factor-erythroid 2-related factor 2 in hypercholesterolemia induced atherosclerosis. Free Radical Biology & Medicine. 2014 doi: 10.1016/j.freeradbiomed.2014.02.017. 24583459 [DOI] [PubMed] [Google Scholar]
- 24.Testa G., Gamba P., Di Scipio F., Sprio A.E., Salamone P., Gargiulo S., Sottero B., Biasi F., Berta G.N., Poli G., Leonarduzzi G. Potentiation of amyloid-β peptide neurotoxicity in human dental-pulp neuron-like cells by the membrane lipid peroxidation product 4-hydroxynonenal. Free Radical Biology & Medicine. 2012;53:1708–1717. doi: 10.1016/j.freeradbiomed.2012.08.581. 22981873 [DOI] [PubMed] [Google Scholar]
- 25.Dolado I., Swat A., Ajenjo N., De Vita G., Cuadrado A., Nebreda A.R. p38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. 17292829 [DOI] [PubMed] [Google Scholar]