

Abstract
Emerging evidence indicates that mitochondrial cardiolipins (CL) are prone to free radical oxidation and this process appears to be intimately associated with multiple biological functions of mitochondria. Our previous work demonstrated that a significant amount of potent lipid electrophiles including 4-hydroxy-nonenal (4-HNE) was generated from CL oxidation through a novel chemical mechanism. Here we provide further evidence that a characteristic class of CL oxidation products, epoxyalcohol-aldehyde-CL (EAA-CL), is formed through this novel mechanism in isolated mice liver mitochondria when treated with the pro-apoptotic protein t-Bid to induce cyt c release. Generation of these oxidation products are dose-dependently attenuated by a peroxidase inhibitor acetaminophen (ApAP). Using a mouse model of atherosclerosis, we detected significant amount of these CL oxidation products in liver tissue of low density lipoprotein receptor knockout (LDLR −/−) mice after Western diet feeding. Our studies highlight the importance of lipid electrophiles formation from CL oxidation in the settings of apoptosis and atherosclerosis as inhibition of CL oxidation and lipid electrophiles formation may have potential therapeutic value in diseases linked to oxidant stress and mitochondrial dysfunctions.
Keywords: Cardiolipin, 4-hydroxy-2-nonenal (4-HNE), Epoxyalcohol-aldehyde-CL (EAA-CL), Mitochondria, Apoptosis, Lipid peroxidation, Atherosclerosis, Liquid chromatography–mass spectrometry (LC–MS)
Abbreviations: 4-ONE, 4-oxo-2-nonenal; 4-HNE, 4-hydroxy-nonena; ApAP, acetaminophen; ALDH2, aldehyde dehydrogenase-2; BHT, butylate hydroxytoluene; CL, cardiolipin cyt c cytochrome c; EAA-CL, epoxyalcohol-aldehyde-CL; ESI, electrospray; ETC, electron transport chain; H2O2, hydrogen peroxide; HODE, hydroxyoctadienoic acid; HpODE, hydroperoxyoctadecadienoic acid; KODE, keto-octadecadienoic acid; L4CL, tetralinoleoyl cardiolipin; L3OCL, trilinoleoyl oleoyl cardiolipin; LA, linoleic acid; LC–MS, liquid chromatography–mass spectrometry; LDLR −/−, low density lipoprotein receptor knockout; M4CL, tetramyristeoyl cardiolipin; MRM, multiple reaction monitoring; PHGPX, hospholipid hydroperoxide glutathione peroxidase; Prdx3/Prx3, peroxiredoxin 3; PUFAs, Polyunsaturated fatty acids; ROS, reactive oxygen species
Highlights
-
•
4-HNE and other electrophilic lipids are formed from mitochondrial cardiolipin.
-
•
Novel electrophilic oxidation products EAA-CL were identified in vitro and in vivo.
-
•
Level of EAA-CL in liver tissue of LDLR −/− mice is higher with Western diet feeding.
-
•
ApAP dose-dependently inhibits EAA-CL formation during t-Bid induced cyt c release.
-
•
CL electrophilic lipid formation is important in apoptosis and atherosclerosis.
Graphical Abstract
Introduction
Cardiolipin (CL) is a class of phospholipids that contain four fatty acid side chains and three glycerol backbones in the same molecule. CLs are a structurally unique class of phospholipids as phospholipids commonly have two fatty acids and one glycerol unit. CLs primarily reside in the inner membrane of mitochondria and are critical for maintaining the structural integrity of mitochondrial membranes and functions of multiple protein complexes in the electron transport chain (ETC) [1]. In most mammalian tissues, a majority of CLs contain four linoleic acid (LA, 18:2, ω-6) chains (tetralinoleoyl CL, L4CL). The fatty acid composition of CL appears to be an important factor for maintaining mitochondrial function [2]. Furthermore, mitochondrion is the major cellular source for reactive oxygen species (ROS) including superoxide and hydrogen peroxide (H2O2). Incorporation of four LA side chains in CLs and their association with mitochondria render CL to be readily oxidized by ROS [3]. The mechanisms for this seemingly paradoxical combination remain to be elucidated. Cardiolipin oxidation has attracted increased research attention in recent years and emerging evidence shows that it is critically involved in regulation of apoptosis [4], mitochondrial dysfunction, mitophagy [5], and several human diseases [6]. Upon apoptotic stimulation, CL interacts with cytochrome c (cyt c) to form a peroxidase complex that catalyzes CL oxidation. Accumulating evidence indicates that these oxidation products of CL play an important role in the mitochondrial stage of the cell death program [1,7].
Oxidative stress-induced lipid peroxidation has been linked to numerous human diseases including atherosclerosis [8,9]. Polyunsaturated fatty acids (PUFAs) such as LA in cell membranes are the primary targets for free radicals attack [8]. Lipid peroxidation generates an array of oxidation products and one class of these products, reactive lipid electrophiles, is increasingly recognized as an important lipid mediator due to its potential to alter protein structure and functions by covalent modification of certain critical nucleophilic amino acid residues. 4-Hydroxy-2-nonenal (4-HNE) is one of the most studied reactive lipid electrophiles and it can trigger multiple signaling pathways in different biological processes [10]. Elevated levels of 4-HNE were observed in atherosclerotic lesions and liver tissues and were positively correlated with cell death in these tissues [11,12]. In contrast to the well-studied pleiotropic biology of 4-HNE, mechanisms responsible for 4-HNE formation in vivo are much less understood [13,14]. Our previous in vitro work demonstrated that oxidation of L4CL by cyt c and H2O2 led to the formation of 4-HNE and other oxidation products via a novel chemical mechanism that involved cross-chain peroxyl radical addition and decomposition [16]. As shown in Fig. 1, oxidation of L4CL by the peroxidase activity of cyt c and CL complex in the presence of H2O2 results in the formation of hydroperoxyoctadecadienoic acid (HpODE). HpODE can be reduced to form hydroxyoctadienoic acid (HODE) or dehydrated to form keto-octadecadienoic acid (KODE). During this process, through intra-molecular peroxyl radical addition and decomposition of an unstable intermediate, several reactive aldehydes are produced including epoxyalcohol-aldehyde-CL (EAA-CL) (1 d), 4-HNE (1e), and 4-oxo-2-nonenal (4-ONE) (1f). 4-HNE and 4-ONE are diffusible electrophiles that can reach protein targets far away from the generation sites but EAA-CL most likely targets only closely associated proteins. Moreover, the formation of this unique EAA-CL can serve as a footprint for this novel chemical mechanism in vivo. However, the biological relevance of these lipid electrophiles EAA-CL from CL oxidation remains to be defined.
Fig. 1.
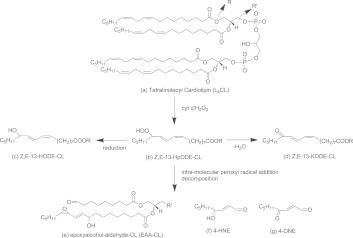
Proposed chemical mechanism for lipid electrophiles formation from L4CL oxidation.Only one regioisomer at carbon 13 (C13) is shown in the figure for simplicity.
In this study, we provide mass spectrometry (MS) evidence for this class of characteristic bioactive lipid electrophiles EAA-CL from cardiolipin oxidation in the context of apoptosis using t-Bid induced cyt c release in isolated mice liver mitochondria. Interestingly, we observed dose-dependent inhibition of CL oxidation and formation of EAA-CL by a widely used analgesic drug acetaminophen (ApAP) that also served as a peroxidase inhibitor [15]. Furthermore, the very mechanism appeared to operate in vivo in the settings of atherosclerosis in a mice model of Western diet-induced atherosclerosis using low density lipoprotein receptor knockout (LDLR −/−) mice. We found significant amounts of EAA-CL present in the mice liver after Western diet feeding. Taking these data together, our current study shed light on CL oxidation and lipid electrophiles production relevant to apoptosis and atherosclerosis, with inhibition of this process appearing to be a viable strategy for preventing cyt c release and apoptosis.
Methods and materials
Materials
Phospholipids, tetralinoleoyl cardiolipin (L4CL) and tetramyristeoyl cardiolipin (M4CL) were purchased from Avanti Polar Lipids (Alabaster, AL, USA) and used without further purification. All other chemicals were purchased from Sigma Aldrich Chemical Company (Milwaukee, WI). HPLC quality solvents, such as methanol, water, 2-propanol, and acetonitrile were purchased from either Fisher Chemical (Phillipsburg, NJ) or EM Science (Gibbstown, NJ).
Animals and diets
LDLR −/− mice were originally purchased from Jackson Laboratory. At 2–3 month of age, these mice were fed a Western diet (TD 88,137, Harlan Teklad, Madison, WI) containing 0.2% cholesterol and 21.2% fat or normal chow diet for 16 weeks. At the end of 16 weeks, the mice were food-deprived for 5 h and sacrificed by an overdose of isoflurane followed by cervical dislocation. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of Shanghai Institutes for Biological Sciences.
Oil Red O staining of liver sections
Oil red O staining of liver section was performed according to a literature procedure [16]. Briefly, liver samples were embedded in an optimal cutting temperature compound (Tissue-Tek), and were frozen at −20 °C. Liver sections were cut and stained with Oil Red O for 4 h. Sections were then counterstained with hematoxylin for 3 min. Images were captured using a Q-Imaging Micropublisher camera mounted on an Olympus upright microscope.
Quantification of oxidation products of CL by liquid chromatography–mass spectrometry (LC–MS)
The MS analysis of oxidized CL was performed according to our previously published methods [15,17]. After addition of 0.75% NaCl to the liver mitochondria pellet or homogenized liver tissue, the total lipids were extracted with chloroform and methanol (2:1, v:v) containing 0.1 mM butylated hydroxytoluene (BHT) and 0.1 mM triphenylphosphine. The separated organic phase was evaporated, re-suspended in methanol:acetonitrile:H2O (60/20/20, v/v/v) and stored at −80 °C until analysis by LC–MS. The extracted lipid fraction was separated online by UPLC using a Waters Acquity UPLC system (Waters Corp., Milford, MA). Mass spectrometry analysis was performed on a Thermo Quantum Ultra or Vantage triple quadrupole mass spectrometer (Thermo Scientific Inc., San Jose, CA, USA). The mass spectrometer was operated in the negative ion mode using selective reaction monitoring (SRM). Nitrogen was used as the sheath gas and auxiliary gas. The capillary temperature was set at 350 °C. The spray voltage was 4.5 kV, and the tube lens voltage was 100 V. The following transitions were monitored in SRM mode: M4CL, m/z 619.6–227.2; L4CL, m/z 723.6–279.2; L3OCL, m/z 724.6–279.2; HODE-L4CL, m/z 731.6–279.2, 295.2; HODE-L3OCL, m/z 732.6–279.2, 295.2; KODE-L4CL, m/z 730.6–279.2, 293.2; KODE-L3OCL, m/z 731.6–279.2, 293.2; EAA-L4CL, m/z 685.6–279.2; EAA-L3OCL, m/z 686.6–279.2. Data acquisition and analysis were performed using X’calibur software, version 2.0.
The area under the curve (AUC) for M4CL, CL, KODE-CL, HODE-CL, EAA-CL and MLCL was determined and the AUC of each CL in each sample was normalized to M4CL AUC. The corrected values were used to calculate the ratio of corresponding CL over the total CL and was expressed as percent of total CL. Data are presented as the mean ± SEM.
t-Bid induces CL oxidation in isolated mitochondria
Mouse liver mitochondria were isolated as previously described [15,18]. Fifteen microgram of mitochondria were incubated in 100 μl experimental buffer (250 mM sucrose, 10 mM Hepes, pH 7.4, 1 mM ATP, 5 mM succinate, 0.08 mM ADP, 1 mM dithiothreitol, and 2 mM K2HPO4, pH 7.4). After addition of 10 ng t-Bid, the samples were incubated for 30 min at room temperature. Mitochondria were pelleted by centrifugation at 8000 rpm for 10 min at 4 °C. The oxidation products of CL in the pellets were extracted as described above.
ApAP inhibits CL oxidation and in isolated mitochondria
Fifteen microgram of energized mitochondria were pre-incubated with 0–400 μM ApAP for 10 min. CL oxidation induced by t-Bid was processed as described above.
2.7. Statistical analysis
Results are expressed as means ± SD. Statistical analysis was performed using t tests. A probability value of p < 0.05 was considered statistically significant.
Results
Formation of Lipid electrophile EAA-CL from cardiolipin oxidation in vitro during t-Bid Induced cyt c release from mitochondria
Mitochondria play a central role in cell survival and apoptosis. The apoptotic pathways are highly orchestrated by pro- and anti-apoptotic Bcl-2 superfamily protein members. Bid is an abundant pro-apoptotic protein of the Bcl-2 family that is crucial for death receptor-mediated apoptosis in many cell types. It has been well established that t-Bid, a truncated form of Bid, facilitates the disruption of the mitochondrial trans-membrane potential and the release of apoptogenic proteins including cyt c. During this process, t-Bid also causes the remodeling of mitochondrial cristae and CL oxidation [19]. To test the biological relevance of lipid electrophile generation from CL oxidation during apoptosis, we treated mice liver mitochondria with t-Bid to induce cyt c release and identified the production of EAA-CL using mass spectrometry (shown in Fig. 2). We performed LC–MS analysis using the collision-induced dissociation (CID) of m/z 685 (doubly charged, Fig. 2A), the putative structure of EAA-CL, in the negative ion mode. A fragment with m/z 279 in the CID spectrum (Fig. 2B) was consistent with the presence of linoleate side chains while m/z 171 and 311 represented the presence of side chains of truncated aldehyde at C9 and epoxyalcohol-aldehyde LA moiety respectively. Other fragments such as m/z 1091.5 and 307 were consistent with the proposed structure. Thus our MS data provided unambiguous evidence for the formation of this novel lipid electrophile from CL oxidation. In addition, other oxidation products such as HODE-CL were also identified by CID experiment (data not shown). We further performed quantitative analysis of the major oxidation products using multiple reaction monitoring (MRM) by monitoring the transition of each parent ion to their respective characteristic fragmentation (Fig. 2C). The MRM results clearly demonstrated the formation of multiple oxidation products including EAA-CL and HODE-CL. Their elution order on a reverse phase LC column was consistent with the polarity of each compound. Furthermore, our previous work demonstrated that ApAP served as an inhibitor of the peroxidase activity of cyt c/CL complex. We then tested the ability of ApAP to inhibit the formation these reactive lipid electrophiles and our data clearly showed that ApAP dose-dependently attenuated the formation of EAA-CL and HODE-CL (Fig. 2D).
Fig. 2.
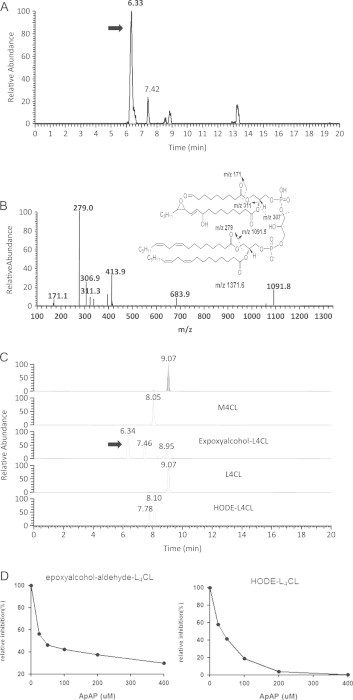
t-Bid induces EAA-L4CL formation in isolated mitochondria and ApAP dose-dependently inhibits L4CL oxidation and epoxyalcohol-aldehyde-CL formation.(A) Total ion chromatogram (TIC) of m/z 685.5 (doubly charged) in isolated mitochondria when treated with t-Bid. Arrow shows the putative EAA-CL peak. (B) MS spectrum of CID of m/z 685.5 at 6.33 min, EAA-CL. (C) MRM analysis of L4CL oxidation products in isolated mitochondria after t-Bid treatment. SRM transitions are shown in Methods. (D) ApAP inhibits L4CL oxidation and EAA-CL formation. L4CL oxidation products are expressed as % of relative inhibition.
Formation of lipid electrophiles from CL oxidation in liver tissue of LDLR KO mice
Oxidative stress is a hallmark of atherogenesis and free radical lipid peroxidation has been associated with every stage of this disease [20]. LDLR −/− is a well-established mouse model used to study atherosclerosis. 4-HNE was identified in mouse atherosclerotic lesions and liver, and levels of 4-HNE were associated with cell death in these tissues [21]. We employed this mouse model to test if EAA-CL and other cardiolipin oxidation products were formed in the liver during the formation of atherosclerotic plaques in the aorta. Western diet feeding for 16 weeks resulted in significant atherosclerotic lesion formation in the aortas as revealed by Red Oil O staining (Fig. 3A). We analyzed the liver tissues in these animals and identified multiple oxidation products from CL oxidation including 4-HNE, EAA-CL, KODE-CL, and HODE-CL (Fig. 3B). Furthermore, levels of EAA-CL from liver tissue of Western diet group were significantly elevated compared to that from the control group. These data represent the first in vivo evidence for the formation of these novel lipid electrophiles, EAA-CL, from mitochondrial cardiolipin during atherosclerosis.
Fig. 3.
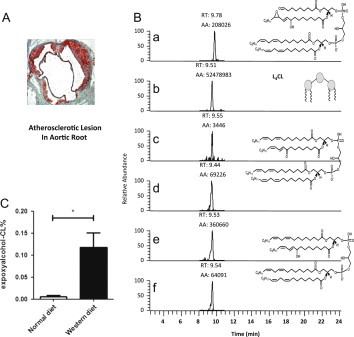
EAA-L4CL and L4CL oxidation present in LDLR KO mice liver after high fat feeding.(A) Oil red O staining of atherosclerotic lesions at the aortic root after Western diet feeding. Images are shown at 10× magnification. (B) Quantification of L4CL oxidation products in LDLR KO mice liver using MRM. MRM transitions: (a) EAA-L4CL, m/z 685.6 to 279.2; (b) L4CL, m/z 723.6 to 279.2; (c and d) KODE- L4CL, m/z 730.6 to 293.2 and 279.2; (e and f) HODE- L4CL, m/z 731.6 to 279.2 and 295.2. (C) Levels of EAA-CL in LDLR KO mice liver fed control diet and western diet. EAA-CL is expressed as % of total CL (means ± SD, n = 4 chow diet vs n = 6 western diet).
Besides the major cardiolipin species L4CL, mouse liver tissue contains other minor cardiolipins such as L3OCL. The structure of L3OCL has one of the LA side chains replaced by an oleic acid. According to our chemical mechanism of 4-HNE formation from CL, cross-chain peroxyl radical reaction occurs between two adjacent side chains; thus the presence of an unreactive oleic acid side chain in L3OCL may disrupt this reaction and lead to less lipid electrophile production through this mechanism. As shown in Fig. 4, our data showed that the formation of similar electrophiles EAA-CL from L3OCL was indeed significantly suppressed compared to those from L4CL, highlighting the importance of this cross-chain reaction during the formation of reactive electrophiles. Interestingly, formation of other major oxidation products, such as HODE-CL, from L3OCL was decreased compared to L4CL but did not reach statistical significance.
Fig. 4.
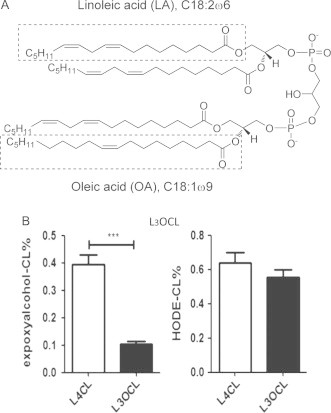
Level of EAA-CL from L3OCL is decreased than that from L4CL in LDLR KO mice liver due to the presence of one un-reactive fatty acid. (A) Chemical structure of L3OCL. (B) Levels of oxidation products of L3OCL and L4CL from LDLR KO mice liver after feeding Western diet. CL oxidation products are expressed as % of total CL (means ± SD, n = 6).
Discussion
Free radical-induced lipid peroxidation has been linked to multiple human diseases including atherosclerosis [8]. Among the complicated oxidation products, lipid electrophiles, including 4-HNE, generated from lipid oxidation have attracted increased attention due to their potential roles in altering protein structures and functions through covalent modification of critical nucleophilic amino acid residues [10,21,22]. Overwhelming evidence indicates that mitochondria play an essential role in ROS generation, lipid peroxidation, and the pleiotropic effects of 4-HNE in various biological processes. In contrast to the well-studied biology of 4-HNE, the chemical mechanisms for 4-HNE formation and cellular locations remain poorly defined [23,24]. In a previous report we proposed a novel chemical mechanism for the formation of 4-HNE and other reactive lipid aldehydes from mitochondrial cardiolipin oxidation [17]. In our current study, we provide evidence that this mechanism operates both in vitro in t-Bid induced cyt c release and in vivo in mice liver of atherosclerosis after Western diet feeding. Furthermore, formation of these bioactive lipid mediators can be dose-dependently inhibited by a widely used clinical reagent ApAP acting as a peroxidase inhibitor, corroborating with previous studies where ApAP was a potent inhibitor for peroxidase activity of cylooxygense (COX)[25], hemoproteins including hemoglobin and myoglobin [26], and peroxidase of cyt c/CL complex [15]. Our findings have significant implications in linking mitochondrial lipid peroxidation and lipid electrophile production to important biological processes, such as apoptosis, and human diseases that are associated with oxidative stress.
The novel chemical mechanisms for the formation of 4-HNE from CL oxidation are based on the fact that the extent of oxidation for L4CL is far greater than other phospholipids. The susceptibility of L4CL to free radical oxidation has been attributed to the presence of four LA side chains in the same molecules and the rate-limiting step reaction of hydrogen atom abstraction by a peroxyl radical may occur through intra-molecular reactions. Furthermore, subsequent peroxyl radical addition reactions that form cross-chain products may be responsible for the formation of 4-HNE. A seminal work reported by Kagan et al. demonstrated that oxidation of CL by the peroxidase function of cyt c/CL complex was required to trigger intrinsic apoptotic pathways [27]. Recent studies from the same group also reported that CL oxidation was linked to traumatic brain injury [28] and mitophagy [5]. However, the role of bioactive lipid electrophiles from CL oxidation remains to be studied. Our studies suggest that 4-HNE formation from this process may also play a significant role in these events because 4-HNE has been shown to induce apoptosis and autophage [10,29,30]. Moreover, it is well established that 4-HNE plays an important role in cardiovascular diseases. On one hand, 4-HNE directly suppresses contractile function, enhances ROS generation, and modulates multiple signal transduction pathways that are known to contribute to atherosclerosis, myocardial ischemia-reperfusion injury, heart failure, and cardiomyopathy [21]. On the other hand, levels of circulating anti-cardiolipin antibody are positively correlated with endothelial dysfunction and atherosclerosis. It is interesting to note that the anti-cardiolipin antibody actually recognized oxidized CL [31]. Formation of these epitopes is most likely through covalent modifications of immune responsive proteins by lipid electrophiles generated from CL oxidation [32]. Moreover, L4CL is the major CL species in the mitochondria of most mammalian tissues and it constitutes more than 70% of mitochondrial CLs in the heart [33]. Thus formation of 4-HNE via this novel mechanism is likely important in the context of cardiovascular diseases. Our current study clearly demonstrates that formation of 4-HNE from CL operates in vivo and has significant pathophysiological relevance with the detection of the characteristic product EAA-CL and other CL oxidation products (Fig. 3). This mechanism is further substantiated by the observation that the presence of only one un-oxidized fatty acid (such as oleic acid) in some minor cardiolipin species significantly decreases the formation of this characteristic bioactive lipid and other oxidation products such as HODE-CL (Fig. 4).
Modulating the formation of 4-HNE and other lipid electrophiles from CL oxidation in mitochondria may represent a viable approach to attenuate the cytotoxicity of these bioactive lipid electrophiles. Several strategies have been explored to inhibit the oxidation of CL in mitochondria [34]. The peroxidase activity of cyt c/CL complex is activated by hydrogen peroxide or lipid hydroperoxides; thus scavenging H2O2 or reducing lipid hydroperoxides may lead to the inhibition of apoptosis [35]. In fact, liver apoptosis and cyt c release were suppressed in transgenic mice over-expression of phospholipid hydroperoxide glutathione peroxidase (PHGPX), the enzyme that reduces lipid hydroperoxides. Levels of cardiolipin oxidation products in these transgenic animals were also suppressed compared to the wild type animals [36]. Furthermore, reduction of mitochondrial hydrogen peroxide by over-expressing mitochondria-specific peroxiredoxin 3 (Prdx3/Prx3) led to reduced levels of F2-IsoPs, a widely accepted standard for oxidative stress in vivo [37,38], and 4-HNE [39]. Inhibition of the peroxidase activity of cyt c/CL complex represents a novel approach for controlling CL oxidation and its related consequences. ApAP is one of the most widely used analgesics. Previous studies show that it inhibited peroxidase activity of COX by reducing the protoporphyrin radical cation in the peroxidase active site of the enzyme, thereby blocking formation of the catalytic tyrosyl radical in the cyclooxygenase site [25,40]. Later studies demonstrated that ApAP could protect the kidney from oxidative damage following rhabdomyolysis by inhibiting the peroxide-driven lipid peroxidation catalyzed by myoglobin and hemoglobin [41]. We reported recently that ApAP inhibited cyt c redox cycling-induced lipid peroxidation [15]. In current studies, in addition to suppression of the CL oxidation products such as HODE-CL, lipid electrophiles including EAA-CL are also significantly inhibited during t-Bid induced cyt c release (Fig. 2). It is still unclear, however, how the phospholipid containing lipid electrophiles, such as EAA-CL, are metabolized or repaired. The metabolic pathways for these novel phospholipid electrophiles and their biological activities are currently under investigation.
In summary, we provide evidence that, in addition to 4-HNE, a novel class of CL containing lipid electrophiles, EAA-CL, are generated from mitochondrial CL oxidation in vitro and in vivo in the context of apoptosis and atherosclerosis. The widely used clinical reagent ApAP can inhibit CL oxidation and generation of those lipid electrophiles. This study suggests a potential link between mitochondrial CL oxidation, lipid electrophile generation, mitochondrial dysfunction, and apoptosis during the progression of atherosclerosis.
Acknowledgements
This work is supported by grants from the National Natural Science Foundation of China (31170809), the Ministry of Science and Technology of China (2012BAK01B00), the National Key Basic Research Program of China (973 Program, # 2012CB524900), and the Hundred Talents Program from CAS (2012OHTP07). H.Y. is an Associate Fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine at Nanjing Medical University. We acknowledge the help from Drs. Ned A Porter, Sandy Zinkel, Jack Roberts and John A Oates at Vanderbilt University, Nashville, TN, USA. We thank Mr. David R Landzberg at Vanderbilt University School of Medicince for assistance with the manuscript writing.
References
- 1.Gonzalvez F., Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis: An international Journal on Programmed Cell Death. 2007;12(5):877–885. doi: 10.1007/s10495-007-0718-8. 17294083 [DOI] [PubMed] [Google Scholar]
- 2.Kiebish M.A., Yang K., Sims H.F., Jenkins C.M., Liu X., Mancuso D.J., Zhao Z., Guan S., Abendschein D.R., Han X. Myocardial regulation of lipidomic flux by cardiolipin synthase: setting the beat for bioenergetic efficiency. Journal of Biological Chemistry. 2012;287(30):25086–25097. doi: 10.1074/jbc.M112.340521. 22584571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin H., Zhu M. Free radical oxidation of cardiolipin: chemical mechanisms, detection and implication in apoptosis, mitochondrial dysfunction and human diseases. Free Radical Research. 2012;46(8):959–974. doi: 10.3109/10715762.2012.676642. 22468920 [DOI] [PubMed] [Google Scholar]
- 4.Kagan V.E., Tyurin V.A., Jiang J., Tyurina Y.Y., Ritov V.B., Amoscato A.A., Osipov A.N., Belikova N.A., Kapralov A.A., Kini V. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature Chemical Biology. 2005;1(4):223–232. doi: 10.1038/nchembio727. 16408039 [DOI] [PubMed] [Google Scholar]
- 5.Chu C.T., Ji J., Dagda R.K., Jiang J.F., Tyurina Y.Y., Kapralov A.A., Tyurin V.A., Yanamala N., Shrivastava I.H., Mohammadyani D. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature Cell Biology. 2013;15(10):1197–1205. doi: 10.1038/ncb2837. 24036476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paradies G., Petrosillo G., Paradies V., Ruggiero F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009;45(6):643–650. doi: 10.1016/j.ceca.2009.03.012. 19368971 [DOI] [PubMed] [Google Scholar]
- 7.Kagan V.E., Bayir H.A., Belikova N.A., Kapralov O., Tyurina Y.Y., Tyurin V.A., Jiang J., Stoyanovsky D.A., Wipf P., Kochanek P.M. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radical Biology and Medicine. 2009;46(11):1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. 19285551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin H., Xu L., Porter N.A. Free radical lipid peroxidation: mechanisms and analysis. Chemical Reviews. 2011;111(10):5944–5972. doi: 10.1021/cr200084z. 21861450 [DOI] [PubMed] [Google Scholar]
- 9.Lee S., Birukov K.G., Romanoski C.E., Springstead J.R., Lusis A.J., Berliner J.A. Role of phospholipid oxidation products in atherosclerosis. Circulation Research. 2012;111(6):778–799. doi: 10.1161/CIRCRESAHA.111.256859. 22935534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West J.D., Marnett L.J. Endogenous reactive intermediates as modulators of cell signaling and cell death. Chemical Research in Toxicology. 2006;19(2):173–194. doi: 10.1021/tx050321u. 16485894 [DOI] [PubMed] [Google Scholar]
- 11.Poli G., Biasi F., Leonarduzzi G. 4-Hydroxynonenal-protein adducts:a reliable biomarker of lipid oxidation in liver diseases. Molecular Aspects of Medicine. 2008;29(1–2):67–71. doi: 10.1016/j.mam.2007.09.016. 18158180 [DOI] [PubMed] [Google Scholar]
- 12.Yun M.R., Im D.S., Lee S.J., Park H.M., Bae S.S., Lee W.S., Kim C.D. 4-Hydroxynonenal enhances CD36 expression on murine macrophages via p38 MAPK-mediated activation of 5-lipoxygenase. Free Radical Biologyand Medicine. 2009;46(5):692–698. doi: 10.1016/j.freeradbiomed.2008.12.013. 19135147 [DOI] [PubMed] [Google Scholar]
- 13.Schneider C., Porter N.A., Brash A.R. Routes to 4-hydroxynonenal: fundamental issues in the mechanisms of lipid peroxidation. Journal of Biological Chemistry. 2008;283(23):15539–15543. doi: 10.1074/jbc.R800001200. 18285327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider C., Tallman K.A., Porter N.A., Brash A.R. Two distinct pathways of formation of 4-hydroxynonenal. Mechanisms of nonenzymatic transformation of the 9- and 13-hydroperoxides of linoleic acid to 4-hydroxyalkenals. Journal of Biological Chemistry. 2001;276(24):20831–20838. doi: 10.1074/jbc.M101821200. 11259420 [DOI] [PubMed] [Google Scholar]
- 15.Yin H., Vergeade A., Shi Q., Zackert W.E., Gruenberg K.C., Bokiej M., Amin T., Ying W., Masterson T.S., Zinkel S.S. Acetaminophen inhibits cytochrome c redox cycling induced lipid peroxidation. Biochemical and Biophysical Research Communications. 2012;423(2):224–228. doi: 10.1016/j.bbrc.2012.05.058. 22634010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saraswathi V., Gao L., Morrow J.D., Chait A., Niswender K.D., Hasty A.H. Fish oil increases cholesterol storage in white adipose tissue with concomitant decreases in inflammation, hepatic steatosis, and atherosclerosis in mice. Journal of Nutrition. 2007;137(7):1776–1782. doi: 10.1093/jn/137.7.1776. 17585030 [DOI] [PubMed] [Google Scholar]
- 17.Liu W., Porter N.A., Schneider C., Brash A.R., Yin H. Formation of 4-hydroxynonenal from cardiolipin oxidation: intramolecular peroxyl radical addition and decomposition. Free Radical Biologyand Medicine. 2011;50(1):166–178. doi: 10.1016/j.freeradbiomed.2010.10.709. 21047551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nature Protocols. 2007;2(2):287–295. doi: 10.1038/nprot.2006.478. 17406588 [DOI] [PubMed] [Google Scholar]
- 19.Scorrano L., Ashiya M., Buttle K., Weiler S., Oakes S.A., Mannella C.A., Korsmeyer S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Developmental Cell. 2002;2(1):55–67. doi: 10.1016/s1534-5807(01)00116-2. 11782314 [DOI] [PubMed] [Google Scholar]
- 20.Steinberg D., Parhasarathy S., Carew T.E., Khoo J.C., Witztum J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. New England Journal of Medicine. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. 2648148 [DOI] [PubMed] [Google Scholar]
- 21.Mali V.R., Palaniyandi S.S. Regulation and therapeutic strategies of 4-hydroxy-2-nonenal metabolism in heart disease. Free Radical Research. 2014;48:251–263. doi: 10.3109/10715762.2013.864761. 24237196 [DOI] [PubMed] [Google Scholar]
- 22.Benedetti A., Comporti M., Esterbauer H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochimica et Biophysica Acta. 1980;620(2):281–296. doi: 10.1016/0005-2760(80)90209-x. 6254573 [DOI] [PubMed] [Google Scholar]
- 23.Schneider C., Boeglin W.E., Yin H., Porter N.A., Brash A.R. Intermolecular peroxyl radical reactions during autoxidation of hydroxy and hydroperoxy arachidonic acids generate a novel series of epoxidized products. Chemical Research in Toxicology. 2008;21(4):895–903. doi: 10.1021/tx700357u. 18324788 [DOI] [PubMed] [Google Scholar]
- 24.Schneider C., Porter N.A., Brash A.R. Routes to 4-hydroxynonenal: fundamental issues in the mechanisms of lipid peroxidation. Journal of Biological Chemistry. 2008;283(23):15539–15543. doi: 10.1074/jbc.R800001200. 18285327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boutaud O., Aronoff D.M., Richardson J.H., Marnett L.J., Oates J.A. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7130–7135. doi: 10.1073/pnas.102588199. 12011469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boutaud O., Moore K.P., Reeder B.J., Harry D., Howie A.J., Wang S., Carney C.K., Masterson T.S., Amin T., Wright D.W. Acetaminophen inhibits hemoprotein-catalyzed lipid peroxidation and attenuates rhabdomyolysis-induced renal failure. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(6):2699–2704. doi: 10.1073/pnas.0910174107. 20133658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kagan V.E., Tyurin V.A., Jiang J., Tyurina Y.Y., Ritov V.B., Amoscato A.A., Osipov A.N., Belikova N.A., Kapralov A.A., Kini V. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature Chemical Biology. 2005;1(4):223–232. doi: 10.1038/nchembio727. 16408039 [DOI] [PubMed] [Google Scholar]
- 28.Ji J., Kline A.E., Amoscato A., Samhan-Arias A.K., Sparvero L.J., Tyurin V.A., Tyurina Y.Y., Fink B., Manole M.D., Puccio A.M. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nature Neuroscience. 2012;15(10):1407–1413. doi: 10.1038/nn.3195. 22922784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji C., Amarnath V., Pietenpol J.A., Marnett L.J. 4-Hydroxynonenal induces apoptosis via caspase-3 activation and cytochrome c release. Chemical Research in Toxicology. 2001;14(8):1090–1096. doi: 10.1021/tx000186f. 11511183 [DOI] [PubMed] [Google Scholar]
- 30.Ma H., Guo R., Yu L., Zhang Y., Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. European Heart Journal. 2011;32(8):1025–1038. doi: 10.1093/eurheartj/ehq253. 20705694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su J., Frostegård A.G., Hua X., Gustafsson T., Jogestrand T., Hafström I., Frostegård J. Low levels of antibodies against oxidized but not nonoxidized cardiolipin and phosphatidylserine are associated with atherosclerotic plaques in systemic lupus erythematosus. Journal of Rheumatology. 2013;40(11):1856–1864. doi: 10.3899/jrheum.121173. 24037548 [DOI] [PubMed] [Google Scholar]
- 32.Wall S.B., Smith M.R., Ricart K., Zhou F., Vayalil P.K., Oh J.-Y., Landar A. Detection of electrophile-sensitive proteins. Biochimica et Biophysica Acta. 2014;1840:913–922. doi: 10.1016/j.bbagen.2013.09.003. 24021887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H.-Y.J., Jackson S.N., Woods A.S. Direct MALDI-MS analysis of cardiolipin from rat organs sections. Journal of the American Society for Mass Spectrometry. 2007;18(3):567–577. doi: 10.1016/j.jasms.2006.10.023. 17157526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoye A.T., Davoren J.E., Wipf P., Fink M.P., Kagan V.E. Targeting mitochondria. Accounts of Chemical Research. 2008;41(1):87–97. doi: 10.1021/ar700135m. 18193822 [DOI] [PubMed] [Google Scholar]
- 35.Belikova N.A., Tyurina Y.Y., Borisenko G., Tyurin V., Samhan Arias A.K., Yanamala N., Furtmuller P.G., Klein-Seetharaman J., Obinger C., Kagan V.E. Heterolytic reduction of fatty acid hydroperoxides by cytochrome c/cardiolipin complexes: antioxidant function in mitochondria. Journal of the American Chemical Society. 2009;131(32):11288–11289. doi: 10.1021/ja904343c. 19627079 [DOI] [PubMed] [Google Scholar]
- 36.Liang H., Ran Q., Jang Y.C., Holstein D., Lechleiter J., McDonald-Marsh T., Musatov A., Song W., Van Remmen H., Richardson A. Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radical Biologyand Medicine. 2009;47(3):312–320. doi: 10.1016/j.freeradbiomed.2009.05.012. 19447173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Milne G.L., Yin H., Morrow J.D. Human biochemistry of the isoprostane pathway. Journal of Biological Chemistry. 2008;283(23):15533–15537. doi: 10.1074/jbc.R700047200. 18285331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milne G.L., Yin H., Hardy K.D., Davies S.S., Roberts L.J. Isoprostane generation and function. Chemical Reviews. 2011;111(10):5973–5996. doi: 10.1021/cr200160h. 21848345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L., Na R., Gu M., Salmon A.B., Liu Y., Liang H., Qi W., Remmen H.V., Richardson A., Ran Q. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell. 2008;7(6):866–878. doi: 10.1111/j.1474-9726.2008.00432.x. 18778410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ouellet M., Percival M.D. Mechanism of acetaminophen inhibition of cyclooxygenase isoforms. Archives of Biochemistry and Biophysics. 2001;387(2):273–280. doi: 10.1006/abbi.2000.2232. 11370851 [DOI] [PubMed] [Google Scholar]
- 41.Boutaud O., Moore K.P., Reeder B.J., Harry D., Howie A.J., Wang S., Carney C.K., Masterson T.S., Amin T., Wright D.W. Acetaminophen inhibits hemoprotein-catalyzed lipid peroxidation and attenuates rhabdomyolysis-induced renal failure. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(6):2699–2704. doi: 10.1073/pnas.0910174107. 20133658 [DOI] [PMC free article] [PubMed] [Google Scholar]