

Abstract
Palmitate overload induces hepatic cell dysfunction characterized by enhanced apoptosis and altered citric acid cycle (CAC) metabolism; however, the mechanism of how this occurs is incompletely understood. We hypothesize that elevated doses of palmitate disrupt intracellular calcium homeostasis resulting in a net flux of calcium from the ER to mitochondria, activating aberrant oxidative metabolism. We treated primary hepatocytes and H4IIEC3 cells with palmitate and calcium chelators to identify the roles of intracellular calcium flux in lipotoxicity. We then applied 13C metabolic flux analysis (MFA) to determine the impact of calcium in promoting palmitate-stimulated mitochondrial alterations. Co-treatment with the calcium-specific chelator BAPTA resulted in a suppression of markers for apoptosis and oxygen consumption. Additionally, 13C MFA revealed that BAPTA co-treated cells had reduced CAC fluxes compared to cells treated with palmitate alone. Our results demonstrate that palmitate-induced lipoapoptosis is dependent on calcium-stimulated mitochondrial activation, which induces oxidative stress.
Keywords: Metabolic flux analysis, Lipotoxicity, Oxidative stress, ER stress, Fatty liver
Abbreviations: APE, atom percent enrichment; BSA, bovine serum albumin; CAC, citric acid cycle; FFA, free fatty acid; GC–MS, gas chromatography–mass spectrometry; H2DCFDA, 2′,7′-dichlorodihydrofluorescein diacetate; MFA, metabolic flux analysis; MUFA, monounsaturated fatty acid; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; OA, oleate; PA, palmitate; PI, propidium iodide; ROS, reactive oxygen species; SERCA, sarcoplasmic-endoplasmic reticulum calcium ATPase; SFA, saturated fatty acid
Enzymes and metabolites
- AcCoA
acetyl-CoA
- Akg
alpha-ketoglutarate
- Ala
alanine
- Asp
aspartate
- Cit
citrate
- Fum
fumarate
- Gln
glutamine
- Glu
glutamate
- Lac
lactate
- Mal
malate
- Pyr
pyruvate
- Suc
succinate
- ADH
alpha-ketoglutarate dehydrogenase
- CS
citrate syntase
- FUS
fumarase
- GDH
glutamate dehydrogenase
- GLS
glutaminase
- IDH
isocitrate dehydrogenase
- LDH
lactate dehydrogenase
- ME
malic enzyme
- PC
pyruvate carboxylase
- PDH
pyruvate dehydrogenase
- PK
pyruvate kinase
- SDH
succinate dehydrogenase
1. Introduction
The obese and steatotic liver is marked by elevated fatty acids, ER stress, and metabolic alterations that give rise to hepatocyte dysfunction [1–5]. Non-alcoholic fatty liver disease (NAFLD) is a chronic condition resulting from excess lipid accumulation, which affects up to 30% of the U.S. population and is the leading cause of referrals to hepatology clinics [1,6]. Although simple steatosis does not always lead to complications, around 10% of NAFLD patients are at increased risk of developing more serious liver injuries such as nonalcoholic steatohepatitis (NASH) and hepatocellular carcinoma [7]. Free fatty acid (FFA) levels are present in higher concentrations in the plasma of these individuals, suggesting that in vivo alterations in FFA metabolism are linked to corresponding changes in disease severity [8,9].
Altered energy metabolism is a defining characteristic of both human and mouse fatty livers, which can be recapitulated in in vitro models of hepatocyte lipotoxicity [2,4,10,11]. However, the mechanism by which FFA overload induces metabolic dysfunction is undefined. In vivo flux analysis using 2H/13C NMR reported a ∼2-fold increase in citric acid cycle (CAC) flux in NAFLD patients compared to patients with normal intrahepatic triglyceride content [2]. Complementary studies performed in high-fat diet (HFD) fed mice revealed similar increases in CAC activity that were associated with elevated markers of oxidative stress [4]. The authors hypothesized that CAC activation was required to meet energetic demands in the face of reduced respiratory efficiency resulting from mitochondrial oxidative damage. However, we have previously shown that saturated fatty acids (SFAs) can enhance mitochondrial metabolism independently of beta-oxidation in cultured hepatic cells through a mechanism that precedes the onset of oxidative damage [10,11]. Consistent with in vivo mouse studies, these changes in CAC fluxes coincided with enhanced reactive oxygen species (ROS) accumulation, suggesting that altered mitochondrial metabolism may be the cause, rather than a consequence, of enhanced oxidative stress observed in obesity and NAFLD. To confirm this, we performed experiments using antioxidants and mitochondrial inhibitors to demonstrate that CAC activation is critical for palmitate lipotoxicity but does not require prior ROS accumulation [10].
In vitro experiments have demonstrated that SFAs are also potent inducers of ER stress in hepatic cells, which precedes the onset of ROS accumulation and apoptosis [13]. It has been shown that markers of ER stress such as CHOP/GADD135 formation and depletion of ER calcium stores appear soon after cells are treated with long-chain SFAs, but not monounsaturated fatty acids (MUFAs) [14]. ER calcium is depleted shortly after SFA exposure, suggesting a mechanism of SFA toxicity that involves rapid disruption of ER homeostasis [12,15,16]. The exact role of this calcium efflux in mediating lipotoxicity is unknown, although intracellular calcium levels impact many critical aspects of cell function. Calcium is integral in two important aspects of cell biology: oxidative metabolism and apoptosis. Calcium ions act as essential cofactors by activating several CAC enzymes, particularly dehydrogenases, and transporters involved in the malate–aspartate redox shuttle [17–20]. Calcium fluxes also initiate mitochondrial apoptotic pathways. Pro- and anti-apoptotic proteins of the Bax, Bcl, and Bim families have been shown to regulate the net movement of calcium into and out of the mitochondria [21–23].
Because of the rapid appearance of ER stress markers in response to palmitate treatment, we hypothesized that disruption of ER homeostasis may be the initial insult that is responsible for subsequent changes in mitochondrial function. We hypothesized that elevated levels of the SFA palmitate would compromise the ability of the ER to maintain calcium stores, resulting in net efflux of ER calcium that would enhance CAC flux, stimulate oxidative metabolism and ROS production, and ultimately lead to cellular dysfunction and apoptosis. To test this, we treated primary rat hepatocytes and immortalized H4IIEC3 hepatic cells with lipotoxic doses of palmitate, either with or without the intracellular calcium chelator BAPTA-AM. Palmitate-treated cells exhibited decreased ER calcium, elevated mitochondrial calcium, reduced mitochondrial potential, and enhanced oxygen consumption, all of which preceded the onset of apoptotic cell death. BAPTA co-treatment abrogated these lipotoxic phenotypes. 13C metabolic flux analysis (MFA) revealed that palmitate-treated cells exhibited enhanced CAC flux and increased mitochondrial glutamine metabolism that were associated with ROS accumulation. BAPTA co-treatment also suppressed glutamine-dependent CAC activation and reduced oxidative stress. These results offer a mechanistic explanation for the close association between ER stress and ROS accumulation reported in prior lipotoxicity studies, in which calcium serves as a critical mediator linking changes in ER homeostasis to the onset of mitochondrial dysfunction.
2. Materials and methods
2.1. Materials
The fluorescent dyes 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), propidium iodide (PI), Fura-2 AM, and JC-1 were purchased from Invitrogen (Carlsbad, CA, USA). The calcium-specific chelator BAPTA-AM was also obtained from Invitrogen. The fatty acids palmitate and oleate, bovine serum albumin (BSA), and low glucose Dulbecco's modified Eagle's medium (DMEM) were purchased from Sigma Aldrich (St. Louis, MO, USA). Primary hepatocytes were cultured on plates coated with Collagen I (Rat Tail) from BD Biosciences (San Jose, CA).
2.2. Primary rat hepatocyte isolation
Primary hepatocytes were isolated from male Sprague–Dawley rats as described previously [24]. The portal vein and inferior vena cava of anesthetized animals were cannulated and perfused with 37 °C oxygenated perfusion media, pH 7.4, containing 118 mM NaCl, 5.9 mM KCl, 1.2 mM MgSO4, 1.2 mM NaH2PO4, 25 mM NaHCO3, 0.2 mM EGTA and 5 mM glucose. After 15 min, the liver was excised from the animal and perfused with liver digest medium (Invitrogen, Grand Island, NY). Then the cells were dispersed, washed four times, and suspended in attachment media, which consisted of 20 mM glucose DMEM supplemented with 30 mg/L proline, 100 mg/L ornithine, 0.544 mg/L ZnCl2, 0.75 mg/L ZnSO4·7H2O, 0.2 mg/L CuSO4·5H2O, 0.25 mg/L MnSO4, 2 g/L bovine serum albumin (Sigma), 5 nM insulin, 100 nM dexamethasone, 100,000 U penicillin, 100,000 U streptomycin, and 2 mM glutamine. After 4 h of incubation in the attachment media, the primary hepatocytes were switched to a maintenance media identical to the attachment media except it had a concentration of 1 nM (instead of 5 nM) insulin.
2.3. H4IIEC3 cell culture
The H4IIEC3 rat hepatoma cell line was purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). The cells were cultured in 5 mM glucose DMEM supplemented with 2 mM glutamine, 10% FBS, and 1% penicillin/streptomycin antibiotic solution.
2.4. Fatty acid preparation
FFA stock solutions were prepared by coupling free fatty acids with BSA. First, palmitate or oleate was dissolved in pure ethanol at a concentration of 195 mM so that the final concentration of ethanol in our FFA stock solutions did not exceed 1.5% by volume. This solution was then added to a prewarmed 10% w/w BSA solution (37 °C) to achieve a final FFA concentration of 3 mM, and this solution was allowed to incubate in a water bath for an additional 10 min. The final ratio of FFA to BSA was 2:1. All vehicle treatments were prepared using stocks of 10% w/w BSA with an equivalent volume of ethanol added to match the concentration in FFA stocks. The final concentration of ethanol in all experimental treatments was less than 0.2% by volume.
2.5. ROS accumulation
Levels of intracellular ROS were assessed using the radical-sensitive dye H2DCFDA, which is oxidized to the fluorescent 2, 7-dicholorofluorescein (DCF) upon exposure to ROS. Following treatment with indicated reagents, the cells were washed with Hank's Balanced Saline Solution (HBSS) twice. Dye was added at a concentration of 10 μM H2DCFDA and incubated for 1 h at 37 °C in darkness. Fluorescence was measured using the excitation/emission wavelengths 485/530 nm with a Biotek FL600 microplate reader.
2.6. Toxicity assays
Propidium iodide (PI), an intercalating dye, was used to measure cell death induced by elevated fatty acids using excitation and emission wavelengths of 530 nm and 645 nm. Fluorescence was measured using the Biotek FL600 microplate reader.
2.7. Apoptosis measurements
To monitor cellular apoptosis as a function of caspase 3 and 7 activities, we utilized the commercial Apo-ONE Homogenous Caspase 3/7 Assay kit that combines a lysis buffer with Z-DEVD-R110, a caspase-3/7 specific substrate. Upon exposure to active caspases, the DEVD peptide is cleaved and the molecule becomes fluorescent (ex/em, 485/530 nm).
2.8. Oxygen consumption flux
To determine overall mitochondrial activity, we measured the direct oxygen uptake flux of cells treated with fatty acids, vehicle, or inhibitors using the Oroboros Oxygraph-2K (Oroboros Instruments, Austria). This instrument uses two separate chambers with individual oxygen probes to detect real-time changes in media oxygen concentration at a constant temperature of 32 °C and stirring speed of 750 rpm. Briefly, H4IIEC3 hepatic cells were cultured on 10-cm dishes until 90% confluent, as previously described [10]. Cells were then treated with specified fatty acid and inhibitor combinations for 3 h. After 3 h, the media was collected and the cells were trypsinized and harvested from the plates. Once harvested, the cells were manually counted using a hemocytometer and resuspended in the same media. Then, two million cells were injected into the Oxygraph instrument to measure oxygen consumption.
2.9. Mitochondrial potential
JC-1 is a dye which exists in a monomeric form in non-polarized mitochondria and fluoresces in the green (em: 530) spectrum when excited at 485 nm. The dye accumulates in the mitochondria based upon the potential. This accumulation is accompanied by formation of dye aggregates shifting the fluorescence to the red (em: 590 nm) spectrum when excited at 485 nm. Therefore red/green ratio indicates alterations in the mitochondrial potential between different cells and treatments.
2.10. ER calcium release assays
To measure ER calcium levels, H4IIEC3 cells were loaded with the ratiometric, cytosolic calcium dye Fura-2 AM following a method developed for pancreatic islets [25]. Twenty-four hours prior to the experiment, cells were plated at 500,000 cells per dish on MatTek imaging dishes. Cells were then loaded with 3 μM Fura-2 for 30 min, washed three times, and then perfused with imaging buffer containing 119 mM NaCl, 25 mM HEPES, 4.7 mM KCl, 2.5 mM CaCl2·[(H2O)6], 1.2 mM MgSO4, 1.2 mM KH2PO4. Fura-2 fluorescence imaging was performed using a Nikon TE2000-U microscope at excitations of 340 and 380 nm every 5 s. Once a baseline was established, 1 μM thapsigargin was perfused across the dish to prevent ER calcium reuptake, and fluorescence was measured every 5 s. The data shown are expressed in normalized fluorescence units with excitation at 340/380 nm for 5 individual plates with 30–50 cells analyzed per plate.
2.11. Mitochondrial calcium
The mitochondrial calcium indicator Rhod-2, AM (Invitrogen) was used to assess mitochondrial calcium in H4IIEC3 cells. Cells were pre-treated with indicated treatments and then loaded with 10 μM Rhod-2. Cells were loaded with the dye for 1 h, washed three times, and given fresh DMEM. Fluorescence was measured at ex/em of 552/581 using a Biotek Synergy plate reader.
2.12. Polar metabolite extraction and GC–MS analysis of 13C enrichment
Intracellular metabolites from H4IIEC3 rat hepatomas were extracted as previously described [11]. To quench cell metabolism, 1 mL of −80 °C methanol was added to cells cultured on 10-cm dishes. Cells were scraped and placed into a separate tube containing water and chloroform, centrifuged, and the polar phase was collected and dried for GC–MS analysis. The tert-butylsilyl derivatives of the polar metabolites were generated by incubating with MBTSTFA + 1% TBDMCS (Pierce). 13C isotopic enrichment of the derivatized sample was analyzed using an Agilent 6890N/5975B GC–MS equipped with a 30 m DB-35ms capillary column.
2.13. Metabolic flux analysis
13C MFA was performed using the INCA software package [26] and a previously developed model of hepatic metabolism comprising glycolysis, CAC, and anaplerotic pathways [10]. Our experiments involved replacing the unlabeled glutamine in normal DMEM with a [U-13C5]glutamine isotope tracer. When consumed by cells in culture, this tracer gives rise to unique 13C enrichment patterns in downstream metabolites that depend on the atom rearrangements that occur within intervening metabolic pathways. These enrichment patterns provide quantitative information on the relative fluxes through intracellular metabolic pathways. By minimizing the lack-of-fit between experimentally measured mass isotopomer abundances and computationally simulated mass isotopomer distributions, the INCA program can be used to calculate metabolic flux maps associated with each of our chosen treatments [27,28]. Fluxes were estimated a minimum of 50 times starting from random initial values to identify a global best-fit solution. Once this solution was achieved, a chi-square test was used to assess the goodness-of-fit. Additionally, 95% confidence intervals were calculated for all estimated parameters by assessing the sensitivity of the sum-of-squared residuals to parameter variations [29]. Comprehensive 13C MFA results and a detailed description of our network model are available in the Appendix A.
2.14. Statistical analysis
Analysis of variance (Model I ANOVA) and Tukey–Kramer methods for multiple comparisons, along with Student's t-test for pair-wise comparisons, were utilized to determine statistical significance amongst the experimental data. Results are presented as ±one standard error of the mean (SEM) unless otherwise indicated.
3. Results
3.1. Elevated concentrations of palmitate, but not oleate, induce cell death marked by loss of mitochondrial potential and apoptosis in hepatic cells
Primary rat hepatocytes treated with elevated concentrations of the SFA palmitate exhibit cell death in a dose-dependent manner as indicated by increasing PI fluorescence (Figure 1A). Elevated doses of the MUFA oleate had no effect on cell viability in primary hepatocytes. Based on these results and previously published studies, we chose 400 μM palmitate as the lipotoxic concentration used in all further experiments [11]. Similar to primary rat hepatocytes, H4IIEC3 rat hepatoma cells also exhibited increased cell death after 24 h of palmitate treatment (Figure 1A). Additionally, primary hepatocyte and H4IIEC3 cells exhibited elevated caspase activity following 12 h of palmitate treatment, while oleate did not induce markers of apoptosis (Figure 1B). While both primary hepatocytes and H4IIEC3 cells exhibited significantly increased apoptosis in response to elevated palmitate, the effects were both more rapid and more pronounced in H4IIEC3 cells.
Figure 1.
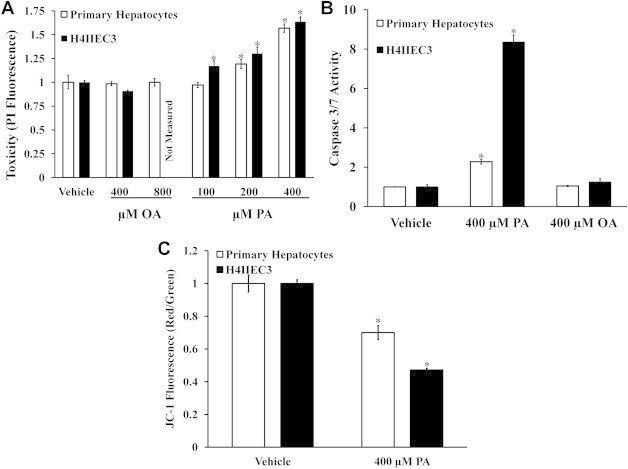
Elevated doses of palmitate, but not oleate, induce lipotoxicity in primary rat hepatocytes and H4IIEC3 hepatic cells. Cells were incubated with increasing doses of palmitate (PA) or oleate (OA), followed by measurements of cell death and mitochondrial potential. (A) Cell death measured by PI fluorescence for primary hepatocytes and H4IIEC3 cells treated with increasing doses of palmitate or oleate for 24 h. (B) 12-h caspase activity measured for primary hepatocytes and H4IIEC3 hepatic cells treated with 400 μM palmitate. (C) JC-1 fluorescence for primary hepatocytes and H4IIEC3 hepatic cells treated with vehicle (BSA) or 400 μM palmitate (PA) for 6 h. JC-1 fluorescence is depicted as the ratio between red and green fluorescent signals. Data represent mean ± S.E., n = 8; *, different from vehicle, p < .05. All fatty acid treatments are normalized to equal volume vehicle (BSA) controls.
To determine if palmitate lipotoxicity was associated with loss of mitochondrial potential, primary rat hepatocytes and H4IIEC3 cells were incubated with FFA treatments for 6 h followed by staining with the mitochondrial potential dye JC-1. Both primary hepatocytes and H4IIEC3 cells exhibited decreased mitochondrial potential after 6 h of palmitate treatment (Figure 1C). While elevated palmitate decreased the mitochondrial potential of both cell types, the effect was more significant in hepatoma cells. Taken together, we posit that H4IIEC3 cells provide a representative model of hepatocyte response to FFA overload, because they recapitulate all of the qualitative features of lipotoxicity observed in primary rat hepatocytes.
3.2. Palmitate lipotoxicity is marked by a redistribution of intracellular calcium
Previous studies have observed reduced ER calcium stores and increased markers of ER stress following palmitate treatment in primary rat hepatocytes and CHO cells [30,31]. To test if our lipotoxicity model exhibits the same decrease in ER luminal calcium, we assessed the relative levels of ER calcium in H4IIEC3 cells treated with 400 μM palmitate for 6 h. Compared to vehicle treatment, cells treated with palmitate exhibited a smaller fold change in Fura-2 fluorescence following thapsigargin treatment, indicating depleted ER calcium stores (Figure 2A). Additionally, we calculated the area under the curve (AUC) from the point of initial thapsigargin treatment to the peak of Fura-2 fluorescence to estimate the relative difference in total ER calcium release between vehicle- and palmitate-treated H4IIEC3 cells (Figure 2B). H4IIEC3 cells treated with 400 μM palmitate had a decreased AUC, confirming that total luminal calcium was diminished by palmitate treatment. Next, we sought to determine if decreased ER calcium was associated with increased mitochondrial calcium. After 6 h of treatment with 400 μM palmitate, H4IIEC3 cells were incubated with the mitochondrial calcium indicator Rhod-2 AM. Palmitate-treated cells exhibited a 39 ± 7% increase in Rhod-2 fluorescence, indicating elevated mitochondrial calcium levels (Figure 2C).
Figure 2.
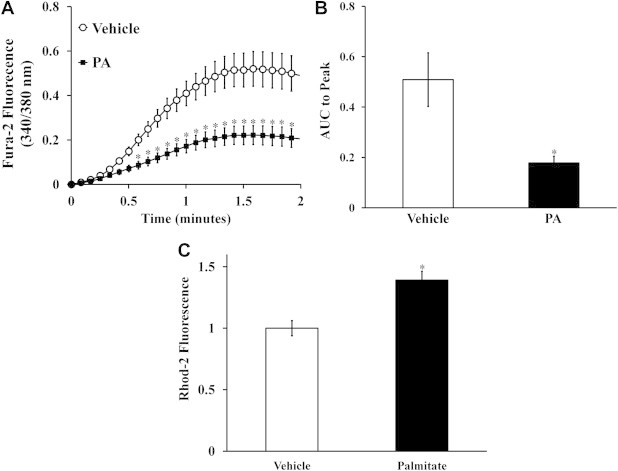
Lipotoxic palmitate redistributes intracellular calcium. (A) To assess ER calcium, H4IIEC3 hepatic cells pre-treated with vehicle (BSA) or 400 μM palmitate (PA) for 6 h were then perfused with thapsigargin (1 μM) while changes in cystolic Fura-2 fluorescence were recorded. (B) Calculated area under curve (AUC) to peak fluorescence was used to quantify relative calcium load released by ER. (C) Relative mitochondrial calcium levels assessed by Rhod-2 fluorescence for H4IIEC3 hepatic cells treated with vehicle (BSA) or 400 μM palmitate (PA) for 6 h. Data represent mean ± S.E., n = 5 plates with 25–30 cells per plate for thapsigargin assays, n = 6 for Rhod-2 measurements; *different from vehicle, p < .05.
3.3. ER calcium release promotes ROS overproduction, enhanced oxygen consumption, and apoptosis in response to a palmitate load
To test whether these other lipotoxic phenotypes were dependent on the observed ER-to-mitochondrial calcium translocation, the cell-permeable calcium chelator BAPTA-AM was administered to H4IIEC3 cells in both the presence and absence of palmitate treatments. Primary hepatocytes and H4IIEC3 hepatic cells co-treated with 40 μM BAPTA and 400 μM palmitate exhibited decreased apoptotic markers relative to cells treated with palmitate alone (Figure 3A). This was associated with reductions in cell death, as measured by PI fluorescence at 24 h (Supplementary Figure S1). The ability of BAPTA to reduce lipotoxic cell death was in agreement with previous reports [30]. While palmitate treatment was characterized by ROS accumulation, reduction in mitochondrial potential, and enhanced oxygen uptake by H4IIEC3 cells, BAPTA co-treatment suppressed all of these palmitate-induced metabolic phenotypes (Figure 3B–D). However, BAPTA co-treatment did not significantly reduce CHOP expression (Supplementary Figure S2). To confirm that BAPTA was rescuing cells by chelating intracellular calcium, cells were also treated with the calcium chelator EGTA, and similar reductions in lipotoxicity were observed in cells co-treated with palmitate (Supplementary Figure S3). These results suggest that palmitate-induced mitochondrial alterations occur downstream of ER stress, and that BAPTA is able to rescue the lipotoxic effects of palmitate by suppressing calcium-dependent activation of mitochondrial metabolism.
Figure 3.
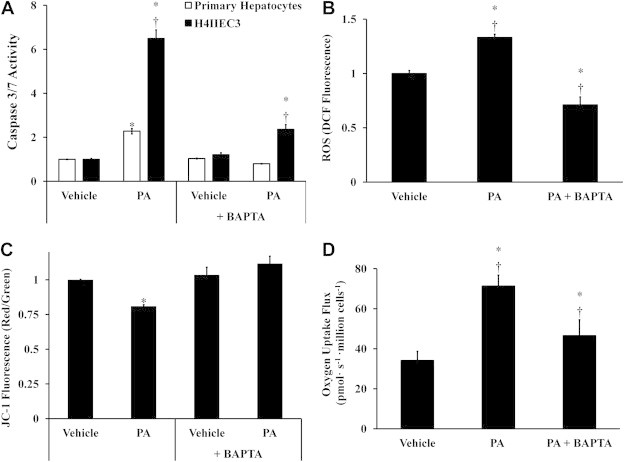
Co-treatment with the intracellular calcium chelator BAPTA-AM reduces the lipotoxic effects of palmitate. Hepatic cells were treated with either vehicle (BSA) or 400 μM palmitate (PA) in the presence or absence of 40 μM BAPTA to examine the role of redistributed calcium stores on apoptosis, ROS accumulation, and mitochondrial metabolism. (A) Caspase 3/7 activity was measured at 12 h to assess the effect of BAPTA treatment on apoptosis in both primary hepatocytes and H4IIEC3 cells. (B) ROS levels at 6 h were measured by DCF fluorescence in H4IIEC3 cells. (C) Mitochondrial membrane potential at 6 h was assessed by JC-1 fluorescence in H4IIEC3 cells. (D) Oxygen uptake measurements of BAPTA- and/or PA-treated H4IIEC3 cells. Data represent mean ± S.E., n = 4 for DCF, n = 3 for oxygen uptake measurements, n = 8 for JC-1, caspase activity, and toxicity assays; *different from vehicle, p < .05; †different from each other, p < .05.
3.4. 13C flux analysis demonstrates that chelation of intracellular calcium reverses metabolic alterations associated with lipotoxicity
Hepatic cells treated with palmitate exhibit an altered metabolic phenotype marked by elevated CAC flux, enhanced glutamate anaplerosis, and increased oxygen consumption [10]. Prior work in our lab has shown that these changes are the primary cause of subsequent ROS accumulation and apoptosis in H4IIEC3 cells, and are not simply byproducts of the apoptosis cascade [10]. To examine how BAPTA affects intracellular metabolism, we performed experiments by replacing unlabeled glutamine in DMEM with the stable isotope tracer [U-13C5]glutamine. First, we examined the 13C atom percent enrichment (APE) of several intermediate metabolites. The APE values represent the fractional contribution of exogenous glutamine to the biosynthesis of these intermediates, relative to other unlabeled sources of carbon (e.g., glucose). GC–MS analysis of intracellular malate and glutamate extracted from palmitate-treated H4IIEC3 cells revealed that their APEs approached 60% and 75%, respectively, compared to 30% and 55% for vehicle-treated cells (Figure 4). BAPTA co-treatment reduced the isotopic enrichment of these metabolites back to vehicle-treated levels. These results demonstrate that palmitate lipotoxicity is characterized by increased glutamine conversion to glutamate and increased relative contribution of glutamate carbon to CAC intermediates. Furthermore, stimulation of this anaplerotic pathway is calcium-dependent.
Figure 4.
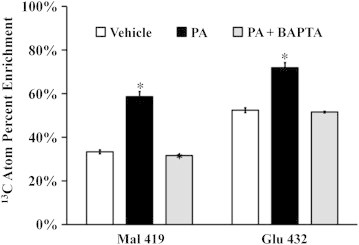
Isotopic enrichment of mitochondrial metabolites. H4IICE3 hepatic cells were incubated with [U-13C5]glutamine and treated with vehicle (BSA), palmitate (PA), or PA + BAPTA for 6 h. Intracellular metabolism was then quenched and metabolites were analyzed using GC–MS. The resulting mass isotopomer distributions were corrected for natural isotope abundance using the method of Fernandez et al. [38]. The atom percent enrichment (APE) of cells was calculated using the formula , where N is the number of carbon atoms in the metabolite and Mi is the fractional abundance of the ith mass isotopomer. APE represents the fractional incorporation of 13C from the labeled isotope tracer (i.e., glutamine) to the measured metabolite fragment ion. The fragment ion Mal 419 contains all four malate carbons. The fragment ion Glu 432 contains all five glutamate carbons. Data represent mean ± S.E., n = 3; *different from vehicle, p < .05.
By combining 13C mass isotopomer measurements of malate, lactate, glutamate, and aspartate fragment ions derived from GC–MS with measured rates of oxygen consumption, we applied 13C MFA to calculate 12 metabolic fluxes (Figure 5) and their associated 95% confidence intervals for vehicle-treated, palmitate-treated, and palmitate + BAPTA co-treated H4IIEC3 cells. Hepatic cells fed 400 μM palmitate were characterized by higher rates of glutamine uptake (Figure 6A), alpha-ketoglutarate dehydrogenase flux (Figure 6C), citrate synthase flux (Figure 6D), and malic enzyme flux (Figure 6E) in comparison to vehicle-treated control cells. BAPTA co-treatment led to reductions in the estimated alpha-ketoglutarate, malic enzyme, and glutaminase fluxes. On the other hand, similar measurements of pyruvate carboxylase (Figure 6F) flux across all treatments suggest that this mode of anaplerosis was not sensitive to palmitate or BAPTA exposure. Rather, the increases in CAC flux and its suppression by BAPTA appear to be glutamine-dependent.
Figure 5.
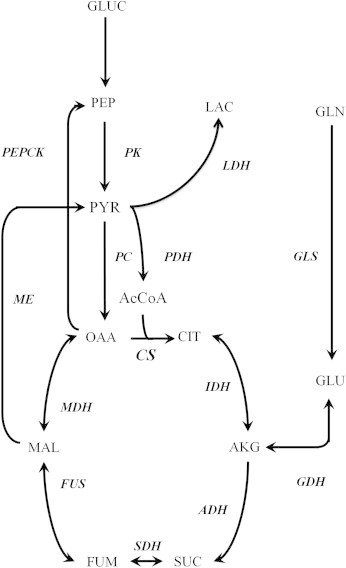
Metabolic network used for 13C MFA. The reactions considered in the metabolic flux analysis are shown.
Figure 6.
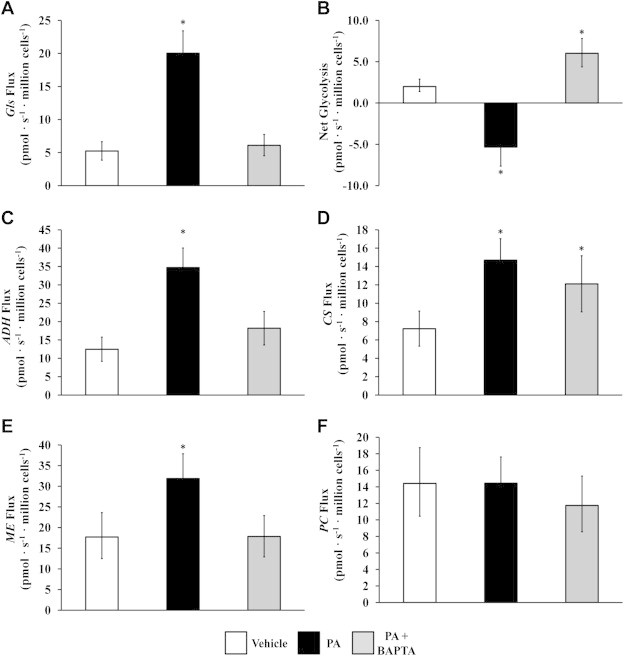
13C flux analysis of mitochondrial metabolism. We performed 13C MFA as detailed in the Methods and the Appendix A. Intracellular CAC and anaplerotic fluxes were calculated for H4IIEC3 cells treated with vehicle (BSA), palmitate (PA), or PA + BAPTA. Calculated fluxes for (A) glutamine uptake, (B) ‘Net glycolysis’ defined as the difference between lactate secretion and glycolytic pyruvate production, (C) alpha-ketoglutarate dehydrogenase, (D) citrate synthase, (E) malic enzyme, and (F) pyruvate carboxylase. Abbreviations: ADH, alpha-ketoglutarate dehydrogenase; CS, citrate synthase; GLN, glutamine uptake; ME, malic enzyme; PC, pyruvate carboxylase. Error bars indicate 95% confidence intervals; *different from vehicle, p < .05.
Our experiments were performed under physiological glucose concentrations and in the absence of glucagon or other hormones that stimulate gluconeogenesis. Therefore, cells exhibited glycolytic metabolism with net conversion of glucose to pyruvate. The 13C MFA calculations allowed us to determine the difference between pyruvate production by glycolysis and its consumption to form lactate, which we denote as ‘net glycolysis’ (Figure 6B). We found that palmitate-treated H4IIEC3 cells were characterized by a negative net glycolytic rate, indicating that excess anaplerotic carbon was exported from the CAC and was excreted as lactate. On the other hand, BAPTA supplementation reverted palmitate-treated cells back to a positive net glycolytic phenotype, similar to that exhibited by vehicle-treated cells, thus demonstrating a suppression of palmitate-induced glutamate anaplerosis by BAPTA.
4. Discussion
Oxidative stress and elevated CAC flux in the liver are characteristics of obesity, NAFLD/NASH, and hepatic lipotoxicity [3,4,11,14,32]. In the current study, we demonstrate that lipotoxic concentrations of the SFA palmitate are associated with the net redistribution of intracellular calcium from the ER to the mitochondria. Our results demonstrate that suppressing intracellular calcium levels can reverse several markers of lipotoxicity in primary rat hepatocytes and H4IIEC3 cells. Experiments co-treating hepatic cells with palmitate plus the calcium chelator BAPTA were able to partially rescue cell death while reducing ROS accumulation and caspase activation. Furthermore, our novel 13C MFA studies revealed that BAPTA prevented the acceleration of CAC metabolism and glutamate anaplerosis associated with palmitate treatment. Our results suggest that altered ER calcium homeostasis provides a critical link between ER stress, ROS accumulation, and altered metabolic phenotypes that contribute to palmitate lipotoxicity. Although mitochondrial lipid composition or respiratory chain coupling could be directly altered by palmitate, it has been previously demonstrated that ER stress and calcium release precede the onset of mitochondrial dysfunction [12]. Taken together with our findings, this indicates that aberrant ER calcium release is the immediate cause of mitochondrial flux alterations that occur in response to palmitate overload.
Perturbed ER homeostasis has been hypothesized as a major initiator of lipid-induced toxicity [1]. Features of ER impairment in these models include activation of the unfolded protein response (UPR) and decreased ER calcium stores. While the UPR can be activated independently of canonical luminal sensors by increased saturation of the ER membrane [33], the role of these ER stress markers in mediating other aspects of lipotoxicity has been unclear. For example, CHOP is a pro-apoptotic protein that is expressed during prolonged periods of UPR. While CHOP is upregulated in response to lipotoxic loads of palmitate, siRNA silencing of CHOP does not prevent apoptosis in H4IIEC3 cells [14]. Additionally, primary hepatocytes from Chop−/− mice exhibited no resistance to elevated palmitate concentrations in vitro. These results confirm that palmitate alters ER function, but do not fully define how ER stress contributes to apoptosis and mitochondrial dysfunction in palmitate-treated hepatic cells. Therefore, CHOP expression is effectively a marker of lipotoxicity but is not required for apoptosis. Collectively, the literature implies that alternate products of ER stress signaling may mediate lipotoxicity, these intermediates have not been previously identified.
Like CHOP expression, ER calcium homeostasis is perturbed in obesity and lipotoxic conditions in both hepatic and non-hepatic cells [1,12,30]. Our use of calcium chelators demonstrates that calcium signaling is critical for palmitate-induced apoptosis, in agreement with previous reports [30]. However, these previous studies did not provide a possible mechanism for how ER calcium release can stimulate apoptosis or promote other markers of lipotoxicity such as oxidative stress or mitochondrial dysfunction. While calcium is a known contributor to the intrinsic apoptotic pathway [21], our experiments demonstrate a unique, direct connection between ER stress and metabolic derangements associated with palmitate lipotoxicity. Our prior work has shown that these metabolic alterations persist even when ROS accumulation and apoptosis are inhibited by antioxidant co-treatments [10], thus implying that they are not simply a byproduct of apoptotic signaling but instead function to promote lipotoxicity. In the current study, we were able to prevent palmitate-induced activation of CAC flux by quenching cytosolic calcium levels with BAPTA co-treatment. Consistent with our prior studies, this normalization of mitochondrial fluxes was associated with reductions in ROS accumulation and caspase activity. Our data therefore demonstrate that alterations in ER calcium storage and trafficking may be an initiating event that causally precedes several downstream aspects of hepatocyte lipotoxicity. Although the effect was not statistically significant, we did observe a partial decrease in CHOP expression in H4IIEC3 hepatic cells co-treated with BAPTA (Supplementary Figure S2), which suggests that oxidative stress could be involved in a positive feedback loop that further enhances ER stress.
Normally, ER calcium is maintained by sarcoendoplasmic reticulum calcium ATPase (SERCA), which functions to pump calcium into the ER lumen from the cytosol. The activity of the SERCA pump is known to be impaired in the obese liver [1] and cholesterol-loaded macrophages [34]. Under normal physiological conditions, the ER membrane is highly fluid due to a low ratio of free cholesterol to phospholipids. Increasing the saturation of ER membranes or perturbing the phosphatidylcholine/phosphatidylethanolamine (PC/PE) ratio has been shown to effectively limit the ability of the SERCA pump to buffer cytosolic calcium. In fact, overexpressing SERCA in vivo is enough to reduce liver ER stress in obese mice, demonstrating that SERCA function is critical to the activation of ER stress in obesity [1]. We hypothesize that saturated fatty acids may be preferentially incorporated into the ER phospholipid membrane and thereby disrupt SERCA function by increasing the membrane saturation. This may, in turn, lead to a net efflux of ER calcium that subsequently translocates to the mitochondria. As shown by the present study, the ER calcium efflux promotes downstream markers of palmitate lipotoxicity, such as ROS accumulation, elevated CAC flux, glutamate anaplerosis, and caspase activation (Figure 7).
Figure 7.
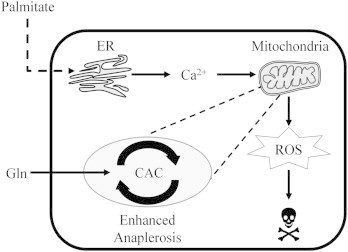
Hypothetical mechanism of palmitate lipotoxicity. Our results demonstrate that lipotoxic concentrations of the saturated fatty acid palmitate alter ER calcium stores and induce mitochondrial dysfunction characterized by elevated glutamine consumption, CAC flux, oxygen consumption, and ROS accumulation. We propose that calcium efflux from ER directly stimulates these altered mitochondrial phenotypes leading to apoptosis. Co-treating hepatic cells with the calcium chelator BAPTA suppresses both PA-induced apoptosis and the associated metabolic abnormalities.
Transient increases in cytosolic calcium due to ER release can directly impact mitochondrial metabolism through two mechanisms, both of which may explain the observed increases in glutamine and O2 consumption by palmitate-treated cells. After uptake through the mitochondrial calcium uniporter, calcium can directly affect the enzymatic activities of pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (IDH), and alpha-ketoglutarate dehydrogenase (ADH) [35,36]. In the case of ADH, calcium increases the enzyme's affinity for its substrate alpha-ketoglutarate (aKG), thus driving the reaction in the forward direction. Activation of ADH may therefore deplete mitochondrial aKG levels and promote increased anaplerosis from glutamate. Our measurement of increased mitochondrial calcium in palmitate-treated H4IIEC3 cells supports this potential mechanism. Alternatively, calcium can amplify O2 consumption without being taken up by the uniporter. Gellerich et al. [19] inhibited mitochondrial calcium uptake in isolated mitochondria and still observed changes in mitochondrial oxygen consumption that were sensitive to extramitochondrial calcium levels. Interestingly, this phenotype is also associated with increased glutamate metabolism. Calcium can alter the malate–aspartate shuttle by enhancing the activity of the glutamate/aspartate antiporter encoded by SLC25A13 and SLC25A12. These are given the common names of citrin (for liver) and aralar (for most other tissues). The malate–aspartate shuttle functions to transport reducing equivalents (e.g., derived from cytosolic NADH) into the mitochondria. Elevated cytosolic levels of calcium could therefore impact mitochondrial metabolic activity by enhancing the capacity of the malate–aspartate shuttle to transport reducing equivalents into the mitochondria, which could result in increased O2 consumption. Although we measured increased mitochondrial calcium in the presence of palmitate, further studies need to be performed to determine the specific role of the mitochondrial calcium uniporter in the lipotoxic phenotype.
Our 13C MFA studies revealed that calcium directly stimulates metabolic alterations, in particular to CAC and associated anaplerotic pathways, in the context of palmitate lipotoxicity. The results indicate that lipotoxic treatments were marked by enhanced oxidative metabolism, which could be abrogated by BAPTA co-treatment. This reduction in CAC flux was dependent on the potential ability of BAPTA to modify glutamate anaplerosis as revealed by the reduction in the 13C enrichment of intracellular glutamate and malate of cells fed [U-13C5]glutamine. Similarly, Noguchi et al. [11] found that glutamate supplementation enhanced palmitate-induced ROS accumulation and apoptosis in palmitate-treated H4IIEC3 cells, and that its effect was strongest of all single amino acids tested. Combined with our current observation that BAPTA co-treatment suppresses glutamine conversion to glutamate and the subsequent entry of glutamate carbon into the CAC, this finding suggests that upregulation of calcium-stimulated anaplerotic metabolism is a critical arm of hepatic lipotoxicity.
Understanding the potential downstream effects of ER stress activation in the context of obesity is important to design potential therapies to prevent the progression of NAFLD toward NASH and other severe liver disorders. This is made particularly relevant by the fact that no pharmacological therapies are currently approved for the treatment of NAFLD or NASH [37]. Because of the rapid appearance of ER stress markers in response to palmitate treatment, we hypothesized that disruption of ER homeostasis may be the initial insult that is responsible for subsequent changes in mitochondrial function. Our experiments outline a novel role for intracellular calcium transport in mediating hepatocyte lipotoxicity. Our cell imaging and 13C MFA results demonstrate that net efflux of ER calcium activates mitochondrial metabolism, thus complementing the findings of Wei et al. [30] that show ER stress playing a central role in palmitate lipotoxicity. For the first time, we show that BAPTA improves hepatic cell viability and reduces caspase activation through the suppression of mitochondrial metabolism and ROS accumulation in the context of a lipotoxic fatty acid load. Specifically, BAPTA co-treatment blunted both ROS accumulation and caspase activation at 6- and 12-h time points. Our unique 13C MFA approach demonstrates that BAPTA suppresses these lipotoxic phenotypes by preventing palmitate-stimulated mitochondrial dysfunction involving enhanced CAC flux and glutamate anaplerosis. Clearly, BAPTA co-treatment is capable of significantly changing and delaying the normal sequence of events in the mechanism of palmitate lipotoxicity. Our data provide a novel mechanism connecting palmitate-induced depletion of ER calcium stores with dysregulated mitochondrial metabolism, indicating an important role for impaired SERCA function and calcium signaling in the initiation of lipotoxic oxidative stress.
Acknowledgments
This research was supported by National Science Foundation CAREER Award CBET-0955251 (to JDY) and the Vanderbilt Diabetes Research and Training Center (NIH DK020593). RAE was supported by the NSF Graduate Research Fellowship Program. We would like to thank David Wasserman lab members Ashley S. Williams and Louise Lantier for their technical assistance with obtaining oxygen uptake measurements using the Oroboros Oxygraph-2K instrument. Additionally, we thank Prasanna Dadi for his time and help with performing ER calcium release assays.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Fu S., Yang L., Li P., Hofmann O., Dicker L., Hide W. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011 May 26;473(7348):528–531. doi: 10.1038/nature09968. [PubMed PMID: WOS:000290951300044] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sunny N.E., Parks E.J., Browning J.D., Burgess S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metabolism. 2011 Dec 7;14(6):804–810. doi: 10.1016/j.cmet.2011.11.004. [PubMed PMID: 22152305. English] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozcan U., Cao Q., Yilmaz E., Lee A., Iwakoshi N., Ozdelen E. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004 Oct 15;306(5695):457–461. doi: 10.1126/science.1103160. [PubMed PMID: ISI:000224626500044. English] [DOI] [PubMed] [Google Scholar]
- 4.Satapati S., Sunny N.E., Kucejova B., Fu X.R., He T.T., Mendez-Lucas A. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. Journal of Lipid Research. 2012 Jun;53(6):1080–1092. doi: 10.1194/jlr.M023382. [PubMed PMID: 22493093. Pubmed Central PMCID: 3351815: WOS:000303913200005. English] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldstein A., Canbay A., Angulo P., Taniai M., Burgart L., Lindor K. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003 Aug;125(2):437–443. doi: 10.1016/s0016-5085(03)00907-7. [PubMed PMID: ISI:000184531200023. English] [DOI] [PubMed] [Google Scholar]
- 6.Angulo P., Lindor K.D. Non-alcoholic fatty liver disease. Journal of Gastroenterology and Hepatology. 2002 Feb;17:S186–S190. doi: 10.1046/j.1440-1746.17.s1.10.x. [PubMed PMID: WOS:000178216300016] [DOI] [PubMed] [Google Scholar]
- 7.Neuschwander-Tetri B.A., Caldwell S.H. Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology. 2003 May;37(5):1202–1219. doi: 10.1053/jhep.2003.50193. [PubMed PMID: 12717402. Epub 2003/04/30. English] [DOI] [PubMed] [Google Scholar]
- 8.Li Z.Z., Berk M., McIntyre T.M., Feldstein A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease role of stearoyl-CoA desaturase. Journal of Biological Chemistry. 2009 Feb 27;284(9):5637–5644. doi: 10.1074/jbc.M807616200. [PubMed PMID: WOS:000263560600027] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puri P., Wiest M.M., Cheung O., Mirshahi F., Sargeant C., Min H.K. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology. 2009 Dec;50(6):1827–1838. doi: 10.1002/hep.23229. [PubMed PMID: 19937697] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egnatchik R.A., Leamy A.K., Noguchi Y., Shiota M., Young J.D. Palmitate-induced activation of mitochondrial metabolism promotes oxidative stress and apoptosis in H4IIEC3 rat hepatocytes. Metabolism. 2014 Feb;63(2):283–295. doi: 10.1016/j.metabol.2013.10.009. [PubMed PMID: 24286856. Pubmed Central PMCID: PMC3946971. English] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noguchi Y., Young J., Aleman J., Hansen M., Kelleher J., Stephanopoulos G. Effect of anaplerotic fluxes and amino acid availability on hepatic lipoapoptosis. Journal of Biological Chemistry. 2009 Nov 27;284(48):33425–33436. doi: 10.1074/jbc.M109.049478. [PubMed PMID: ISI:000272028500049. English] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borradaile N.M., Han X., Harp J.D., Gale S.E., Ory D.S., Schaffer J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. Journal of Lipid Research. 2006 Dec;47(12):2726–2737. doi: 10.1194/jlr.M600299-JLR200. [PubMed PMID: WOS:000242100800013] [DOI] [PubMed] [Google Scholar]
- 13.Leamy A.K., Egnatchik R.A., Young J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Progress in Lipid Research. 2013 Jan;52(1):165–174. doi: 10.1016/j.plipres.2012.10.004. [PubMed PMID: 23178552] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfaffenbach K., Gentile C., Nivala A., Wang D., Wei Y., Pagliassotti M. Linking endoplasmic reticulum stress to cell death in hepatocytes: roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. American Journal of Physiology – Endocrinology and Metabolism. 2010 May;298(5):E1027–E1035. doi: 10.1152/ajpendo.00642.2009. [PubMed PMID: ISI:000276635200014. English] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Padilla A., Descorbeth M., Almeyda A.L., Payne K., De Leon M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Research. 2011 Jan 25;1370:64–79. doi: 10.1016/j.brainres.2010.11.013. [PubMed PMID: WOS:000287065900006] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei Y., Wang D., Topczewski F., Pagliassotti M. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. American Journal of Physiology – Endocrinology and Metabolism. 2006 Aug;291(2):E275–E281. doi: 10.1152/ajpendo.00644.2005. [PubMed PMID: ISI:000238841200010. English] [DOI] [PubMed] [Google Scholar]
- 17.Brookes P.S., Yoon Y.S., Robotham J.L., Anders M.W., Sheu S.S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology – Cell Physiology. 2004 Oct;287(4):C817–C833. doi: 10.1152/ajpcell.00139.2004. [PubMed PMID: WOS:000223762000001] [DOI] [PubMed] [Google Scholar]
- 18.Barron J.T., Gu L.P., Parrillo J.E. Malate-aspartate shuttle, cytoplasmic NADH redox potential, and energetics in vascular smooth muscle. Journal of Molecular and Cellular Cardiology. 1998 Aug;30(8):1571–1579. doi: 10.1006/jmcc.1998.0722. [PubMed PMID: WOS:000076003600011. English] [DOI] [PubMed] [Google Scholar]
- 19.Gellerich F.N., Gizatullina Z., Trumbeckaite S., Nguyen H.P., Pallas T., Arandarcikaite O. The regulation of OXPHOS by extramitochondrial calcium. Biochimica et Biophysica Acta – Bioenergetics. 2010 Jun–Jul;1797(6–7):1018–1027. doi: 10.1016/j.bbabio.2010.02.005. [PubMed PMID: WOS:000279663200049] [DOI] [PubMed] [Google Scholar]
- 20.Contreras L., Satrustegui J. Calcium signaling in brain mitochondria interplay of malate aspartate NADH shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. Journal of Biological Chemistry. 2009 Mar 13;284(11):7091–7099. doi: 10.1074/jbc.M808066200. [PubMed PMID: WOS:000263919000057] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gyorgy H., Gyrogy C., Das S., Garcia-Perez C., Saotome M., Roy S. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006 Nov–Dec;40(5–6):553–560. doi: 10.1016/j.ceca.2006.08.016. [PubMed PMID: ISI:000242558800013. English] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nutt L., Chandra J., Pataer A., Fang B., Roth J., Swisher S. Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. Journal of Biological Chemistry. 2002 Jun 7;277(23):20301–20308. doi: 10.1074/jbc.M201604200. [PubMed PMID: ISI:000176204500028. English] [DOI] [PubMed] [Google Scholar]
- 23.White C., Li C., Yang J., Petrenko N., Madesh M., Thompson C. The endoplasmic reticulum gateway to apoptosis by Bcl-X-L modulation of the InsP(3)R. Nature Cell Biology. 2005 Oct;7(10):1021–1028. doi: 10.1038/ncb1302. [PubMed PMID: ISI:000232356100018. English] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiota M., Inagami M., Fujimoto Y., Moriyama M., Kimura K., Sugano T. Cold acclimation induces zonal heterogeneity in gluconeogenic responses to glucagon in rat liver lobule. American Journal of Physiology. 1995;268(6 Part 1):E1184–E1191. doi: 10.1152/ajpendo.1995.268.6.E1184. [PubMed PMID: BIOSIS: PREV199598402818] [DOI] [PubMed] [Google Scholar]
- 25.Jacobson D.A., Weber C.R., Bao S.Z., Turk J., Philipson L.H. Modulation of the pancreatic islet beta-cell-delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. Journal of Biological Chemistry. 2007 Mar;282(10):7442–7449. doi: 10.1074/jbc.M607858200. [PubMed PMID: WOS:000245080900056] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young J.D. INCA: a computational platform for isotopically nonstationary metabolic flux analysis. Bioinformatics. 2014;30(9):1333–1335. doi: 10.1093/bioinformatics/btu015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antoniewicz M.R., Kelleher J.K., Stephanopoulos G. Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metabolic Engineering. 2007 Jan;9(1):68–86. doi: 10.1016/j.ymben.2006.09.001. [PubMed PMID: WOS:000243832000007] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Young J., Walther J., Antoniewicz M., Yon H., Stephanopoulos G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnology and Bioengineering. 2008 Feb 15;99(3):686–699. doi: 10.1002/bit.21632. [PubMed PMID: ISI:000252511800021. English] [DOI] [PubMed] [Google Scholar]
- 29.Antoniewicz M.R., Kelleher J.K., Stephanopoulos G. Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metabolic Engineering. 2006;8:324–337. doi: 10.1016/j.ymben.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 30.Wei Y., Wang D., Gentile C., Pagliassotti M. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Molecular and Cellular Biochemistry. 2009 Nov;331(1–2):31–40. doi: 10.1007/s11010-009-0142-1. [PubMed PMID: ISI:000271308000005. English] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borradaile N.M., Harp J.D., Schaffer J.E. Palmitate-induced changes in endoplasmic reticulum structure and function: a central role for the ER in lipotoxicity. Arteriosclerosis Thrombosis and Vascular Biology. 2006 May;26(5):E49. [PubMed PMID: WOS:000236942400073] [Google Scholar]
- 32.Puri P., Mirshahi F., Cheung O., Natarajan R., Maher J.W., Kellum J.M. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 33.Volmer R., van der Ploeg K., Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proceedings of the National Academy of Sciences. 2013;110(12):4628–4633. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y.K., Ge M.T., Ciani L., Kuriakose G., Westover E.J., Dura M. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids – implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. Journal of Biological Chemistry. 2004 Aug 27;279(35):37030–37039. doi: 10.1074/jbc.M405195200. [PubMed PMID: WOS:000223453600106] [DOI] [PubMed] [Google Scholar]
- 35.Denton R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochimica et Biophysica Acta. 2009 Nov;1787(11):1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [PubMed PMID: 19413950. English] [DOI] [PubMed] [Google Scholar]
- 36.McCormack J.G., Denton R.M. Intracellular calcium ions and intramitochondrial Ca2+ in the regulation of energy metabolism in mammalian tissues. The Proceeding of the Nutrition Society. 1990 Feb;49(1):57–75. doi: 10.1079/pns19900009. [PubMed PMID: 2190228. English] [DOI] [PubMed] [Google Scholar]
- 37.Sanyal A.J., Chalasani N., Kowdley K.V., McCullough A., Diehl A.M., Bass N.M. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. New England Journal of Medicine. 2010 May 6;362(18):1675–1685. doi: 10.1056/NEJMoa0907929. [PubMed PMID: WOS:000277311200005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandez C.A., Des Rosiers C., Previs S.F., David F., Brunengraber H. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. Journal of Mass Spectrometry. 1996 Mar;31(3):255–262. doi: 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO;2-3. [PubMed PMID: 8799277] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.