

Abstract
GLP-1R agonists improve outcomes in ischemic heart disease. Here we studied GLP-1R-dependent adaptive and cardioprotective responses to ventricular injury. Glp1r−/− hearts exhibited chamber-specific differences in gene expression, but normal mortality and left ventricular (LV) remodeling after myocardial infarction (MI) or experimental doxorubicin-induced cardiomyopathy. Selective disruption of the cardiomyocyte GLP-1R in Glp1rCM−/− mice produced no differences in survival or LV remodeling following LAD coronary artery occlusion. Unexpectedly, the GLP-1R agonist liraglutide still produced robust cardioprotection and increased survival in Glp1rCM−/− mice following LAD coronary artery occlusion. Although liraglutide increased heart rate (HR) in Glp1rCM−/− mice, basal HR was significantly lower in Glp1rCM−/− mice. Hence, endogenous cardiomyocyte GLP-1R activity is not required for adaptive responses to ischemic or cardiomyopathic injury, and is dispensable for GLP-1R agonist-induced cardioprotection or enhanced chronotropic activity. However the cardiomyocyte GLP-1R is essential for the control of HR in mice.
Keywords: Glucagon-like peptide-1, Glucagon-like peptide-1 receptor, Ischemia, Myocardial infarction, Cardiomyopathy, Heart failure, Incretin
Non-standard abbreviations and acronyms: GLP-1, glucagon-like peptide-1; GLP-1R, glucagon-like peptide-1 receptor; tGLP-1, truncated forms of GLP-1 such as GLP-1(9–36) or GLP-1(28–36); HR, heart rate; LAD, left anterior descending; MI, myocardial infarction
1. Introduction
Type 2 diabetes mellitus is treated using pharmacotherapy with agents acting through distinct anti-diabetic mechanisms that may be associated with unexpected adverse effects on cardiovascular outcomes, independent of glycemic control [1]. For example, thiazolidinediones increase fluid retention and peripheral edema in diabetic subjects with heart failure [2] whereas some dipeptidyl peptidase 4 (DPP-4) inhibitors increase the rate of hospitalization for heart failure [3]. The first glucagon-like peptide-1 (GLP-1) receptor (GLP-1R) agonist was approved for clinical use in 2005 and results of several studies suggest that native GLP-1 or degradation-resistant GLP-1R agonists may be beneficial in subjects with ischemic cardiac injury or heart failure [4–6]. The largest randomized controlled trial demonstrated that a 6 h infusion of exenatide significantly improved the myocardial salvage index and reduced infarct size relative to the ischemic area at risk in human subjects with acute myocardial infarction (MI) [7].
Although pre-clinical studies demonstrate that GLP-1R agonists preserve ventricular function and reduce infarct size [8,9], the physiological importance of the endogenous GLP-1R for the response to cardiac injury has not been elucidated. Furthermore, the surprising demonstration that ventricular cardiomyocytes do not express the GLP-1R [10] raises important questions about mechanisms linking GLP-1R signaling to ventricular function and cardioprotection. We have now examined the physiological importance of endogenous GLP-1R signaling for the response to ischemic injury or doxorubicin-induced cardiomyopathy in Glp1r deficient (Glp1r−/−) mice and in newly generated Glp1rCM−/− mice with cardiomyocyte-specific inactivation of the Glp1r. Surprisingly, global or cardiomyocyte-specific disruption of GLP-1R signaling in mice does not influence the extent of injury or survival after MI or experimental cardiomyopathy.
As GLP-1R agonists are administered to humans prior to or following the development of ischemic myocardial injury, we also studied the actions of GLP-1R agonists in mice when administered before or after coronary artery ligation. Surprisingly, administration of exendin-4 after the onset of MI did not modify infarct size or survival. Unexpectedly, the GLP-1R agonist liraglutide continued to produce robust cardioprotection in Glp1rCM−/− mice. Although the cardiomyocyte GLP-1R was not required for liraglutide-mediated increases in heart rate (HR), basal HR was significantly lower in Glp1rCM−/− mice. Taken together, these findings demonstrate that the cardiomyocyte GLP-1R is not essential for i) the endogenous physiological response to ischemic or cardiomyopathic injury, ii) GLP-1R-dependent cardioprotection or iii) the pharmacological GLP-1R-dependent increase in HR. In contrast, basal signaling through the atrial cardiomyocyte GLP-1R is essential for control of HR in mice.
2. Methods
2.1. Animal care
Animal experiments were carried out using protocols approved by Mt. Sinai Hospital and The Toronto Centre for Phenogenomics (TCP; Toronto, ON, Canada). Mice were housed under a 12-h light/dark cycle in the TCP animal facility with free access to standard rodent diet (2018, 18% kcal from fat; Harlan Teklad, Mississauga, ON, Canada) and water, unless otherwise noted. Experiments were carried out in male mice acclimatized to handling. Glp1r−/− mice have been described [11]. To generate Glp1rCM−/− mice, MerCreMer transgenic mice expressing tamoxifen-inducible Cre driven by the α-myosin heavy chain (αMHC) promoter were bred with floxed Glp1r mice [12]. Cre-induced inactivation of the Glp1r gene was carried out via 6 intraperitoneal (i.p.) injections of tamoxifen (50 mg/kg) over 8 days (Supplementary Figure 1). As induction of Cre in cardiac myocytes induces a transient, reversible cardiomyopathy [13], mice were allowed 5 weeks to recover before experimentation.
2.2. Permanent left anterior descending (LAD) coronary artery occlusion
Experimental MI was induced via permanent ligation of the LAD coronary artery in 10–12-week-old male Glp1r−/− mice and Glp1r+/+ littermates, or 16–20-week-old Glp1rCM−/− mice and their αMHC-Cre littermates as described [14]. Cardiac examinations were performed on all deceased mice. The presence of a large amount of blood or clot around the heart and in the thoracic cavity, in addition to a perforation of the infarct or peri-infarct area was indicative of cardiac rupture.
2.3. Experimental cardiomyopathy
Experimental cardiomyopathy was induced via single i.p. injection of doxorubicin (20 mg/kg) in Glp1r−/− mice and Glp1r+/+ littermates, or in C57BL/6J mice [15]. Mice were followed for 10 days and hearts from surviving mice underwent histological assessment, or analysis of gene and protein expression.
2.4. Treatment with GLP-1R agonists
In subsets of experiments involving experimental MI or cardiomyopathy, groups of mice were treated with either liraglutide (30 μg/kg i.p. twice daily, Novo Nordisk), the GLP-1R agonist, exendin-4 (5 nmol/kg i.p. twice daily, CHI Scientific), or saline. All injections took place between 7:00–8:00 am and 4:00–5:00 pm. To assess consequences of GLP-1R activation before induction of ischemia, liraglutide was administered twice daily for 1 week before MI [14]. To assess the effects of activating the GLP-1R following induction of ischemia or cardiomyopathy, exendin-4 injections were initiated concurrent with induction of MI or cardiomyopathy.
2.5. Histology and assessment of left ventricle (LV) infarct scar formation
Animals were anesthetized using avertin (250 mg/kg i.p. injection). The chest was opened and an apical injection of 1 M KCl arrested the heart in diastole. Hearts were perfusion-fixed with 4% buffered formalin at physiological pressure, post-fixed in formalin, embedded in paraffin, sectioned at 6 μm, and stained with Masson's Trichrome or hematoxylin and eosin (H&E). Cardiac morphometry was performed with Aperio ImageScope Viewer software (Aperio Technologies) using digital planimetry [14,16]. Infarcted/scarred LV area was calculated as a % of total LV area. Cardiac hypertrophy was quantified as the heart weight-to or ventricular weight-to-body weight or tibia length ratio. LV atrial natriuretic peptide (ANP) expression was determined via immunohistochemistry utilizing anti-ANP (Santa Cruz) antibody.
2.6. Assessment of heart rate (HR) via telemetry
HR was assessed in conscious, freely moving mice via implantation of radiotelemetry devices (PA-C10 from DSI) as previously described [10]. Mice were allowed 1 week to recover following device implantation prior to data collection. In some experiments, mice were injected with liraglutide (30 μg/kg i.p.) or saline, or subjected to fasting–refeeding.
2.7. Determination of plasma ANP
Plasma ANP levels were quantified using commercially available enzyme-linked immunosorbent assays (ELISA) (Ray Bio, USA) [10].
2.8. Glucose tolerance
Oral and i.p. glucose tolerance tests were performed in fasted mice using a glucose dose of 2 g/kg. During i.p. glucose tolerance testing, mice were injected with either exendin-4 (24 nmol/kg), or saline, 15 min prior to glucose injection.
2.9. Plasma insulin
Plasma was collected from mice via tail bleed at 15 min post-glucose administration during both oral and i.p. glucose tolerance, and plasma insulin levels were determined by ELISA (Alpco Diagnostics).
2.10. Real time quantitative PCR
First-strand cDNA was synthesized from total RNA using the SuperScript III synthesis system (Invitrogen, Carlsbad, CA). Real time PCR was carried out with the ABI Prism 7900 Sequence Detection System using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA). Relative mRNA transcript levels were quantified with the 2−ΔΔCt method [17] using cyclophilin as an internal control. PCR primers are shown in Supplementary Table 1.
2.11. Immunoblot analysis
Frozen powdered ventricular tissue (20 mg) was homogenized as described [18] and following gel electrophoresis, immunoblotting was carried out using antibodies listed in Supplementary Table 2. Immunoblots were visualized with the enhanced chemiluminescence Western blot detection kit (Perkin Elmer) and quantified with Carestream Molecular Imaging Software (Kodak).
2.12. Statistical analysis
All values are presented as mean ± SE (n observations). The significance of differences was determined by a Kaplan Meier survival analysis, an unpaired 2-tailed Student's t-test, a two-way analysis of variance (ANOVA), or a one-way ANOVA followed by a Bonferroni post-hoc analysis where appropriate. Differences were considered significant when P < 0.05.
3. Results
3.1. Glp1r−/− mice do not exhibit enhanced susceptibility to ischemia-induced mortality or experimental cardiomyopathy
Although the cardiovascular consequences of pharmacological activation of the GLP-1R have been extensively studied [8], little is known about the endogenous physiological importance of basal GLP-1R signaling for the response to ventricular injury. We first backcrossed Glp1r−/− mice, originally on a CD1 background [11,19], for 6 generations on to the C57BL/6 background, and observed normal cardiac structure in C57BL/6 Glp1r−/− mice (Supplementary Figure 2). To determine whether loss of basal GLP-1R signaling impairs the response to cardiac injury, Glp1r−/− and littermate Glp1r+/+ mice were subjected to permanent occlusion of the LAD coronary artery. Although results with Glp1r−/− mice trended towards increased mortality, these differences were not statistically significant (Figure 1A). Levels of Tnfα and Ccl2 mRNA transcripts were reduced, whereas Gdf5 expression was increased in ventricular RNA from Glp1r−/− mice (Supplementary Figure 3), however no differences in LV infarct scar formation or cardiac hypertrophy were detected (Figure 1B and C).
Figure 1.
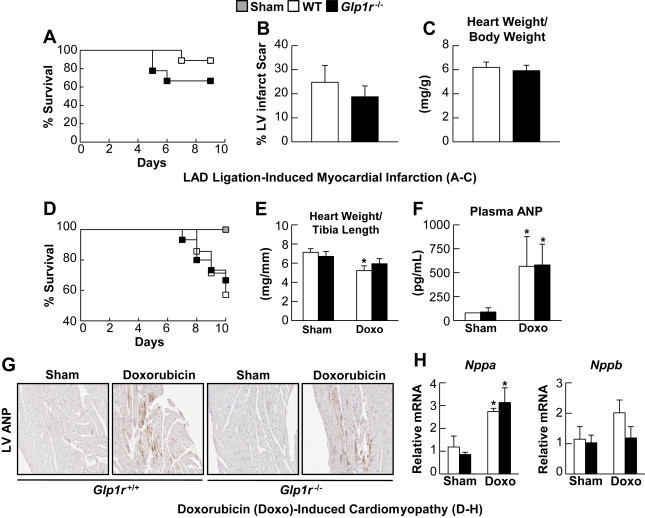
Whole-body GLP-1R deficiency does not influence cardiovascular outcomes following LAD-ligation induced MI and Doxorubicin-induced cardiomyopathy. A: Survival in Glp1r−/− mice and their Glp1r+/+ littermates over 9 days following permanent LAD coronary artery occlusion (n = 9). B: % LV infarct scar formation from Masson's Trichrome fixed heart sections from Glp1r−/− and wild-type littermates at day 9 post-LAD ligation (n = 6). C: Cardiac hypertrophy at day 9 post-LAD ligation (n = 6–8). D: Survival in Glp1r−/− and Glp1r+/+ littermates following treatment with a single dose of doxorubicin (20 mg/kg i.p. injection) (n = 14–15). E: Heart weight/tibia length ratios at 10 days post-doxorubicin (n = 4–5). F: Plasma ANP levels 4 days post-doxorubicin treatment (Shams, n = 3, Doxorubicin, n = 5–7). G: Histology for ANP expression in LV from sham and doxorubicin-treated mice. H: Nppa and Nppb mRNA expression (normalized to Ppia) in ventricles (n = 3–5). Values represent mean ± SE. The significance of differences was determined by a two-way ANOVA followed by a Bonferroni post-hoc analysis. *significantly different from sham counterpart.
As GLP-1R agonists ameliorate the severity of experimental and clinical ventricular dysfunction [4,20,21], we assessed whether loss of basal GLP-1R signaling modified outcomes in mice with doxorubicin-induced cardiomyopathy. Glp1r−/− mice exhibited no differences in survival following doxorubicin administration (Figure 1D). Although the extent of cardiac atrophy was attenuated (Figure 1E) and expression of inflammation-associated genes such as Tnfα, Ccl2, Hmox1, and Tgfβ2 was reduced in ventricles from Glp1r−/− mice (Supplementary Figure 4), plasma levels of ANP (Figure 1F), LV ANP expression (Figure 1G), and levels of Nppa and Nppb mRNA (Figure 1H) were similar in Glp1r−/− vs. Glp1r+/+ mice. Hence, whole body deletion of the Glp1r does not impair the adaptive response to ischemic or cardiomyopathic ventricular injury.
3.2. Systemic GLP-1R activation with exendin-4 does not modify cardiovascular outcomes following induction of ischemia or experimental cardiomyopathy
Treatment of rodents with GLP-1R agonists prior to induction of ischemia produces robust cardioprotection [14], however it remains unclear whether GLP-1R activation commenced after induction of ischemic injury is similarly beneficial in rodents in vivo [8]. Importantly, exendin-4 (exenatide) has been administered to human subjects after the onset of ischemic injury or MI, with promising or indeterminate results [5,7,22]. Hence, we asked whether administration of exendin-4 to mice with ischemic cardiac injury might be similarly cardioprotective. Treatment of C57BL/6J mice with exendin-4 for 1 week starting after LAD coronary artery ligation (5 nmol/kg i.p. twice daily, a dose that improved glucose tolerance in separate groups of mice (data not shown), but did not perturb body weight or random fed glycemia, (Supplementary Figure 5) did not improve survival (Figure 2A), nor improve MI-induced LV remodeling, as evidenced by similar LV infarct scar formation and MI-induced cardiac hypertrophy (Figure 2B and C).
Figure 2.
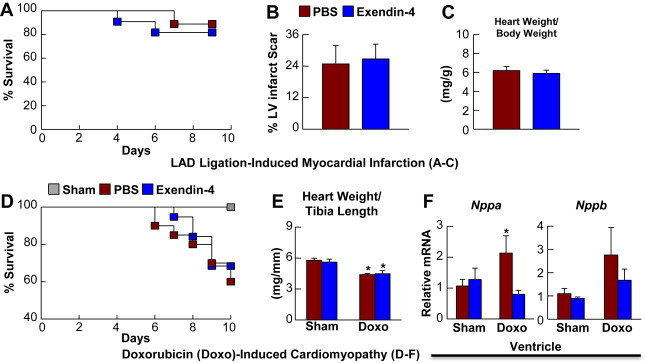
Systemic GLP-1R activation does not influence cardiovascular outcomes following LAD-ligation induced MI and Doxorubicin-induced cardiomyopathy. A: Survival in C57BL/6J mice treated with saline (PBS) or exendin-4 (5 nmol/kg BW i.p., twice daily) for 9 days following permanent LAD coronary artery occlusion (n = 9–11). B: % LV infarct scar formation in heart sections analyzed at day 9 post-LAD ligation (n = 6–7). C: Cardiac hypertrophy analyzed at day 9 post-LAD ligation (n = 8–9). D: Survival following treatment with a single dose of doxorubicin (20 mg/kg i.p. injection) (n = 19–20). Mice were injected for 7 days with saline (PBS) or exendin-4 (5 nmol/kg BW i.p. injection twice daily) with the first injection taking place concurrently with the doxorubicin treatment. E: Heart weight/tibia length ratios at 10 days post-doxorubicin (n = 4–6). F: Nppa and Nppb mRNA expression (normalized to Ppia) in ventricles from saline- and exendin-4-treated C57BL/6J mice at 10 days post-doxorubicin treatment (n = 4–5). Values represent mean ± SE. The significance of differences was determined by a two-way ANOVA followed by a Bonferroni post-hoc analysis. *significantly different from sham counterpart.
As both native GLP-1 and GLP-1R agonists such as exendin-4 have yielded variable outcomes when administered in experimental models and clinical trials of heart failure [6,21,23,24], we asked whether exendin-4 would improve outcomes in mice with experimental cardiomyopathy. Treatment of C57BL/6J mice with exendin-4 for 1 week did not improve survival following doxorubicin-induced cardiomyopathy (Figure 2D), nor the extent of cardiac atrophy (Figure 2E). In contrast, the increase in LV Nppa mRNA transcripts in saline-treated mice was absent in exendin-4-treated mice (Figure 2F). Systemic exendin-4 treatment also reduced ventricular inflammatory gene expression (Il1β and Hmox1) 48 h post-LAD coronary artery occlusion (Supplementary Figure 6A), whereas exendin-4 had less robust effects on ventricular or atrial inflammatory gene expression profiles in the setting of experimental cardiomyopathy (Supplementary Figure 7A). Despite localization of Glp1r expression to cardiac atria [10], exendin-4 did not produce major changes in levels of atrial mRNA transcripts after LAD coronary artery occlusion (Supplementary Figure 6B), or following doxorubicin administration (Supplementary Figure 7B). Hence, systemic GLP-1R activation produces modest changes in cardiac gene expression but does not modify outcomes after ischemic or cardiomyopathic ventricular injury in WT mice.
3.3. Mice with cardiomyocyte-specific inactivation of the Glp1r (Glp1rCM−/−) have normal cardiac structure
As Glp1r−/− mice may exhibit developmental or compensatory adaptive metabolic changes that could indirectly influence their response to cardiac injury [25–27], we generated mice with inducible inactivation of the Glp1r in cardiac myocytes (Glp1rCM−/−; Supplementary Figure 1). Tamoxifen-induction of Cre expression resulted in ∼90% reduction in atrial Glp1r mRNA expression with no change in lung or pancreas Glp1r mRNA expression in Glp1rCM−/− mice (Figure 3A). Oral glucose tolerance (Figure 3B) was normal and the glucoregulatory actions of exendin-4 (Figure 3C and D) were preserved in Glp1rCM−/− mice, consistent with selective inactivation of the Glp1r in cardiomyocytes. Cardiac structure (5 weeks after the last tamoxifen injection) was normal (Figure 3E–G) indicating that selective reduction of Glp1r expression in adult cardiomyocytes does not produce unexpected changes in cardiac chamber development or glucose homeostasis.
Figure 3.
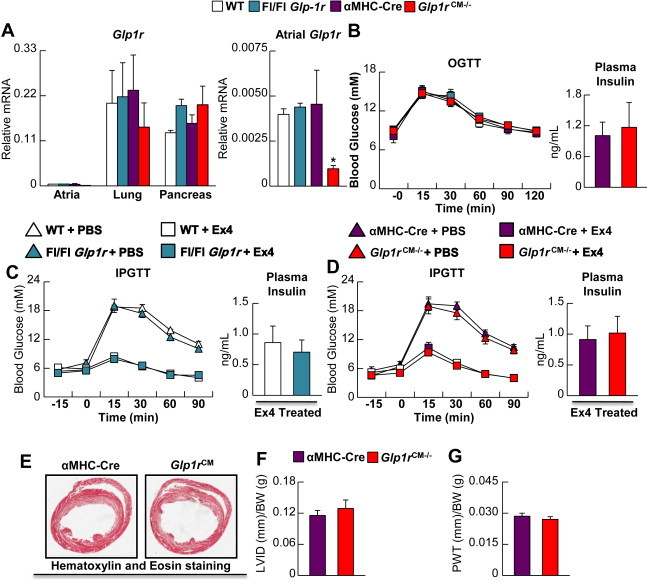
Characterization of cardiomyocyte-specific Glp1r knockout mice (Glp1rCM−/−). A: Glp1r mRNA (normalized to Ppia) in atria, lung and pancreas from Glp1rCM−/− mice 5 weeks following tamoxifen administration (n = 4). The right hand panel for atrial RNA depicts an expanded relative scale for assessment of levels of Glp1r mRNA transcripts. B–D: Glycemic control and plasma insulin levels in Glp1rCM−/− mice and control littermates during oral (B) glucose tolerance testing (OGTT, n = 6–8) and intraperitoneal (C–D; IPGTT, n = 5–7) glucose tolerance testing. During the IPGTT, mice were treated with exendin-4 (24 nmol/kg i.p.) (n = 5–7) or saline (PBS). E: Representative H&E heart cross sections depicting LV structure in αMHC-Cre littermate control vs. Glp1rCM−/− mice (n = 3), F: LV internal diameter (LVID) and G: LV posterior wall thickness (PWT). Values represent mean ± SE. Statistical significance was determined by a one-way ANOVA followed by a Bonferroni post-hoc analysis. *significantly different from all other groups.
3.4. Glp1rCM−/− mice do not exhibit enhanced mortality or adverse LV remodeling after MI
To determine whether selective loss of the cardiomyocyte GLP-1R impaired cardiomyocyte gene or protein expression or the response to ischemic injury, we induced experimental MI by coronary artery ligation in Glp1rCM−/− mice. Viable ventricular myocardium adjacent to the site of infarct from Glp1rCM−/− mice 48 h post-LAD coronary artery occlusion revealed few differences in expression of proteins previously implicated in GLP-1R-dependent cardioprotection, with the exception of heme-oxygenase 1 expression and 5′ AMP activated protein kinase phosphorylation, which were lower in Glp1rCM−/− mice (Figure 4). Similarly, RNA analyses did not reveal major differences in ventricular expression of key genes involved in inflammation, extracellular matrix remodeling, or natriuresis in Glp1rCM−/− mice, other than an exacerbation of the MI-induced increase in Ccl2, Mmp9, and Nppb mRNA expression (Supplementary Figure 8A–C). In contrast, we observed significant differential expression of genes important for inflammation (Il-6), extracellular matrix remodeling (Mmp9, Timp1), and natriuresis (Nppb), and attenuation of MI-induced increases in IL1β, Ccl2, Hmox1, and Gdf15 (Supplementary Figure 8D–F) in atrial RNA from Glp1rCM−/− hearts 48 h post-LAD coronary artery occlusion.
Figure 4.
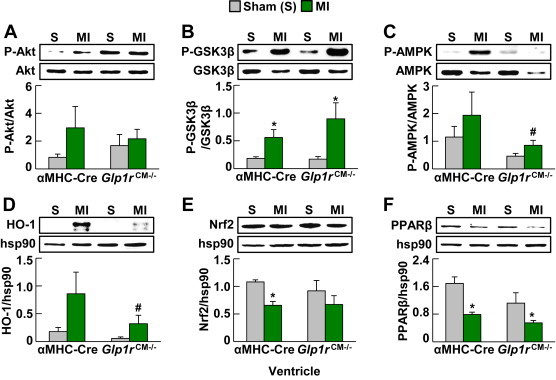
Analysis of candidate cardioprotective proteins in hearts from Glp1rCM−/mice following MI. A: Akt phosphorylation, B: Glycogen synthase kinase 3β (GSK3β) phosphorylation, C: 5′ AMP activated protein kinase (AMPK) phosphorylation, D: Heme oxygenase-1 (HO-1) expression, E: Nuclear respiratory factor 2 (Nrf2) expression, and F: Peroxisome proliferator activated receptor β (PPARβ) expression in ventricles (viable myocardium) extracted from Glp1rCM−/− mice and their αMHC-Cre littermates 48 h post-LAD ligation (n = 3–4). Values represent mean ± SE. The significance of differences was determined by a two-way ANOVA followed by a Bonferroni post-hoc analysis. *significantly different from sham counterpart. #significantly different from corresponding αMHC-Cre littermates.
Despite multiple chamber-specific differences in mRNA and protein expression, Glp1rCM−/− mice exhibited no alterations in survival following LAD coronary artery occlusion (Figure 5A). Ventricular ANP expression, an indirect indicator of heart failure was not dysregulated (Figure 5B), and LV remodeling, assessed by cardiac hypertrophy (ventricular weight/body weight ratios), LV infarct scar formation, LV chamber diameter, and LV wall thinning (Figure 5C–G) were also similar in Glp1rCM−/− vs. αMHC-Cre control littermate hearts. Hence selective loss of the cardiomyocyte GLP-1R results in significant changes in cardiac gene expression but has minimal impact on outcomes and the adaptive remodeling response after experimental MI.
Figure 5.
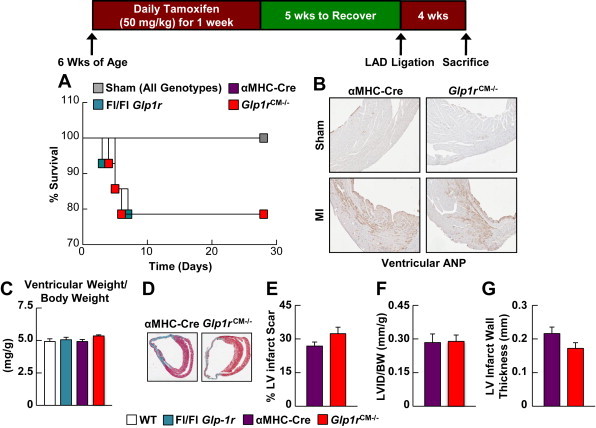
Cardiovascular outcomes following ischemic injury in Glp1rCM−/− mice. A: Survival in Glp1rCM−/− mice and control littermates following permanent LAD coronary artery occlusion (n = 14). B: Histology of LV cross sections for ANP expression in Glp1rCM−/− mice and their αMHC-Cre littermates at day 28 post-LAD coronary artery occlusion. C: Cardiac hypertrophy (ventricular weight/body weight) in Glp1rCM−/− mice and control littermates at day 28 post-LAD coronary artery occlusion (n = 4–9). D: Representative Masson's Trichrome stained heart sections depicting, E: % LV infarct scar formation, F: LV internal diameter (LVID), and G: LV infarct wall thickness in Glp1rCM−/− mice and their αMHC-Cre littermates at day 28 post-LAD coronary artery occlusion (n = 4–5). Values represent mean ± SE.
3.5. The cardiomyocyte GLP-1R is not required for the cardioprotective actions of liraglutide
We previously demonstrated that GLP-1R activation with liraglutide prior to MI produced robust cardioprotection in wild-type mice [14]. To determine whether cardioprotective effects of systemic GLP-1R agonists are mediated through the cardiomyocyte GLP-1R, we treated Glp1rCM−/− mice and their αMHC-Cre littermates with liraglutide for 1 week (Figure 6A) with a dose (30 μg/kg i.p. twice daily) that does not induce weight loss [14] (Supplementary Figure 9). Unexpectedly, cardioprotection with liraglutide was as potent in Glp1rCM−/− mice as in αMHC-Cre littermates (Figure 6B). Furthermore, liraglutide improved cardiac hypertrophy (ventricular weight/body weight ratios), and reduced LV infarct scar formation and LV wall thinning to a similar extent in Glp1rCM−/− vs. αMHC-Cre mice (Figure 6C–G). These findings demonstrate that cardiomyocyte GLP-1R activity is not essential for physiological or pharmacological cardioprotective responses engaged by GLP-1R signaling.
Figure 6.
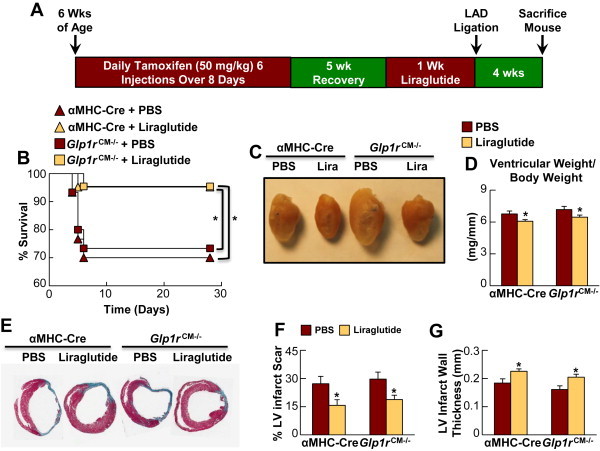
Liraglutide produces cardioprotection in Glp1rCM−/− mice. A: Experimental protocol for liraglutide treatment and LAD coronary artery ligation. B: Survival in PBS- and liraglutide-treated (30 μg/kg BW i.p. twice daily for 1 wk) Glp1rCM−/− mice and αMHC-Cre littermates following LAD coronary artery occlusion (n = 21–30). C–D: Cardiac hypertrophy in PBS and liraglutide-treated Glp1rCM−/− mice and control littermates at day 28 post-LAD ligation (n = 6–10). E: Representative Masson's Trichrome stained heart sections depicting, F: % LV infarct scar formation, and G: LV infarct wall thickness in PBS- and liraglutide-treated Glp1rCM−/− mice and αMHC-Cre littermates at day 28 post-LAD ligation (n = 4–6). Values represent mean ± SE. The significance of differences was determined by a Kaplan Meier survival analysis or a two-way ANOVA followed by a Bonferroni post-hoc analysis. *significantly different from corresponding PBS treated counterpart.
3.6. The cardiomyocyte GLP-1R controls basal HR
As the GLP-1R is predominantly expressed in the atria, and not the ventricle, of rodents and primates [10,28,29], we asked whether atrial GLP-1R expression, perhaps in a subset of pacemaker cells [29], might be important for the pharmacological or physiological control of HR. As GLP-1R agonists increase HR in rodents and humans [8], we assessed HR after acute administration of liraglutide. Despite marked reduction in atrial Glp1r expression, Glp1rCM−/− mice remained equally sensitive to acute liraglutide-induced increases in HR, compared to responses measured in αMHC-Cre control littermates (Figure 7A–D). As levels of GLP-1 rise in the postprandial state, and HR increases following food ingestion [30], we asked whether cardiomyocyte GLP-1R signaling transduces a component of the meal-stimulated increase in HR. Although HR increased briskly in control mice after refeeding, the HR response to refeeding was similar in Glp1rCM−/− mice (Figure 7E). Finally, we asked whether loss of cardiomyocyte Glp1r expression might affect basal HR. Assessment of HR in freely moving conscious mice via radiotelemetry revealed a significant reduction in basal HR in Glp1rCM−/− mice (Figure 7F). Hence, while the atrial GLP-1R is not required for the acute chronotropic response to liraglutide or refeeding, selective loss of GLP-1R signaling in cardiomyocytes disrupts the normal control of HR in mice.
Figure 7.
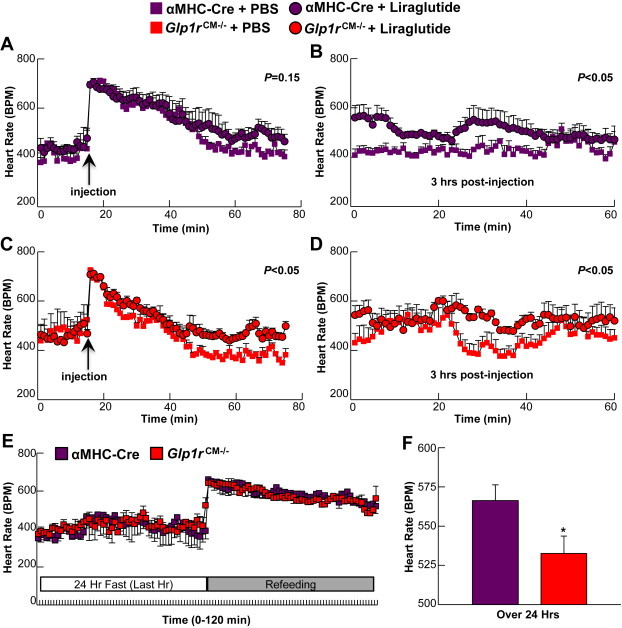
Heart rate in Glp1rCM−/− mice. A–B: HR in αMHC-Cre littermates following an acute liraglutide injection monitored for the first 60 min (A) or 3 h after injection (B) (30 μg/kg BW i.p., n = 3). C–D: HR in Glp1rCM−/− mice following an acute liraglutide injection for the first 60 min (C) or 3 h after injection (D) (30 μg/kg BW i.p., n = 4). Values represent mean ± SE. E: HR in Glp1rCM−/− mice and their αMHC-Cre littermates following a 24 h fast-refeeding protocol; data is presented for the final hr of the 24 h fast and the first hr immediately upon refeeding (n = 3–4). F: Baseline HR over 24 h in freely moving conscious Glp1rCM−/− mice and their αMHC-Cre littermates (n = 3–4). Statistical significance was determined by the use of an unpaired, two-tailed Student's t-test, or a repeated measures two-way ANOVA followed by a Bonferroni post-hoc analysis. *significantly different from αMHC-Cre littermate mice.
4. Discussion
Our results provide multiple new insights that redefine the pharmacology and physiology of GLP-1R-dependent actions in the cardiovascular system. First, complete loss of GLP-1R activity in Glp1r−/− mice has no impact on cardiovascular outcomes after experimental MI or cardiomyopathy. Second, selective disruption of the cardiomyocyte GLP-1R produces alterations in atrial gene expression after experimental MI, however cardiac structure, left ventricular remodeling, infarct size, and survival are not perturbed in Glp1rCM−/− mice. Third, although clinical studies suggest GLP-1R agonists such as exenatide may ameliorate ischemic cardiac injury when administered after the onset of ischemia, we did not observe improvement in outcomes following exendin-4 administration to mice after LAD coronary artery ligation. Fourth, despite putative benefits of GLP-1R agonists in heart failure [8], whole body loss of Glp1r expression does not modify outcomes after induction of cardiomyopathy with doxorubicin, and activating GLP-1R signaling produced no improvement in outcomes in doxorubicin-treated mice. Fifth, although the cardioprotective actions of liraglutide in mice require the GLP-1R [10,14], cardioprotection with liraglutide in mice with experimental MI is independent of the cardiomyocyte GLP-1R. Sixth, whereas the cardiomyocyte GLP-1R is not required for the increase in HR following a) refeeding and b) liraglutide administration, basal HR is significantly lower in Glp1rCM−/− mice.
Glp1r−/− mice studied in the C57BL/6 background exhibit normal cardiac structure and function, and contrary to our initial hypothesis, loss of the GLP-1R did not increase the susceptibility to ischemic or cardiomyopathic injury. Although it is possible that studying larger numbers of Glp1r−/− mice under conditions of insulin resistance, obesity or experimental diabetes might have produced different outcomes, we focused our immediate efforts on verifying this result in a second mouse model enabling loss of GLP-1R signaling in the heart, To eliminate the possibility that germline inactivation of the Glp1r is associated with subtle defects due to developmental compensation, we defined key cardiovascular phenotypes in Glp1rCM−/− mice with conditional inactivation of the Glp1r in adult mice. Notably, the response to coronary artery occlusion was similar in Glp1rCM−/− vs. αMHC-Cre littermate control mice. Hence, the highly concordant data from Glp1r−/− and Glp1rCM−/− mice clearly demonstrate that loss of the cardiomyocyte GLP-1R does not modify the susceptibility to experimental cardiac injury.
The lack of physiological importance of the endogenous cardiomyocyte GLP-1R in the setting of ischemia or doxorubicin-induced cardiomyopathy was surprising given evidence demonstrating robust cardioprotective properties of degradation-resistant GLP-1R agonists [14,31–34]. Greatly complicating interpretation of the existing literature are reports illustrating cardioprotective actions of native GLP-1, which may act through the known GLP-1 receptor, or through GLP-1R-independent pathways via generation of GLP-1(9–36) or GLP-1(28–36) [4,32,35,36]. Importantly, data generated using native GLP-1 or GLP-1(9–36) in the cardiovascular system cannot be inferred to be relevant to mechanisms activated by degradation-resistant GLP-1R agonists [8]. Indeed the available genetic evidence using Glp1r−/− mice demonstrates that the key metabolic and cardiovascular actions of exendin-4 and liraglutide are mediated through the known GLP-1R [10,11,14,37].
A unifying explanation for our results and published literature may lie in the demonstration [10,28,29,38], that the cardiac GLP-1R is localized to atrial, and not ventricular cardiomyocytes. As the majority of experimental models of MI and heart failure encompass direct insults to the LV, basal endogenous atrial GLP-1R activity may not be important for survival and function of ventricular cardiomyocytes. Similarly, we showed that blood pressure or plasma ANP levels were not substantially different in normotensive or hypertensive Glp1r−/− vs Glp1r+/+ mice [10]. Our new experiments extend these findings by demonstrating that Glp1r−/− and Glp1rCM−/− mice do not exhibit defective ANP responses following coronary artery occlusion or doxorubicin-induced cardiomyopathy. Thus, while activation of atrial GLP-1R signaling induces ANP secretion in hypertensive mice, the increase in ANP levels during progression of MI and heart failure is normal in the absence of a functional GLP-1R.
Multiple studies demonstrate that systemic administration of native GLP-1 or exenatide [5,7,39] or GLP-1 infusion directly into the coronary circulation produces cardioprotection [32,40]. Under some scenarios, ANP itself exerts cardioprotective actions [41]. Surprisingly however, the GLP-1R agonist liraglutide continues to exhibit robust cardioprotection following coronary artery occlusion in Glp1rCM−/− mice, indicating that the atrial GLP-1R is not required for GLP-1R agonist-mediated cardioprotection in vivo.
Our current data necessitates reassessment of how GLP-1R agonists exert their cardioprotective actions [8,9,36]. It seems likely that GLP-1R-dependent cardioprotection in vivo arises through indirect mechanisms, perhaps through effects on metabolism, or changes in neural transmission or blood flow. Evidence for the possible importance of indirect metabolic changes arises from studies in rats treated with albiglutide and subjected to ischemia-reperfusion injury; these hearts exhibited increased myocardial carbohydrate oxidation and decreased fatty acid oxidation [42], a metabolic profile associated with improved efficiency of contractile function and consistent with indirect mechanisms transduced through elevations in plasma insulin and activation of the cardiac insulin receptor. Thus, we focused our initial studies in normoglycemic non-diabetic Glp1rCM−/− mice for several reasons. First, the development of hyperglycemia is associated with multiple metabolic and cardiovascular abnormalities [43], which may be partially corrected by administration of GLP-1R agonists, confounding attribution of direct vs. indirect mechanisms. Second, the cardioprotective actions of GLP-1R agonists are preserved in normoglycemic and diabetic mice and humans [14,44]. Third, GLP-1R agonists markedly increase insulin secretion under conditions of hyperglycemia, which may indirectly activate myocardial signaling pathways [43]. Indeed, we observed a significant increase in Akt/GSK3β phosphorylation in ventricular extracts from Glp1rCM−/− mice treated with a much higher 200 μg/kg dose of liraglutide, consistent with increased insulin secretion and activation of myocardial insulin signaling pathways (Supplementary Figure 10). Notably, these effects were absent in Glp1r−/− mice and restored in Pdx1-hGLP1R:Glp1r−/− mice (Supplementary Figure 10) previously shown to exhibit selective restitution of GLP-1R agonist-induced insulin secretion [45].
Our studies in normoglycemic animals raise important questions as to whether systemic GLP-1R activation will similarly confer protection against ischemic injury in obese, hyperglycemic and hyperinsulinemic Glp1rCM−/− mice. Although actions of GLP-1R agonists to increase insulin and enhance cardiac glucose uptake, or engage neural circuits regulating cardiovascular function are important considerations [8,46], these mechanisms would not explain the direct cardioprotective actions ascribed to GLP-1R agonists in ischemia-reperfusion studies ex vivo [8].
Hence, we hypothesize that GLP-1R signaling in cardiac blood vessels, perhaps in endothelial cells or smooth muscle cells, may also contribute in part to modulation of blood flow and cardioprotection. Indeed, Richards et al. used expression of a fluorescent reporter protein under control of endogenous Glp1r regulatory sequences to localize reporter expression within ventricular blood vessels in cells that also-co-expressed smooth muscle actin [38]. A vascular target for GLP-1 action in the heart would also be consistent with studies demonstrating that GLP-1 increases microvascular blood volume and microvascular muscle blood flow in rats [47] and recruits cardiac muscle microvasculature in healthy humans [48]. Furthermore, GLP-1 enhances both mesenteric and coronary blood flow [32,49]. Although the actions of native GLP-1 on blood vessels may potentially be ascribed to GLP-1(9–36), the degradation-resistant GLP-1R agonist exenatide robustly increased myocardial blood flow in human subjects with T2D [50]. Hence it seems likely that clinically utilized degradation-resistant agonists such as exenatide, liraglutide, and lixisenatide, may modulate myocardial blood flow through the known GLP-1R, however whether such increases in blood flow arise independent of changes in heart rate is difficult to ascertain.
The localization of the GLP-1R to sinoatrial nodal cells in primates [29], raises the possibility that GLP-1R agonists increase HR through direct activation of atrial pacemaker cells. Nevertheless, we did not observe differences in the extent of HR elevation following liraglutide administration in control vs. Glp1rCM−/− mice. The mechanisms underlying GLP-1R-dependent increases in HR in rodents are complex, and involve integration of neural signals from both the sympathetic and parasympathetic nervous system [51,52]. Hence, it may not be surprising that GLP-1R agonists exemplified by liraglutide remain capable of increasing HR in mice despite marked attenuation of atrial cardiomyocyte GLP-1R signaling. Nevertheless, our findings reveal an important role for basal cardiomyocyte GLP-1R signaling in regulation of chronotropic activity as we observed a significant reduction in baseline HR in Glp1rCM−/− mice. Hence future studies should aim to elucidate how atrial GLP-1R activity provides signals that integrate with neural and cardiac mechanisms linked to overall control of HR in vivo.
In summary, although GLP-1R agonists produce multiple indirect and direct cardioprotective actions in the cardiovascular system, complete whole body inactivation of the GLP-1R in Glp1r−/− mice, or selective reduction of cardiac GLP-1R expression in Glp1rCM−/− mice does not impair the physiological response to experimental cardiac injury. Moreover, GLP-1R agonists still induce potent cardioprotection despite ablation of cardiomyocyte GLP-1R activity. Furthermore, although the cardiomyocyte GLP-1R is not required for increases in HR following systemic GLP-1R activation, basal HR is significantly reduced in Glp1rCM−/− mice. Our findings reorient the field towards future studies of a) indirect mechanisms linking GLP-1R activation in non-cardiomyocyte cell types to robust cardioprotection and enhancement of ventricular function and b) the importance of atrial GLP-1R signaling for control of HR, in both pre-clinical and clinical studies.
Sources of funding
JRU was supported by fellowships from the Canadian Institutes of Health Research and the Alberta Innovates – Health Solutions. JEC has received fellowships from the Banting and Best Diabetes Centre, University of Toronto and the Canadian Institutes of Health Research. MK and EEM have received fellowships from the Canadian Diabetes Association. DJD has received operating grant support from the Heart and Stroke Foundation of Canada (NA6997), from Novo Nordisk Inc, and is supported by a Canada Research Chair in Regulatory Peptides and a Banting and Best Diabetes Centre Novo Nordisk Chair in Incretin Biology.
Author details
JRU, LLB, JEC, EEM, MK, MGK, XC, JB carried out experiments, analyzed data and contributed to writing of manuscript. DAS, RJS, and DJD planned experiments, analyzed data and contributed to writing of manuscript. Daniel J. Drucker is the primary person (guarantor) taking responsibility for the contents of the article.
Footnotes
John R. Ussher and Laurie L. Baggio contributed equally to this work.
Minsuk Kim, Current address, Department of Pharmacology, School of Medicine, Ewha Womans University, Seoul, South Korea
Conflict of interest
LLB has served as a panel member and/or received a speaker's honorarium for symposia sponsored by Novo Nordisk, Merck Frosst, and GlaxoSmithKline LLC. JRU has received a speaker's honorarium for symposia sponsored by Novo Nordisk. DJD has served as an advisor or consultant to Arisaph Pharmaceuticals, Intarcia, Merck Research Laboratories, Novo Nordisk, Sanofi, Takeda, and Transition Pharmaceuticals. The other authors have no other potential conflicts of interest relevant to this article to disclose.
Appendix A. Supplementary data
References
- 1.Gerstein H.C., Miller M.E., Byington R.P., Goff D.C., Jr., Bigger J.T., Buse J.B. Effects of intensive glucose lowering in type 2 diabetes. New England Journal of Medicine. 2008;358(24):2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindenfeld J., Masoudi F.A. Fluid retention with thiazolidinediones: does the mechanism influence the outcome? Journal of the American College of Cardiology. 2007;49(16):1705–1707. doi: 10.1016/j.jacc.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 3.Scirica B.M., Bhatt D.L., Braunwald E., Steg P.G., Davidson J., Hirshberg B. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. New England Journal of Medicine. 2013;369(14):1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 4.Nikolaidis L.A., Mankad S., Sokos G.G., Miske G., Shah A., Elahi D. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004;109(8):962–965. doi: 10.1161/01.CIR.0000120505.91348.58. [DOI] [PubMed] [Google Scholar]
- 5.Woo J.S., Kim W., Ha S.J., Kim J.B., Kim S.J., Kim W.S. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(9):2252–2260. doi: 10.1161/ATVBAHA.113.301586. [DOI] [PubMed] [Google Scholar]
- 6.Sokos G.G., Nikolaidis L.A., Mankad S., Elahi D., Shannon R.P. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. Journal of Cardiac Failure. 2006;12(9):694–699. doi: 10.1016/j.cardfail.2006.08.211. [DOI] [PubMed] [Google Scholar]
- 7.Lonborg J., Vejlstrup N., Kelbaek H., Botker H.E., Kim W.Y., Mathiasen A.B. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. European Heart Journal. 2012;33(12):1491–1499. doi: 10.1093/eurheartj/ehr309. [DOI] [PubMed] [Google Scholar]
- 8.Ussher J.R., Drucker D.J. Cardiovascular biology of the incretin system. Endocrine Reviews. 2012;33(2):187–215. doi: 10.1210/er.2011-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sivertsen J., Rosenmeier J., Holst J.J., Vilsboll T. The effect of glucagon-like peptide 1 on cardiovascular risk. Nature Reviews Cardiology. 2012;9(4):209–222. doi: 10.1038/nrcardio.2011.211. [DOI] [PubMed] [Google Scholar]
- 10.Kim M., Platt M., Shibasaki T., Quaggin S., Backx P.H., Seino S. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nature Medicine. 2013;19(5):567–575. doi: 10.1038/nm.3128. [DOI] [PubMed] [Google Scholar]
- 11.Scrocchi L.A., Brown T.J., MacLusky N., Brubaker P.L., Auerbach A.B., Joyner A.L. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide receptor gene. Nature Medicine. 1996:21254–21258. doi: 10.1038/nm1196-1254. [DOI] [PubMed] [Google Scholar]
- 12.Wilson-Pérez H.E., Chambers A.P., Ryan K.K., Li B., Sandoval D.A., Stoffers D. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like peptide-1 receptor deficiency. Diabetes. 2013;62(7):2380–2385. doi: 10.2337/db12-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koitabashi N., Bedja D., Zaiman A.L., Pinto Y.M., Zhang M., Gabrielson K.L. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circulation Research. 2009;105(1):12–15. doi: 10.1161/CIRCRESAHA.109.198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noyan-Ashraf M.H., Momen M.A., Ban K., Sadi A.M., Zhou Y.Q., Riazi A.M. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes. 2009;58(4):975–983. doi: 10.2337/db08-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neilan T.G., Blake S.L., Ichinose F., Raher M.J., Buys E.S., Jassal D.S. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation. 2007;116(5):506–514. doi: 10.1161/CIRCULATIONAHA.106.652339. [DOI] [PubMed] [Google Scholar]
- 16.Sauve M., Ban K., Momen A., Zhou Y.-Q., Henkelman R.M., Husain M. Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes following myocardial infarction in mice. Diabetes. 2010;59(4):1063–1073. doi: 10.2337/db09-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Ussher J.R., Folmes C.D., Keung W., Fillmore N., Jaswal J.S., Cadete V.J. Inhibition of serine palmitoyl transferase I reduces cardiac ceramide levels and increases glycolysis rates following diet-induced insulin resistance. PLoS One. 2012;7(5):e37703. doi: 10.1371/journal.pone.0037703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gros R., You X., Baggio L.L., Kabir M.G., Sadi A.M., Mungrue I.N. Cardiac function in mice lacking the glucagon-like peptide-1 receptor. Endocrinology. 2003;144(6):2242–2252. doi: 10.1210/en.2003-0007. [DOI] [PubMed] [Google Scholar]
- 20.Poornima I., Brown S.B., Bhashyam S., Parikh P., Bolukoglu H., Shannon R.P. Chronic glucagon-like peptide-1 infusion sustains left ventricular systolic function and prolongs survival in the spontaneously hypertensive, heart failure-prone rat. Circulation Heart Failure. 2008;1(3):153–160. doi: 10.1161/CIRCHEARTFAILURE.108.766402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q., Anderson C., Broyde A., Polizzi C., Fernandez R., Baron A. Glucagon-like peptide-1 and the exenatide analogue AC3174 improve cardiac function, cardiac remodeling, and survival in rats with chronic heart failure. Cardiovascular Diabetology. 2010;9:76. doi: 10.1186/1475-2840-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernink F.J., Timmers L., Diamant M., Scholte M., Beek A.M., Kamp O. Effect of additional treatment with EXenatide in patients with an acute myocardial infarction: the EXAMI study. International Journal of Cardiology. 2013;167(1):289–290. doi: 10.1016/j.ijcard.2012.09.204. [DOI] [PubMed] [Google Scholar]
- 23.Vyas A.K., Yang K.C., Woo D., Tzekov A., Kovacs A., Jay P.Y. Exenatide improves glucose homeostasis and prolongs survival in a murine model of dilated cardiomyopathy. PLoS One. 2011;6(2):e17178. doi: 10.1371/journal.pone.0017178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nathanson D., Ullman B., Lofstrom U., Hedman A., Frick M., Sjoholm A. Effects of intravenous exenatide in type 2 diabetic patients with congestive heart failure: a double-blind, randomised controlled clinical trial of efficacy and safety. Diabetologia. 2012;55(4):926–935. doi: 10.1007/s00125-011-2440-x. [DOI] [PubMed] [Google Scholar]
- 25.Pederson R.A., Satkunarajah M., McIntosh C.H., Scrocchi L.A., Flamez D., Schuit F. Enhanced glucose-dependent insulinotropic polypeptide secretion and insulinotropic action in glucagon-like peptide 1 receptor −/− mice. Diabetes. 1998;47(7):1046–1052. doi: 10.2337/diabetes.47.7.1046. [DOI] [PubMed] [Google Scholar]
- 26.Hansotia T., Maida A., Flock G., Yamada Y., Tsukiyama K., Seino Y. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. Journal of Clinical Investigation. 2007;117(1):143–152. doi: 10.1172/JCI25483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayala J.E., Bracy D.P., James F.D., Burmeister M.A., Wasserman D.H., Drucker D.J. Glucagon-like peptide-1 receptor knockout mice are protected from high-fat diet-induced insulin resistance. Endocrinology. 2010;151(10):4678–4687. doi: 10.1210/en.2010-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wohlfart P., Linz W., Hubschle T., Linz D., Huber J., Hess S. Cardioprotective effects of lixisenatide in rat myocardial ischemia-reperfusion injury studies. Journal of Translational Medicine. 2013;11:84. doi: 10.1186/1479-5876-11-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pyke C., Heller R.S., Kirk R.K., Orskov C., Reedtz-Runge S., Kaastrup P. GLP-1 receptor localization in monkey and human tissue; novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. 2014;155(4):1280–1290. doi: 10.1210/en.2013-1934. [DOI] [PubMed] [Google Scholar]
- 30.Fagan T.C., Sawyer P.R., Gourley L.A., Lee J.T., Gaffney T.E. Postprandial alterations in hemodynamics and blood pressure in normal subjects. American Journal of Cardiology. 1986;58(7):636–641. doi: 10.1016/0002-9149(86)90291-2. [DOI] [PubMed] [Google Scholar]
- 31.Ban K., Kim H., Cho J., Diamandis E., Backx P.H., Drucker D.J. GLP-1(9-36) protects cardiomyocytes and endothelial cells from ischemia-reperfusion injury via cytoprotective pathways independent of the GLP-1 receptor. Endocrinology. 2010;151(4):1520–1531. doi: 10.1210/en.2009-1197. [DOI] [PubMed] [Google Scholar]
- 32.Ban K., Noyan-Ashraf M.H., Hoefer J., Bolz S.S., Drucker D.J., Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117(18):2340–2350. doi: 10.1161/CIRCULATIONAHA.107.739938. [DOI] [PubMed] [Google Scholar]
- 33.Noyan-Ashraf M.H., Shikatani E.A., Schuiki I., Mukovozov I., Wu J., Li R.K. A glucagon-like peptide-1 analogue reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation. 2013;127(1):74–85. doi: 10.1161/CIRCULATIONAHA.112.091215. [DOI] [PubMed] [Google Scholar]
- 34.Timmers L., Henriques J.P., de Kleijn D.P., Devries J.H., Kemperman H., Steendijk P. Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. Journal of the American College of Cardiology. 2009;53(6):501–510. doi: 10.1016/j.jacc.2008.10.033. [DOI] [PubMed] [Google Scholar]
- 35.Nikolaidis L.A., Elahi D., Shen Y.T., Shannon R.P. Active metabolite of GLP-1 mediates myocardial glucose uptake and improves left ventricular performance in conscious dogs with dilated cardiomyopathy. American Journal of Physiology. Heart and Circulatory Physiology. 2005;289(6):H2401–H2408. doi: 10.1152/ajpheart.00347.2005. [DOI] [PubMed] [Google Scholar]
- 36.Abu-Hamdah R., Rabiee A., Meneilly G.S., Shannon R.P., Andersen D.K., Elahi D. Clinical review: the extrapancreatic effects of glucagon-like peptide-1 and related peptides. Journal of Clinical Endocrinology and Metabolism. 2009;94(6):1843–1852. doi: 10.1210/jc.2008-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tatarkiewicz K., Sablan E.J., Polizzi C.J., Villescaz C., Parkes D.G. Long-term metabolic benefits of exenatide in mice are mediated solely via the known glucagon-like peptide 1 receptor. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2014;306(7):R490–R498. doi: 10.1152/ajpregu.00495.2013. [DOI] [PubMed] [Google Scholar]
- 38.Richards P., Parker H.E., Adriaenssens A.E., Hodgson J.M., Cork S.C., Trapp S. Identification and characterisation of glucagon-like peptide-1 receptor expressing cells using a new transgenic mouse model. Diabetes. 2014;63(4):1224–1233. doi: 10.2337/db13-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikolaidis L.A., Elahi D., Hentosz T., Doverspike A., Huerbin R., Zourelias L. Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation. 2004;110(8):955–961. doi: 10.1161/01.CIR.0000139339.85840.DD. [DOI] [PubMed] [Google Scholar]
- 40.Bose A.K., Mocanu M.M., Carr R.D., Brand C.L., Yellon D.M. Glucagon-like peptide-1 (GLP-1) can directly protect the heart against ischemia/reperfusion injury. Diabetes. 2005;54(1):146–151. doi: 10.2337/diabetes.54.1.146. [DOI] [PubMed] [Google Scholar]
- 41.Kitakaze M., Asakura M., Kim J., Shintani Y., Asanuma H., Hamasaki T. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370(9597):1483–1493. doi: 10.1016/S0140-6736(07)61634-1. [DOI] [PubMed] [Google Scholar]
- 42.Bao W., Aravindhan K., Alsaid H., Chendrimada T., Szapacs M., Citerone D.R. Albiglutide, a long lasting glucagon-like peptide-1 analog, protects the rat heart against ischemia/reperfusion injury: evidence for improving cardiac metabolic efficiency. PLoS One. 2011;6(8):e23570. doi: 10.1371/journal.pone.0023570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bugger H., Abel E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57(4):660–671. doi: 10.1007/s00125-014-3171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lønborg J., Vejlstrup N., Kelbæk H., Nepper-Christensen L., Jørgensen E., Helqvist S. Impact of acute hyperglycemia on myocardial infarct size, area at risk and salvage in patients with ST elevation myocardial infarction and the association with exenatide treatment – results from a randomized study. Diabetes. 2014 doi: 10.2337/db13-1849. [DOI] [PubMed] [Google Scholar]
- 45.Lamont B.J., Li Y., Kwan E., Brown T.J., Gaisano H., Drucker D.J. Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice. Journal of Clinical Investigation. 2012;122(1):388–402. doi: 10.1172/JCI42497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell J.E., Drucker D.J. Pharmacology physiology and mechanisms of incretin hormone action. Cell Metabolism. 2013;17(4):819–837. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Chai W., Dong Z., Wang N., Wang W., Tao L., Cao W. Glucagon-like peptide 1 recruits microvasculature and increases glucose use in muscle via a nitric oxide-dependent mechanism. Diabetes. 2012;61(4):888–896. doi: 10.2337/db11-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subaran S.C., Sauder M.A., Chai W., Jahn L.A., Fowler D.E., Aylor K.W. GLP-1 at physiological concentrations recruits skeletal and cardiac muscle microvasculature in healthy humans. Clinical Science (London) 2014;127(3):163–170. doi: 10.1042/CS20130708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dokken B.B., Hilwig W.R., Teachey M.K., Panchal R.A., Hubner K., Allen D. Glucagon-like peptide-1 (GLP-1) attenuates post-resuscitation myocardial microcirculatory dysfunction. Resuscitation. 2010;81(6):755–760. doi: 10.1016/j.resuscitation.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 50.Gejl M., Sondergaard H.M., Stecher C., Bibby B.M., Moller N., Botker H.E. Exenatide alters myocardial glucose transport and uptake depending on insulin resistance and increases myocardial blood flow in patients with type 2 diabetes. Journal of Clinical Endocrinology and Metabolism. 2012;97(7):E1165–E1169. doi: 10.1210/jc.2011-3456. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto H., Lee C.E., Marcus J.N., Williams T.D., Overton J.M., Lopez M.E. Glucagon-like peptide-1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. Journal of Clinical Investigation. 2002;110(1):43–52. doi: 10.1172/JCI15595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffioen K.J., Wan R., Okun E., Wang X., Lovett-Barr M.R., Li Y. GLP-1 receptor stimulation depresses heart rate variability and inhibits neurotransmission to cardiac vagal neurons. Cardiovascular Research. 2011;89(1):72–78. doi: 10.1093/cvr/cvq271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.