

Abstract
Non-alcoholic fatty liver disease (NAFLD) results from increased hepatic lipid accumulation and steatosis, and is closely linked to liver one-carbon (C1) metabolism. We assessed in C57BL6/N mice whether NAFLD induced by a high-fat (HF) diet over 8 weeks can be reversed by additional 4 weeks of a dietary methyl-donor supplementation (MDS). MDS in the obese mice failed to reverse NAFLD, but prevented the progression of hepatic steatosis associated with major changes in key hepatic C1-metabolites, e.g. S-adenosyl-methionine and S-adenosyl-homocysteine. Increased phosphorylation of AMPK-α together with enhanced β-HAD activity suggested an increased flux through fatty acid oxidation pathways. This was supported by concomitantly decreased hepatic free fatty acid and acyl-carnitines levels. Although HF diet changed the hepatic phospholipid pattern, MDS did not. Our findings suggest that dietary methyl-donors activate AMPK, a key enzyme in fatty acid β-oxidation control, that mediates increased fatty acid utilization and thereby prevents further hepatic lipid accumulation.
Keywords: Obesity, Hepatic steatosis, One-carbon metabolism, AMP-activated protein kinase, β-oxidation, Acyl-carnitines
Abbreviations: ACC, acetyl-CoA carboxylase; AMPK, AMP-activated protein kinase; ANT, adenine nucleotide translocase; Bhmt, betaine-homocysteine methyltransferase; C1, one-carbon; CACT, carnitine-acylcarnitine transporter; Cpt1a, carnitine palmitoyltransferase-1a; Cbs, cystathionine β-synthase; C, control diet; CMS, methyl-donor supplemented control diet; DIO, diet-induced obesity; Fasn, fatty acid synthase; Gapdh, glyceraldehyde 3-phosphate dehydrogenase; GNMT, glycine N-methyltransferase; HSP90, heat shock protein 90; HF, high-fat diet; HFMS, methyl-donor supplemented high-fat diet; HMW adiponectin, high molecular weight adiponectin; Hcy, homocysteine; β-HAD, β-hydroxyacyl CoA dehydrogenase; 3-HB, β-hydroxybutyrate; Hprt1, hypoxanthine phosphoribosyltransferase 1; LDL, low density lipoprotein; MAT, methionine adenosyltransferase; MCD, malonyl-CoA decarboxylase; MDS, methyl-donor supplementation; MTR, methionine synthase; NAFLD, non-alcoholic fatty liver disease; NEFA, non-esterified fatty acids; PC, phosphatidylcholine; Pemt, phosphatidylethanolamine methyltransferase; PGC1α, peroxisome proliferator-activated receptor-γ co-activator-1α; PL, phospholipids; PPARα, peroxisome proliferator-activated receptor-α; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SM, sphingomyelin; SREBP1c, sterol regulatory element-binding protein-1c; TG, triacylglycerol; VAT, visceral adipose tissue; VLDL, very low density lipoprotein
1. Introduction
The generic term non-alcoholic fatty liver disease (NAFLD) describes an incremental metabolic dysfunction of the liver starting with hepatic steatosis and steatohepatitis that can progress to end-stage liver diseases like cirrhosis and hepatocellular cancer [1]. Obesity is one of the principal causes of hepatic steatosis [2,3], which can be described as the manifestation of the metabolic syndrome in the liver. 33% of NAFLD patients have the entire characteristic of the metabolic syndrome and nearly 90% develop one particular feature of the syndrome [4]. Conditions like obesity, type 2 diabetes mellitus and hyperlipidemia therefore promote NAFLD [5]. Hepatic triacylglycerol (TG) accumulation results from an imbalance of free fatty acid (non-esterified fatty acids, NEFA) uptake from circulation, de novo lipogenesis, fatty acid oxidation and TG export via very low density lipoprotein (VLDL) particles to peripheral fat stores [6].
C1-metabolism is of prime importance in hepatic lipid homeostasis. This appears to originate primarily from its role in providing methyl-groups for biosynthesis of phosphatidylcholine via the phosphatidylethanolamine methyltransferase (PEMT) pathway. Evidence for this close association of lipid homeostasis and C1-metabolism comes mainly from studies in mice lacking PEMT, glycine N-methyltransferase (GNMT) or methionine adenosyltransferase (MAT) enzymes or models with dietary methionine and choline deficiency [7–10]. Especially the analysis of lipoprotein metabolism in PEMT−/− mice demonstrated the prime importance of C1-metabolism in hepatic lipid homeostasis. Cultured hepatocytes derived from PEMT−/− mice show reduced secretion of VLDL and reduced VLDL plasma levels when animals are fed a HF/high cholesterol diet [11]. Moreover, an attenuation of atherosclerosis in LDL receptor-deficient mice was observed by PEMT deletion [12]. Central to C1-metabolism is the methionine cycle. It maintains the homeostasis of the essential amino acid methionine from which the methyl-group donor S-adenosylmethionine (SAM) is generated. SAM is a universal methyl-group provider for more than 100 methylation reactions [13]. The transfer of the methyl-group to various acceptors leads to the formation of S-adenosylhomocysteine (SAH) followed by hydrolysis to homocysteine (Hcy). Hcy represents the branch point molecule of the methionine cycle and the transsulfuration pathway [14]. In the latter, Hcy is irreversibly converted by cystathionine β-synthase (CBS) to cystathionine which can be catabolized to cysteine [14]. But Hcy can also be remethylated to preserve methionine via i) betaine-homocysteine methyltransferase (BHMT) dependent oxidation of choline and betaine in the sarcosine pathway or via ii) the methionine synthase (MTR) with methyl-group delivery from 5-methyltetrahydrofolate [14,15].
A strategy to prevent or attenuate fatty liver disease is based on dietary supplementation with lipotropic compounds of C1-metabolism. In 1964 Ball [16] described for the first time that lipotropes such as choline and betaine can prevent hepatic fat infiltration. Improvements of disease state by MDS could also be observed in models of alcoholic fatty liver disease [17,18] most likely by restoring a repression of phosphatidylcholine (PC) biosynthesis [19]. An alleviation of hepatic lipid accumulation by providing methyl-donors has been reported in a variety of nutritional models [20–22]. However, whether a dietary methyl-donor supplementation (MDS) can also reverse a pre-established NAFLD has rarely been studied [23]. We here describe the effects of MDS on the progression of hepatic lipid accumulation combined with the analysis of hepatic amino acid status and phospholipid (PL) signatures. Furthermore, we provide evidence that extra methyl-donors elevate the activation state of hepatic AMP-activated protein kinase (AMPK) and thereby increase fatty acid oxidation with a major reduction in acyl-carnitine levels contributing to a reduced hepatic lipid accumulation.
2. Material and methods
2.1. Animal housing, dietary treatments, and sample collection
Animal experiments were conducted in conformity with the Public Health Service policy and German guidelines for animal care. The study was approved by the Department of Veterinary Affairs of the government of Oberbayern/Germany. The design of feeding trial is shown in Supplemental Figure S1. In brief, eight week-old male C57BL/6NCrl mice (n = 46) obtained from Charles River Laboratories were maintained in 12 h light/dark cycle with unlimited access to food and water during the trial. After feeding a standard laboratory chow for 2 weeks, mice were divided into a control (C, n = 23) and HF group (n = 23) with similar mean body weight. The following 12-weeks feeding trial was bipartite including at first a 8-weeks period to achieve diet-induced obesity (DIO) associated with NAFLD in mice (DIO-phase) followed by a 4-weeks period of MDS (MDS-phase). During the DIO-phase, the HF group was fed a beef-tallow based diet with 34% fat (w/w) (Ssniff GmbH, no. E15741-34; Supplemental Table S1) while the control group was fed a carbohydrate/starch based diet comprising 4.2% fat (Ssniff GmbH, no. E15000-04; Supplemental Table S1) for 8 weeks. At the end of the DIO-phase (time-point 8 weeks), five animals of each dietary group were analyzed as described below to determine the status of obesity and NAFLD. For the MDS-phase, C mice were divided into a control group (C) and a control/methyl-donor supplemented group (CMS), and the HF mice were divided into a high-fat group (HF) and a HF/methyl-donor supplemented (HFMS) group, respectively (n = 9 in each group) with similar mean body weights in all groups. C and HF diets were identical except that the MS diets contained additionally 15 g/kg choline chloride, 15 g/kg betaine, 7.5 g/kg methionine, 15 mg/kg folic acid, 1.5 mg/kg vitamin B12 and 150 mg/kg ZnSO4 (see Supplemental Table S1; supplementation with methyl-donors and cofactors was according to Wolff et al. [24]). Subsequently to the DIO-phase the MDS-phase started by feeding the remaining animals of the C and HF groups either the parent diet or the corresponding diet supplemented with the methyl-donors and cofactors [24]. Body weight and food intake were measured during the entire feeding trial and the corresponding intakes of energy, methionine, cystine, choline chloride, folate, vitamin B12 and vitamin B6 were calculated for the specific feeding intervals and are provided in Supplemental Tables S2 and S3, respectively. For the analyses at the end of the feeding trials (8 weeks and 12 weeks, respectively), mice in a non-fasting state were anesthetized using isoflurane, blood was taken from the retro-orbital sinus, and after cervical dislocation, corresponding organ samples, such as liver and adipose tissues were collected, snap-frozen in liquid nitrogen and stored at −80 °C until analysis.
2.2. Measurements of basal clinical parameters
For measuring blood glucose levels, mice were deprived from food for 6 h and blood glucose levels were measured from the tail vein with an Accu-Check blood glucose meter (Roche Diagnostics). At the end of the DIO and MDS-phases, plasma glucose, plasma insulin and hepatic TG, PL and NEFA concentrations were analyzed. Plasma insulin levels in non-fasting state were determined applying an Ultra-Sensitive Mouse Insulin ELISA Kit (Crystal Chem Inc.) according to the manufacturer's instructions. Intra-assay variation was generally <10%. Standard curve calculation was performed by Prism4 software using 4-parameter logistic fit model [sigmoidal dose-response curve (variable slope)]. Analysis of total and high molecular weight (HMW) adiponectin in blood plasma was performed by using the Adiponectin (Mouse) Total, HMW ELISA Kit (Alpco Diagnostics) according to the manufacturer's instructions. Standard curve calculation was performed by Prism5 software using third order polynomial (cubic) fit model.
For analysis of hepatic TG and total PL, liver tissues were ground in liquid nitrogen and dissolved in 0.9% NaCl. NEFA and PL were determined from the supernatant using the commercial enzymatic colorimetric kits NEFA-HR(2) (NEFA, Wako Chemicals GmbH) and Phospholipid C (PL, Wako Chemicals GmbH), respectively, according to the manufacturer's instructions. TG were isolated by alkalinous hydrolytic glycerol extraction in alcoholic KOH and 1 ml 0.15 M magnesium sulfate at 70 °C for 30 min. Isolated glycerol in the supernatant was determined by the commercial enzymatic colorimetric kit Triglycerides liquicolormono (Human GmbH) according to the manufacturer's instructions.
2.3. Histology
Hepatic TG accumulation, was visualized in liver cryosections (5 μm) fixed in 10% formalin for 1 h, rinsed three times in water, equilibrated in 60% isopropanol and stained for 1 h in 0.3% Oilred O (Sigma-Aldrich) staining solution. Sections were again rinsed in 60% isopropanol, counterstained by hematoxylin (Medite) for 1 min and rinsed in water. Stained sections were mounted in Aquatex® (Merck). Light microscopic analysis was performed by using the Leica DMI 4000 B light microscope and the Leica DFC 490 camera (Leica Microsystems).
2.4. RNA extraction and RNA expression analyses
Isolation of total RNA was performed using Trizol reagent (Invitrogen) and the RNeasy Mini Kit (Qiagen GmbH). RNA was extracted from homogenized liver tissue by phenol/chloroform extraction. RNA from the aqueous phase was isolated by RNeasy Mini Kit columns according to the manufacturer's instructions. During the isolation residual DNA was digested using RNase-Free DNase Set (Qiagen GmbH). RNA concentration was determined with a NanoDrop ND-1000 UV-Vis spectrophotometer and RNA integrity was assessed with the 2100 Bioanalyzer (Agilent Technologies) using the RNA 6000 Nano Chip Kit (Agilent Technologies).
Gene expression was measured by Real-time quantitative PCR (RT-qPCR) using 10 ng of isolated total RNA, corresponding gene-specific primers, QuantiTect® SYBR Green RT-PCR kit (Qiagen GmbH, Hilden, Germany) and a Mastercycler ep realplex apparatus (Eppendorf, Hamburg, Germany) following the manufacturer's instructions. The following gene-specific primers were applied:
carnitine palmitoyltransferase 1a (Cpt1a),
fwd. 5′-GTCCCAGCTGTCAAAGATACCG-3′, rev. 5′-ATGGCGTAGTAGTTGCTGTTAACC-3′;
fatty acid synthase (Fasn),
fwd. 5′-ATGAAGCTGGGCATGCTCA-3′, rev. 5′-CCGGCATTCAGAATCGTGGC-3′;
sterol regulatory element-binding protein-1c (Sreb1c),
fwd. 5′-ATGGATTGCACATTTGAAGACATG-3′, rev. 5′-AGAGGAGGCCAGAGAAGCAG-3′;
acetyl-CoA carboxylase 1 (Acc1), Qiagen order no. QT01554441;
acetyl-CoA carboxylase 2 (Acc2),
fwd. 5′-AGGGTCATAGAGAAGGTGCTCA-3′, rev. 5′-AGATCCTCGGGCGTCACCAT-3′.
The following amplification conditions and melting curve analysis were used: cDNA synthesis at 50 °C for 30 min terminated by reverse transcriptase enzyme inactivation at 95 °C for 15 min; subsequently, 40 cycles of denaturation, annealing and elongation (at 95 °C for 15 s, 61 °C for 30 s and 72 °C for 30 s, respectively) followed by melting curve analysis (1.75 °C/min). Relative gene expression was calculated by the 2−ΔΔCq method using the geometric mean of the invariant genes hypoxanthine phosphoribosyltransferase 1 (Hprt1) and glyceraldehyde 3-phosphate dehydrogenase (Gapdh) for normalization [25,26]. The following gene-specific primers were applied:
Gapdh, fwd. 5′-CCTGGAGAAACCTGCCAAGTATG-3′,
rev. 5′-GAGTGGGAGTTGCTGTTGAAGTC-3′;
Hprt1, fwd. 5′-GTCGTGATTAGCGATGATGAACC-3′,
rev. 5′-GTCTTTCAGTCCTGTCCATAATCAG-3′.
2.5. Analysis of metabolites
Amino acid analysis: liver tissues were ground in liquid nitrogen and 100 mg of the homogenates were dissolved in 150 μl H2O/methanol (50:50), vortexed and centrifuged. 40 μl of the supernatant or 40 μl of blood plasma samples were used to determine the concentration of amino acids and derivatives following the iTRAQ-labeling method using the AA45/32™ Kit (Applied Biosystems). Samples were treated according to the manufacturer's instructions and analyzed via LC-MS/MS (3200QTRAP LC/MS/MS, Applied Biosystems). Spectra were processed using the Analyst® 1.5 Software [27]. HPLC-MS/MS detection of hepatic SAM and SAH levels was performed using a Waters 2795 alliance HT, coupled to a Quattro Micro API tandem mass spectrometer (Waters Corporation) according to the previously described methodology with minor modifications [28]. 1 N acetic acid was added to a given weight of any tissue and was followed by homogenization in a Micro-Dismembrator U (Sartoris AG) and centrifugation at 12,000 g for 10 min. 25 μl of the supernatant were added to 40 μl of internal standard (SAM and SAH) and 375 μl of 20 mM ammonium acetate buffer (pH 7.4), followed by cleaning on SPE column and analysis by HPLC-MS/MS. Plasma total homocysteine (tHcy) was determined using the Homocysteine HPLC Kit (Immundiagnostik AG) and the Agilent 1100 Series HPLC System (Agilent Technologies) with fluorescence detection (Excitation 385 nm/Emission 515 nm) according to the manufacturer's instructions and integration of peak areas of internal standard and plasma samples for quantification. Liver Hcy and cystathionine levels were determined by gas chromatography mass spectrometry (GC-MS) according to Stabler et al., with minor modifications [29]. Briefly, homogenization of liver tissue samples was performed with 0.1 M perchloric acid followed by centrifugation at 12,000 g for 10 min and neutralization with 1 M K3PO4. Samples were stored for further preparation as described by Stabler et al. [29].
Lipid analysis: quantification of acyl-carnitines, phosphatidylcholine, lysophosphatidylcholine and sphingolipid species was performed with the Absolute IDQ™ Kit p180 (Biocrates Life Sciences AG) with modifications for tissue analysis [30]. Samples were extracted from 60 mg frozen ground liver using a 1/15 extraction ratio (w/v) at −18 °C (ice-methanol mixture) followed by shaking at 4 °C for 10 min. Samples were centrifuged (10 min 14,000 g at 4 °C) and dried by evaporation of methanol in a vacuum centrifuge (SpeedVac SPDIIIV, Thermo Savant) by applying 1 mbar pressure. According to weight of tissue samples, the pellets were dissolved in 5% phenylisothiocyanate (PITC) derivatization reagent (v/v) to extract analytes (120 μl for 60 mg tissue). Samples were finally diluted 1:30 for flow injection analysis (FIA) using FIA coupled MS (AB SCIEX, QTRAP5500). Data were analyzed with MetIQ software v1.2.2 (Biocrates Life Sciences AG). Slightly differing sample weights were corrected after Biocrates METiQ quantification.
3-hydroxybutyrate analysis: 3-hydroxybutyrate (3-HB) was analyzed by cyclic enzymatic colorimetric measurement using the Autokit 3-HB (Wako Chemicals GmbH) according to the manufacturer's instruction. For quantification, 2 μl of liver extract in H2O/methanol (50:50) and the Ketone Body Calibrator 300 (300 μM, Wako Chemicals GmbH) was used.
2.6. Analysis of proteins
Enzyme activity measurements: activity of β-hydroxyacyl CoA dehydrogenase (β-HAD) was measured as previously described by Molero et al. [31]. Briefly, ground liver samples were homogenized (1:20 – wt/vol) in 50 mM Tris-HCl, 1 mM EDTA, 0.1% Triton X-100, pH 7.2, and were subjected to three freeze-thaw cycles. For measurements of β-HAD enzyme activity, 10 μl of six-fold diluted extracts were used at 30 °C as previously described [31]. The reactions were monitored at 340 nm using a Varioskan-plate spectrophotometer (Thermo Fisher Scientific).
Nuclear protein extraction: for nuclear protein extraction, liver tissue was homogenized in 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 20 mM NaF, 0.5 mM DTT by dounce homogenization. After centrifugation (3,300 g, 15 min), 150 μl low salt buffer (20 mM HEPES, 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA, 20 mM NaF, 25% glycerol (v/v), 0.5 mM DTT) and 150 μl high salt buffer (20 mM HEPES, 1.5 mM MgCl2, 1.2 M KCl, 0.2 mM EDTA, 20 mM NaF, 25% glycerol (v/v), 0.5 mM DTT) was added to cell nuclei, followed by 30 min of shaking. After an additional centrifugation (25,000 g, 30 min) nuclear protein extracts were isolated for further analysis.
Western blot analysis: gelelectrophoresis was performed by denaturing liver protein extracts in Laemmli sample buffer (310 mM Tris, pH 6.8, 5% SDS, 12.5% glycerol, 2.5 mM EDTA, 50 mM dithiothreitol and 0.01% bromophenol blue) at 95 °C for 5 min. 30 μg and 50 μg of samples were separated on 10% and 7.5% SDS-polyacrylamide gels, respectively. Protein-transfer to a nitrocellulose membrane (Whatman GmbH) was performed using the semidry Transblot B44 or Tankblot Eco-Mini system (Biometra). For immunodetection of specific proteins, membranes were blocked in 2% ECL advance blocking reagent (GE Healthcare) in TBS (20 mM Tris, 140 mM NaCl, pH 7.6) for 1 h and incubated with the following primary antibodies at 1:500–1:20,000 dilutions in TBS-T (20 mM Tris, 140 mM NaCl, 0.1% Tween 20, pH 7.6) at 4 °C over night: goat anti-BHMT, rabbit anti-SREBP1 activated form (Novus Biologicals); rabbit anti-CBS (Aviva Systems Biology); rabbit anti-ACC1, rabbit anti-phospho-ACC1 (Ser76) (Millipore); rabbit anti-phospho-ACC2 (Ser219/Ser221) (Santa Cruz Biotechnology); rabbit anti-ACC1/2, rabbit anti-AMPKα, rabbit anti-phospho-AMPKα (Thr172) and rabbit anti-β-ACTIN (Cell Signaling); mouse anti-heat shock protein 90 (HSP90) (StressMarq Biosciences Inc.). After several washing steps, secondary donkey anti-goat or anti-mouse, and goat anti-rabbit conjugated with IRDye 800 (Li-cor) at 1:10,000–1:50,000 dilutions in TBS-T were exposed at room temperature for 1 h. Signal detection and quantification of fluorescence intensity was performed with the Odyssey infrared imaging system (Li-cor) and by using the Odyssey Application Software 3.0 (Li-cor) for calculation of integrated intensity. β-actin and HSP90 were used for normalization of protein abundance.
2.7. Statistical analysis
Statistical analysis was performed by using Prism 4.01 software (GraphPad Software). In case of inhomogeneous variances, data were analyzed using Student's t-test with Welch's correction. Data from C, CMS, HF and HFMS groups were tested by one-way ANOVA followed by Newman-Keuls Multiple Comparison Test. In case of unequal variances data were logarithmic transformed. In addition, outliers were detected by using Grubb's test and excluded from analysis. Analysis of body weight development was performed with SigmaStat 3.5 (Systat Software, Inc.) using the two-way ANOVA with repeated measures and the Holm-Sidak method. For all tests, the bilateral alpha risk was α = 0.05. Differences in liver acyl-carnitines and plasma or hepatic amino acid concentrations were tested using the R software package [32]. The p-values were adjusted for multiple testing using the p.adjust function within the R-library limma and the Benjamini–Hochberg method [33]. Correlations were determined using the rcorr function within the Hmisc package [34]. Multivariate data analysis of liver PL subclasses was done with the TIGR MeV software version 4.1 using normalized metabolite data [x = (value − average)/sd] for hierarchical clustering analysis. Data were tested by one-way ANOVA with bilateral alpha risk of α = 0.01 and Bonferroni adjustment.
3. Results
3.1. Feeding HF diets causes NAFLD while MDS alleviates the progression of hepatic steatosis and reduces free fatty acid levels
At the end of the first 8 weeks of feeding, HF animals showed a ∼35% increased body weight (p < 0.001) compared to the control group (Figure 1A), with liver and visceral adipose tissue (VAT) weight increased by ∼31% (p = 0.001) and ∼150% (p = 0.002), respectively. HF feeding also increased basal blood glucose (p = 0.012) and plasma insulin levels (p = 0.161) as compared to mice on C diet (Table 1). The observed body weight difference between control and HF fed animals remained over the next 4 weeks of MDS feeding (Figure 1A, p < 0.001). Liver weight increased further in all animals over the additional 4 weeks with highest levels in animals on HF diets (Table 2). However, animals of the HFMS group displayed the lowest % liver to body weight ratio. Glucose and insulin levels also further increased over time and showed highest levels in HF and HFMS mice (Table 2), while plasma total or HMW adiponectin levels did not reveal any differences.
Figure 1.
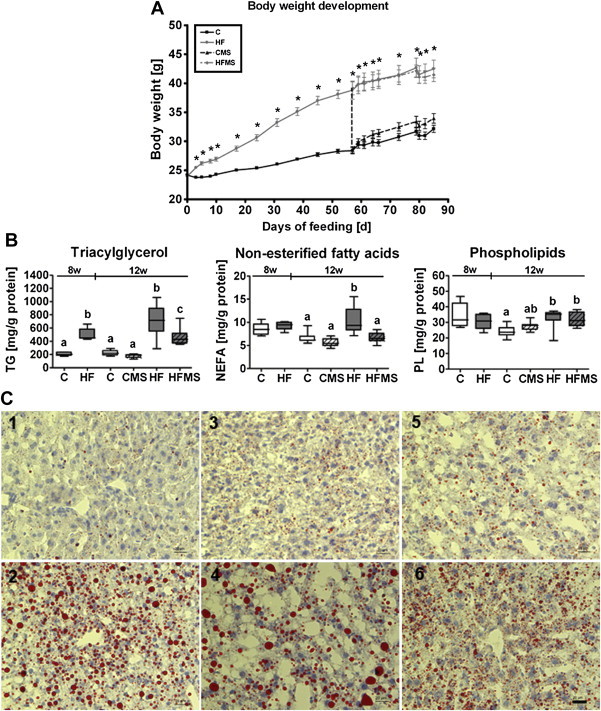
Body weight development (A), hepatic triacylglycerol, non-esterified fatty acids and total phospholipids (B) and histological analysis of fat accumulation in liver tissues (C) from C, CMS, HF and HFMS mice. The bipartite 12-weeks feeding trial includes a 8-weeks DIO-phase to achieve DIO associated with NAFLD in mice (part 8 w) followed by a 4-weeks MDS-phase (part 12 w). The vertical line between part 8 w and part 12 w delineates the transition between the two phases. Data are presented as mean ± SEM (8 w = 8 weeks, n = 5; 12 w = 12 weeks, n = 8–9). (A) Asterisk indicates statistical significance (p < 0.05) of HF and HFMS mice compared to C and CMS mice (p < 0.05). (B) Open and gray boxes represent data from C and HF animals, respectively, and open lined and gray lined boxes represent data from CMS and HFMS animals, respectively. Data are presented as box and whisker plot. Different subscript letters indicate statistical significance (p < 0.05). Data from the 8-weeks DIO-phase (C, HF) and 4-weeks MDS-phase (C, CMS, HF, HFMS) were analyzed separately using Student's t-test and one-way ANOVA followed by Newman-Keuls Multiple Comparison Test, respectively. (C) Shown are Oil red O-stained mouse liver cryosections, labeled by numbers respective the diet and feeding time: control diet fed mice (1) and HF mice (2) after the 8-weeks feeding phase; mice fed control diet (3), HF diet (4), CMS diet (5) and HFMS diet (6) after the 12-weeks feeding period, which included the 4-weeks MDS-phase. Tissue sections from mice. Scale bar: 20 μm. NEFA, non-esterified fatty acids; PL, phospholipids; TG, triacylglycerol.
Table 1.
Basal parameters of control and HF C57BL/6N male mice after 8 weeks of dietary treatment.
| C | HF | |
|---|---|---|
| Liver weight [g] | 1.24 ± 0.04a | 1.62 ± 0.04b |
| % Liver/body weight | 4.59 ± 0.06 | 4.34 ± 0.11 |
| VAT weight [g] | 1.10 ± 0.05a | 3.86 ± 0.38b |
| % VAT/body weight | 4.09 ± 0.11a | 10.22 ± 0.74b |
| Blood glucose [mg/dl] | 123.4 ± 3.4a | 169.4 ± 10.1b |
| Plasma insulin [ng/ml] | 1.23 ± 0.25 | 2.72 ± 0.77 |
Data are presented as mean ± SEM (n = 5). Different subscript letters indicate statistical significance (p < 0.05). Visceral adipose tissue (VAT) was calculated by summing up the weight of epididymal, perirenal and mesenteric adipose tissue.
Table 2.
Basal parameters of C, CMS, HF and HFMS mice after 4 weeks of therapeutic MDS.
| C | CMS | HF | HFMS | |
|---|---|---|---|---|
| Liver weight [g] | 1.40 ± 0.04a | 1.56 ± 0.04ac | 1.83 ± 0.11b | 1.70 ± 0.08bc |
| % Liver/body weight | 4.37 ± 0.12ab | 4.58 ± 0.07a | 4.29 ± 0.13ab | 4.08 ± 0.10b |
| VAT weight [g] | 1.84 ± 0.15a | 2.15 ± 0.17a | 4.80 ± 0.37b | 4.56 ± 0.27b |
| % VAT/body weight | 5.67 ± 0.35a | 6.28 ± 0.36a | 11.19 ± 0.52b | 10.94 ± 0.38b |
| Blood glucose [mg/dl] | 136.9 ± 4.2a | 146.8 ± 4.2a | 171.6 ± 5.5b | 167.7 ± 1.9b |
| Plasma insulin [ng/ml] | 1.42 ± 0.13a | 2.15 ± 0.48ab | 3.68 ± 0.70b | 3.01 ± 0.57b |
| Plasma total adiponectin [μg/ml] | 22.77 ± 1.46 | 20.64 ± 1.13 | 19.84 ± 0.76 | 18.93 ± 0.71 |
| Plasma HMW adiponectin [μg/ml] | 5.46 ± 0.34 | 5.32 ± 0.32 | 5.16 ± 0.46 | 4.91 ± 0.31 |
| HMW/total adiponectin [%] | 24.08 ± 0.82 | 25.89 ± 1.20 | 25.81 ± 1.71 | 25.81 ± 0.80 |
Data are presented as mean ± SEM (n = 7–9). Different subscript letters indicate statistical significance (p < 0.05). Visceral adipose tissue (VAT) was calculated by summing up the weight of epididymal, perirenal and mesenteric adipose tissue.
For mice analyzed after the 8-weeks feeding phase, TG content in liver of animals fed HF compared to mice fed control diet increased significantly (p = 0.003), while hepatic free fatty acids (NEFA) and total PL levels remained unaffected by diet (Figure 1B, see respective parts 8 w). For mice analyzed after the 12-weeks feeding phase, which included additional 4 weeks of control and HF diet feeding and in parallel MDS, TG levels in HF mice displayed still significantly higher levels than in control and CMS mice (part 12 w of Figure 1B), whereas TG levels in HFMS were significantly lower than in HF mice, but not as low as in control and CMS mice (p < 0.001). Oil red O-stained cryosections of livers from control and HF mice after 8-weeks (Figure 1C1 and C2, respectively) and 12-weeks feeding (Figure 1C3 and C4, respectively), as well as from CMS (Figure 1C5) and HFMS (Figure 1C6) mice after 12-weeks feeding, including a MDS-phase, confirmed the changes in hepatic TG content: increased TG accumulation in livers of HF mice reflected by intense staining with increased lipid droplet sizes after 12-weeks HF (Figure 1C4) compared to 8-weeks HF (Figure 1C2), reduced staining intensity and a major reduction in lipid droplet size in livers of HFMS animals (Figure 1C6) compared to HF (Figure 1C4), control (Figure 1C3) or CMS mice (Figure 1C5). For hepatic free fatty acid (NEFA) and PL levels after the 4-weeks MDS-phase as shown in part 12 w of Figure 1B, significantly increased NEFA concentrations were detected in HF mice compared to C and CMS mice, whereas this increase was not observed in HFMS animals. PL concentrations were found significantly elevated in both HF and HFMS mice compared to control mice.
3.2. Amino acid profiling in plasma and liver reveals a specific signature of MDS
Concentrations of 31 amino acids and their derivatives were analyzed in plasma (Supplemental Table S4) and livers (Supplemental Table S5) by LC-MS/MS. As shown in Figure 2, in plasma of CMS and HFMS mice compared to control mice, l-serine levels significantly increased (p = 0.007), while glycine levels significantly decreased (p < 0.001). For sarcosine, only CMS mice displayed significantly elevated concentrations compared to control and HF mice (p = 0.001). l-methionine was only slightly increased in plasma of HFMS mice (p = 0.064). For systemic total (reduced) Hcy levels, in comparison to controls, concentrations increased significantly in CMS animals (p < 0.001), whereas they were significantly decreased in HF mice, and no differences were observed in HFMS animals.
Figure 2.
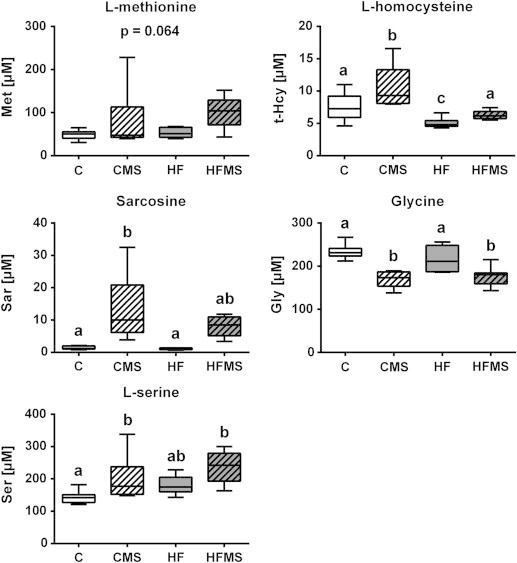
Analysis of blood plasma for selected metabolite concentrations after 4 weeks of dietary MDS. Data are presented as box and whisker plot (n = 8–9). Open and gray boxes represent data from C and HF mice, respectively, and open lined and gray lined boxes represent data from CMS and HFMS mice, respectively. Different subscript letters indicate statistical significance (p < 0.05). Gly, Glycine; Hcy, reduced (total) l-homocysteine; Met, l-methionine; Sar, Sarcosine; Ser, l-serine.
Hepatic amino acid levels (Figure 3A) in MDS animals compared to C showed significant increases for sarcosine (p < 0.001), while glycine (p < 0.001) concentrations significantly decreased, and l-serine levels in CMS and HFMS mice did not change. However, both HF and HFMS mice showed significantly lower serine levels than CMS mice. l-methionine levels remained unaltered regardless of diet. MDS however changed hepatic SAM, SAH, Hcy and cystathionine concentrations as shown in Figure 3B. MDS significantly increased hepatic SAM (p = 0.002) and cystathionine, levels (p = 0.001) in HFMS mice compared to all other diet groups, including CMS mice. SAH levels were significantly increased in CMS and HFMS mice and decreased in HF mice compared to control (p < 0.001). Furthermore, the data analysis revealed that MDS significantly increased hepatic Hcy levels in CMS mice (p = 0.009) and marginally in HFMS mice compared to control.
Figure 3.
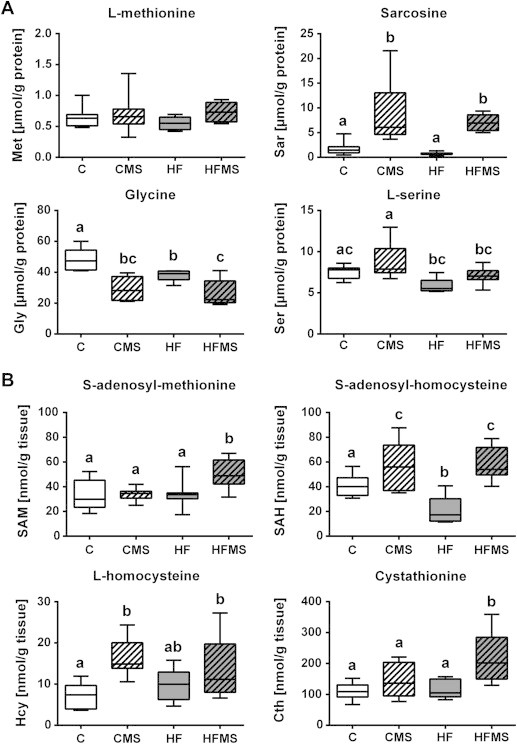
Analysis of liver tissues for selected hepatic metabolites after 4 weeks of dietary MDS. (A and B) Data are presented as box and whisker plot (n = 7–9). Open, gray, open lined and gray lined boxes show data from C, HF, CMS and HFMS animals, respectively. Different subscript letters indicate statistical significance (p < 0.05). Cth, Cystathionine; Gly, Glycine; Hcy, l-Homocysteine; Met, l-methionine; SAH, S-adenosyl-homocysteine; SAM, S-adenosyl-methionine; Sar, Sarcosine; Ser, l-serine.
3.3. Proteins of hepatic C1-metabolism are as well affected by MDS
To assess diet effects on key enzymes of methyl-group utilization, protein levels of BHMT and CBS were determined by Western blot analysis. Comparing mice fed control diet to mice fed extra methyl-donors (CMS, HFMS), significantly increased BHMT and reduced CBS protein levels were detected (Figure 4). Similar changes were also found in HF mice demonstrating a strong diet effect.
Figure 4.
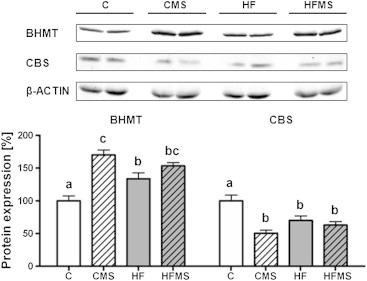
Influence of dietary treatment on hepatic BHMT and CBS protein expression in C, CMS, HF and HFMS mice. Data are presented as mean ± SEM (n = 8–9). Open, gray, open lined and gray lined boxes show data from C, HF, CMS and HFMS animals, respectively. Different subscript letters indicate statistical significance (p < 0.05). Bhmt, Betaine–homocysteine methyltransferase; Cbs, Cystathionine β-synthase.
3.4. Changes in expression for mRNAs and proteins and levels of metabolites involved in de novo lipogenesis and mitochondrial β-oxidation
The reduced TG accumulation in HFMS mice suggested that hepatic lipid handling is altered. This could either be by reduced fatty acid import, diminished de novo fatty acid synthesis, increased fatty acid oxidation or by enhanced VLDL mediated TG export.
To determine whether de novo lipogenesis is affected, gene expression for hepatic sterol regulatory element-binding protein1c (SREBP1c), fatty acid synthase (FASN) (Figure 5A), and acetyl-CoA carboxylase 1 and 2 (ACC1 and ACC2) (Figure 6A) was analyzed by qRT-PCR. In addition, protein expression of SREBP1 (Figure 5B and Supplemental Figure S2), total ACC1 (265 kDa) and ACC2 (280 kDa) and their phosphorylated forms P-ACC1 (P-Ser76) and P-ACC2 (P-Ser219/P-Ser221) (Figure 6B) were determined by Western blot analysis. In addition, quantification of the ratios for P-ACC1 and total ACC1 protein (P-ACC1/ACC1) and P-ACC2 and total ACC2 (P-ACC2/ACC2) were determined.
Figure 5.
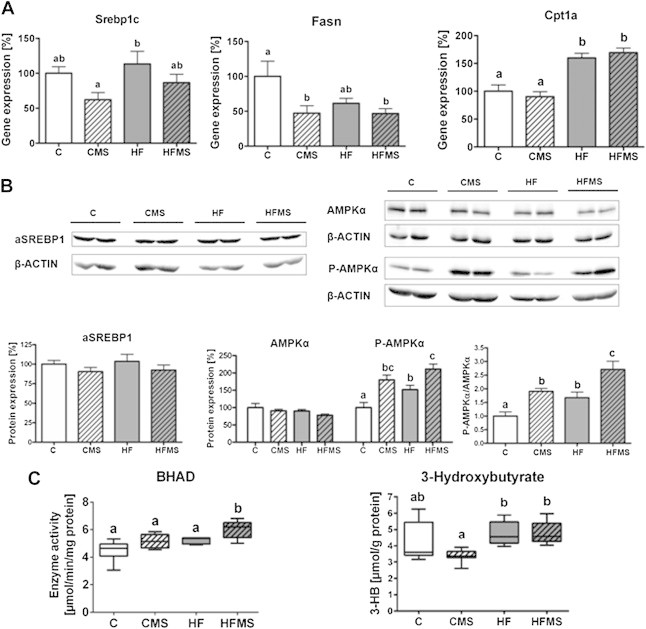
Influence of 4-weeks dietary methyl-donor supplementation on de novo lipogenesis, pivotal regulators, specific enzyme activity of fatty acid oxidation and ketone body production in the liver. (A) Gene expression of Srebp1c, Fasn and Cpt1a. (B) Protein expression of activated SREBP1 (aSREBP1), and AMPKα expression (AMPKα) and phosphorylation (P-AMPKα). (C) β-hydroxyacyl CoA dehydrogenase (BHAD) enzyme activity and 3-hydroxybutyrate concentrations. Separate gels were run for quantification of total AMPKα and P-AMPKα. β-ACTIN of the respective membranes were used for protein normalization. Data are presented as mean ± SEM (n = 5–9). Open, gray, open lined and gray lined boxes show data from C, HF, CMS and HFMS animals, respectively. Different subscript letters indicate statistical significance (p < 0.05). 3-HB, 3-hydroxybutyrate; AMPKα, AMP-activated protein kinase α; P-AMPKα, phosphorylated AMP-activated protein kinase α (Thr172); aSREBP1, soluble activated SREBP1; β-HAD, β-hydroxyacyl-CoA dehydrogenase; Cpt1a, carnitine palmitoyltransferase 1a; Fasn, fatty acid synthase.
Figure 6.
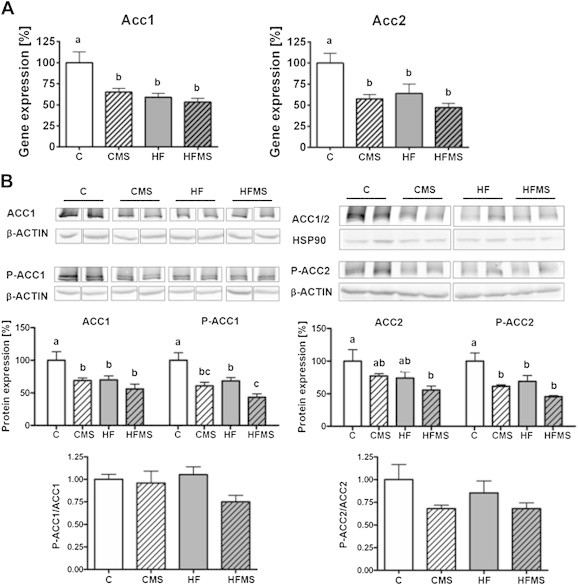
Influence of 4-weeks dietary methyl-donor supplementation on genes regulating fatty acid metabolism in the liver. (A) Gene expression of acetyl-CoA carboxylase 1 and 2 (ACC1 and ACC2). (B) Shown are protein expression levels for ACC1 and ACC2 and their phosphorylated form (P-ACC1 and P-ACC2) relative to control in %, and the ratio for P-ACC1/ACC1 and P-ACC2/ACC2 (normalized values were used). Separate gels were run for quantification of total ACC1 and P-ACC1, as well as for ACC2 and P-ACC2. β-ACTIN or HSP90 of the respective membranes were used for protein normalization. Data are presented as mean ± SEM (n = 7–9). Open, gray, open lined and gray lined boxes show data from C, HF, CMS and HFMS animals, respectively. Different subscript letters indicate statistical significance (p < 0.05).
With regard to Srebp1c gene expression, no obvious changes upon HF feeding or MDS compared to control diet were observed, except that HF animals compared to CMS fed mice showed elevated SREBP1c mRNA levels (p = 0.045). However, for Fasn, a target gene of SREBP1c, significantly decreased mRNA levels in MDS mice (p = 0.013) were revealed (Figure 5A). Furthermore, mRNA expression for ACC1 and 2 in CMS, HF and HFMS mice compared to control mice was significantly lower (ACC1: p < 0.0003; ACC2: p = 0.0029; Figure 6A).
Analysis of activated and nuclear SREBP1 protein did not show expression differences in activated and nuclear SREBP1 protein (Figure 5B, Supplemental Figure S2) which suggests that SREBP1 proteolytical cleavage and activation remain unaffected in liver. Interestingly, significantly reduced expression for both total (p = 0.0063) and phosphorylated ACC1 (p < 0.0001) protein were measured in mice fed CMS (ACC1: p < 0.05; P-ACC1: p < 0.01), HFMS (ACC1: p < 0.01; P-ACC1: p < 0.001) and HF (ACC1: p < 0.05; P-ACC1: p < 0.01) compared to controls, whereas P-ACC1/ACC1 ratios did not change. For hepatic ACC2 protein expression, in comparison to control mice significantly lower levels for total ACC2 were only found in HFMS mice, but not in CMS and HF mice which showed only a tendency for lower levels (p = 0.0503), whereas for P-ACC2 significantly reduced levels (p = 0.0005) were determined for CMS (P-ACC2: p < 0.01), HF (P-ACC2: p < 0.05) and HFMS (P-ACC2: p < 0.001) mice. However, P-ACC2/ACC2 ratios were not significantly different between the dietary groups (Figure 6B). Our findings indicate that de novo lipogenesis might be diminished by transcriptional downregulation of ACC1 upon MDS and HF diets, leading to reduced ACC1 protein, rather than by enzymatic inhibition of ACC1 upon phosphorylation; similar regulation appears to apply for ACC2, which is involved in the control of mitochondrial import of fatty acids for β-oxidation.
Degradation of fatty acids in liver occurs mainly via mitochondrial β-oxidation [35] and the rate limiting step is the transfer of acyl-CoA via the carnitine–acylcarnitine transporter (CACT) across the inner mitochondrial membrane. The acylation of carnitine is catalyzed by carnitine palmitoyltransferase 1 (CPT1) on the outer mitochondrial membrane [4]. Higher Cpt1a mRNA levels were detected in HF and HFMS mice than C and CMS mice (p < 0.001), but no further increase by MDS was observed (Figure 5A).
CPT1 is known to be regulated allosterically by malonyl-CoA generated by ACC2 causing an inhibition of the enzyme and leading to reduced fatty acid import into mitochondria. Malonyl-CoA levels generated by ACCs are mainly controlled by phosphorylation of ACC by AMPK as a central regulator of cellular energy homeostasis [36]. We therefore also determined protein levels for total AMPKα and phospho-AMPKα (P-AMPKα; P-Thr172) in liver tissues by Western blot analysis, and calculated the P-AMPKα/AMPKα ratio. Although total AMPKα protein levels remained unaffected upon HF or MDS, P-AMPKα levels increased significantly in both HF and MDS animals as compared to controls, and significant higher levels were found in HFMS than HF livers (p < 0.0001; Figure 5B). For P-AMPKα/AMPKα ratio, HFMS mice showed a 2.7-fold higher ratio compared to control mice, which was the highest significant value compared to the respective ratio of CMS (1.9) and HF (1.7) mice as shown in Figure 5B. Interestingly, the comparison of P-AMPKα/AMPKα ratios between HFMS and HF mice, that reflects the effect of methyl-donor supplementation of HF diet, revealed a 1.6-fold increased P-AMPKα/AMPKα ratio. Based on previous observations [37], our data suggested that AMPK activity is increased, and consequently, hepatic β-oxidation capacity is elevated in HFMS mice as compared to HF mice.
To proof this further, we determined in the liver the enzyme activity of β-HAD (oxidation of β-hydroxyacyl CoA dehydrogenase), which is a specific enzyme of β-oxidation. The analysis revealed a significantly increased β-HAD activity in HFMS mice (p < 0.001) compared to C, CMS and HF mice (Figure 5C). Furthermore, we analyzed hepatic β-hydroxybutyrate (3-HB) as typical metabolite of ketogenesis, but this did not reveal any changes in CMS and HFMS mice compared to the respective control and HF diet (Figure 5C). We next determined free carnitine and acyl-carnitine concentrations in liver tissues via FIA-MS (Supplemental Table S6). As shown in Figure 7, significantly reduced concentrations of dl-carnitine, short chain acyl-carnitines as well as long-chain acyl-carnitines including C16:0, C16:1, and C18:0 were detected in MDS mice. Interestingly, levels of medium-chain acyl-carnitines such as C7-DC, C8, C9, C10, C10:1, and C12 were not significantly affected by MDS, except for C12:1, which was slightly increased upon CMS (Supplemental Table S6). However, these medium-chain acyl-carnitines showed higher levels (p < 0.001) in HF and HFMS mice than control and CMS mice, except for C7-DC and C12 in CMS mice (Supplemental Table S6), suggesting a generalized obesity effect in elevating medium-chain acyl-carnitines in liver.
Figure 7.
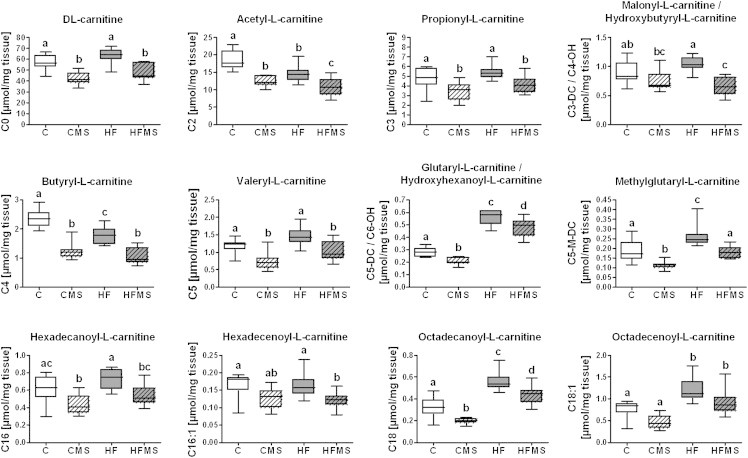
Analysis of C, CMS, HF and HFMS liver tissues for selected hepatic carnitine and acyl-carnitine concentrations after 12-weeks feeding, including 4-weeks dietary methyl-donor supplementation. Data are presented as box and whisker plot (n = 8–9). Data from C, HF, CMS and HFMS animals are shown as open, gray, open lined and gray lined boxes, respectively. Different subscript letters indicate statistical significance (p < 0.05). C0, dl-carnitine; C2, acetyl-l-carnitine; C3, propionyl-l-carnitine; C3-DC/C4-OH, malonyl-l-carnitine/hydroxybutyryl-l-carnitine; C4, butyryl-l-carnitine; C5, valeryl-l-carnitine; C5-DC/C6-OH, glutaryl-l-carnitine/hydroxyhexanoyl-l-carnitine; C5-M-DC, methylglutaryl-l-carnitine; C16, hexadecanoyl-l-carnitine; C16:1, hexadecenoyl-l-carnitine; C18, octadecanoyl-l-carnitine; C18:1, octadecenoyl-l-carnitine.
3.5. Diet-induced obesity (DIO) affects phospholipid subclasses in liver of HF mice while MDS reveals no effects on phospholipid status
We also quantified a large number of phosphatidylcholine (PC) species as well as selected sphingomyelins (SM) and lyso-PC species in liver tissues via LC-MS/MS. Figure 8 shows a heat-map based on the metabolites analyzed in liver samples of 8–9 animals in each of the different dietary groups of trial. The corresponding data are provided in Supplemental Table S7. Out of 105 lipid species analyzed, 52 revealed significant differences between C and HF mice, but clustering did not provide evidence for an effect of the MDS. Amongst the metabolites that discriminated most between animals from C and HF diets were predominantly PC species with medium-chain fatty acids revealing reduced levels in livers from HF and HFMS mice such as PC aa C30:0, PC aa C32:0, PC aa C32:1, PC aa C32:2, PC aa C34:3 or PC aa C34:4. In contrast, PC species with long-chain fatty acids, including PC aa C36:1, PC aa C38:0, PC aa C38:3, PC aa C38:4, PC aa C38:5 or PC aa C40:6, were increased in liver samples of HF and HFMS mice. Furthermore, some ether-linked PC subclasses (PC ae) showed significantly increased hepatic concentrations in HF and HFMS mice as compared to control liver tissues (Supplemental Table S7). These ether-PC species contained also fatty acids with higher numbers of double-bonds. Amongst the lyso-PC subclass, especially lysoPC a C18:0 showed elevated levels in NAFLD liver while lysoPC a C18:2 decreased in those samples (Supplemental Table S7). Regarding changes in hydroxyl-sphingomyelin concentrations in NAFLD tissues, SM (OH) C16:1 and SM (OH) C24:1 levels were increased, whereas other sphingomyelins as for example SM C24:0 and SM C24:1 showed reduced levels (Supplemental Table S7). Taken together, all lipid subclasses covered in the analysis revealed marked alterations in hepatic tissues when HF and HFMS samples were compared to C and CMS samples. Yet, a significant alteration in the lipid profiles by MDS was not observed.
Figure 8.
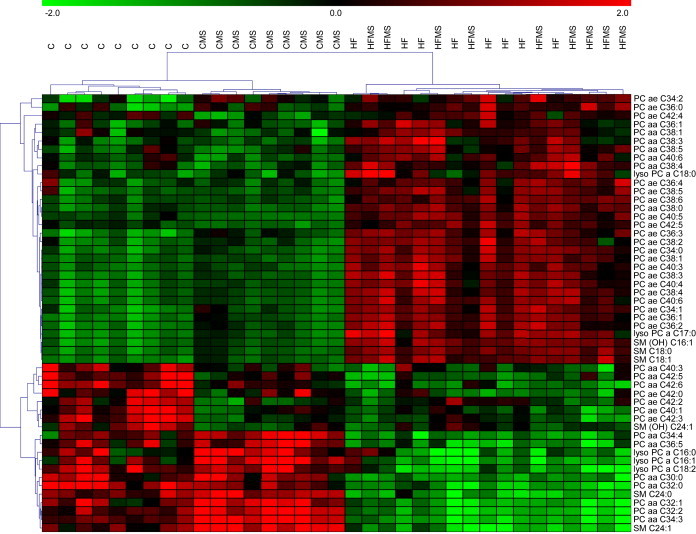
Heat-map diagram of differentially expressed phospholipid subclasses in the liver of C, CMS, HF and HFMS animals. A standard score of 52 regulated phosphatidylcholine, lysophosphatidylcholine, sphingomyelin and hydroxysphingomyelin (n = 8–9, p < 0.01) is shown. Green and red indicate down and upregulation, respectively.
4. Discussion
Excess fat deposition in liver, referred to as NAFLD, is commonly observed in DIO models in rodents and mimics the condition in humans with metabolic syndrome. Methyl-donors such as choline and betaine act as lipotropic compounds and were shown to attenuate NAFLD and steatosis [16,20,21]. In addition, it has been demonstrated that feeding mice and rats with methionine and choline deficient diet led to the development of steatohepatitis [38,39]. However, whether dietary MDS can reverse an established NAFLD has not been proven. We therefore studied the effects of a dietary supplementation of methyl-donors on NAFLD progression when animals after 8-weeks HF feeding received a HF diet enriched with methyl-donors for additional 4 weeks (Figure 9). The study included as well animals receiving a low fat/carbohydrate-rich control diet either supplemented or not with the methyl-donors.
Figure 9.
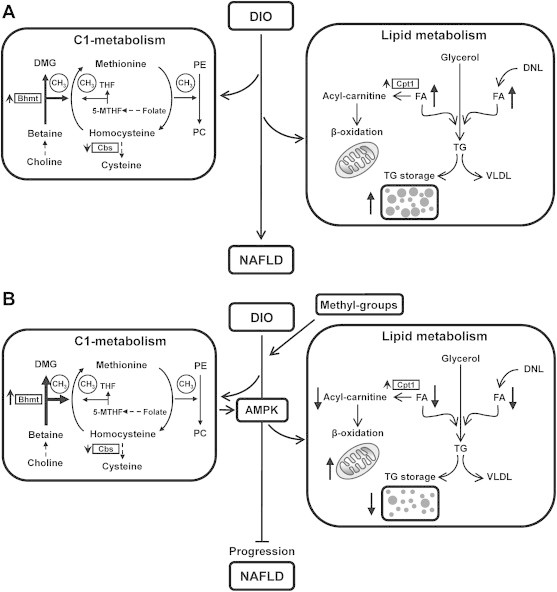
Schematic presentation of the interrelationship of identified changes in gene and protein expression, as well as in metabolite levels in methyl-group donor supplemented DIO mice. (A) DIO-mediated NAFLD results in increased hepatic free fatty acid concentrations and triacylglycerol accumulation accompanied by changes of BHMT and CBS in the C1-metabolism. (B) MDS down-regulates DNL (decreased ACC1 and 2 mRNA and protein levels; not depicted) and enhances AMPK activity which induces elevated hepatic β-oxidation, thereby preventing DIO mediated NAFLD progression and further increasing BHMT expression. Observed changes of mRNA (e.g. Cpt1a), proteins (BHMT, CBS), protein phosphorylation (P-AMPKα) and metabolites (acyl-carnitine, FA) are depicted. Upward- and downward-oriented arrows indicate up-regulation or down-regulation, respectively. AMPK, AMP-activated protein kinase; C1-metabolism, one-carbon metabolism; Cpt1a, carnitine palmitoyltransferase-1a; DIO, diet-induced obesity; DMG, dimethylglycine; DNL, de novo lipogenesis; FA, free fatty acids; 5-MTHF, 5-methyl-tetrahydrofolate; NAFLD, non-alcoholic fatty liver disease; PC, phosphatidylcholine; PE, phosphatidylethanolamine; CH3, methyl group transfer via SAM; TG, triacylglycerol; THF, tetrahydrofolate; VLDL, very low density lipoprotein.
4.1. Reduction of elevated hepatic free fatty acid levels in HF diet fed animals upon methyl-donor supplementation
8-weeks HF feeding was sufficient to cause high hepatic TG deposition as well as increased liver and VAT weight associated with the body weight gain. Animals displayed also plasma hyperglycaemia and started to develop hyperinsulinemia. Compared to mice from 12-week HF feeding, HFMS animals, which received first HF diet for 8 weeks followed by HFMS diet for 4 weeks, showed significant reduced hepatic TG and NEFA levels. This was also visible in Oil red O-stained liver sections. Although MDS could not completely reverse the NAFLD state, it prevented completely further TG accumulation. This is crossly in line with findings of Kathirvel et al. [40] describing reduced hepatic fat deposition and improved insulin sensitivity in a murine DIO model by betaine supplementation for the last 6 weeks of a trial. According to the glucolipotoxicity hypothesis, increased plasma and hepatic NEFA levels combined with hyperglycaemia are the main drivers for NAFLD [41,42]. In hepatocytes, an increase in free fatty acids and acyl-CoA levels is considered to result from a reduced ability to enhance fatty acid oxidation by a simultaneously reduced peroxisome proliferator-activated receptor-α (PPARα) activation, enhanced SREBP1c expression and reduced CPT1 activity [42,43]. In this respect, our finding that methyl-donor administration can reduce the elevated hepatic free fatty acid levels in animals on HF diet to normal levels, as in controls, is of particular importance.
4.2. Diminished hepatic CBS protein level but increased BHMT protein status in mice upon HF diet
A high dietary fat intake and increased hepatic lipid processing requires phospholipids for storage as well as for VLDL secretion into circulation. Our metabolite analysis in liver clearly demonstrates that MDS increased hepatic SAH levels and most importantly, increased the levels of SAM in HFMS mice needed for PC synthesis. MDS also raised Hcy and cystathionine levels. In plasma, methyl-supplemented animals displayed as well elevated serine and decreased glycine levels and most prominently increased concentrations of sarcosine which is the prime metabolite for methyl-group excretion. Despite a lack of MDS effect on hepatic methionine concentrations, they tended to be higher in supplemented animals. In all mice on HF diet with or without MDS a significantly diminished hepatic CBS protein level but increased BHMT protein status was observed. This confirms literature findings in which extra dietary betaine or choline upregulates BHMT mRNA expression and activity on the background of either methionine-deficient or methionine-adequate diets [44–46]. The importance of BHMT in Hcy homeostasis and its association to NAFLD was recently demonstrated in a Bhmt−/− mouse model [47] displaying increased hepatic TG concentrations and altered choline and phosphatidylcholine concentrations associated with reduced VLDL secretion emphasizing the crucial role of BHMT in hepatic lipid homeostasis [47].
4.3. Changes of acyl-carnitine levels in all methyl-supplemented animals
In the present study, HF diets caused significant alterations in liver phosphatidylcholines, lyso-PC and sphingomyelins, but no effects of MDS on the concentrations of these metabolite species could be observed. However, amongst the lipid subclasses analyzed, acyl-carnitine levels changed significantly in all methyl-supplemented animals. Both, short-chain acyl-carnitines (C2–C6) as well as long-chain derivatives (>C16) showed significantly reduced concentrations while medium-chain acyl-carnitines (>C8 < C14) remained unaffected. Although the latter, as for example C8, C9 or C10:1 revealed increased levels in liver tissues of animals on HF diets, MDS was without any effects. There is convincing evidence from a comparative analysis that the acyl-carnitines in tissues mirror in both, concentration and pattern the status of the corresponding acyl-CoA species [48]. Acyl-CoA esters are known as regulators of energy metabolism [49]. Long-chain acyl-CoA act as inhibitors for the mitochondrial adenine nucleotide translocase (ANT), which is considered to be the overall rate limiting step in energy metabolism [49]. In addition, malonyl-CoA has been identified as a crucial allosteric regulator of CPT1, the rate-limiting enzyme for transfer of long-chain acyl-CoA into mitochondria for β-oxidation.
4.4. Expression of genes and proteins involved in de novo lipogenesis and mitochondrial β-oxidation
ACC1 and 2 catalyze the synthesis malonyl-CoA from acetyl-CoA, whereas malonyl-CoA is degraded by a malonyl-CoA decarboxylase (MCD). ACC1-generated malonyl-CoA is utilized by FASN for cytosolic fatty acid synthesis. In contrast, the mitochondrial bound ACC2 generates malonyl-CoA which operates as inhibitor of CPT1, the rate-limiting protein for mitochondrial fatty acid import. ACC is under the control of insulin leading to increased malonyl-CoA levels, whereas phosphorylation of ACCs decreases their activity resulting in a reduction of malonyl-CoA [37]. Phosphorylation of ACC [36] and MCD [37] is mainly regulated by AMPK leading to reduced malonyl-CoA levels. Stimulation of AMPK results in the activation of catabolic processes, e.g. fatty acid uptake and transport into mitochondria, β-oxidation and the repression of many anabolic processes, e.g. fatty acid and cholesterol synthesis, gluconeogenesis [50].
In contrast, to the expected classical inhibition of ACCs upon increased AMPK-mediated phosphorylation, and the enhanced AMPK phosphorylation upon HF and MDS as observed in our study (see also discussion below), it was surprising that ACC1 and 2 were rather regulated on transcriptional level upon MDS and HF diets, leading to reduced hepatic ACC1 and 2 mRNA and protein levels without obvious changes in ratios for P-ACC1/ACC1 and P-ACC2/ACC2. In addition, hepatic FASN mRNA levels were reduced in mice by MDS. Together, these changes strongly suggest that de novo lipogenesis is altered.
Interestingly, the transcription factor SREBP1c, known to alter gene transcription of lipogenic enzymes and a prime candidate to be involved in the transcriptional regulation of ACCs and FASN [50], did not reveal any diet or supplementation effects on its own hepatic mRNA or protein expression level. However, in this study we did not assess whether the binding of SREBP1c to the promoter of these target genes was reduced upon the diets. Moreover, one could speculate that the regulation of other transcription factors, known to be involved in the complex transcriptional control of lipogenesis and TG synthesis, such as ChREBP (carbohydrate-response element-binding protein), LXR (liver X receptor), PGC1α (peroxisome proliferator activated receptor-γ co-activator-1α) and PGC1β or XBP1 (X-box binding protein 1), or epigenetic mechanism, such as DNA methylation, histone modifications or microRNA, might have changed the mRNA and protein expression of ACCs and FASN or other genes, which are functionally involved in the reduction of TG and NEFA levels in HFMS mice [50].
Regarding mitochondrial β-oxidation, increased CPT1a transcript levels were found in both HF groups, but no change by added methyl-donors was observed. However, when total and P-AMPKα levels were determined, it became obvious that total AMPKα protein levels remained unaffected by diet, but the threonine172 (Thr172)-phosphorylation state of AMPKα, and consequently the P-AMPKα/AMPKα ratio, which enhances the AMPK activity, increased strongly. For both the HF and CMS diet compared to the control diet, increased P-AMPKα and P-AMPKα/AMPKα levels were found suggesting that both the high fat and methyl-donor supplementation can lead to AMPK activation in liver. Importantly, for HFMS compared to CMS and HF diet, P-AMPKα/AMPKα levels were significantly 1.4-fold and 1.6-fold higher, respectively, indicating synergistic effects for MDS combined with HF diet feeding on AMPKα phosphorylation.
Considering increased phosphorylation of AMPKα as enhanced enzymatic AMPK activity, consequently, both mitochondrial fatty acid oxidation and mitochondrial biogenesis could be affected via the ‘master regulator’ of mitochondrial biogenesis PGC1α. It has been shown that PGC1α enhances the activity of several transcription factors for mitochondrial genes [50]. Interestingly, AMPK can directly phosphorylate PGC1α, which can cause activation of its own transcription via a positive feedback loop [51]. An alternative mechanism for PGC1α activation by AMPK is through promotion of PGC1α deacetylation by increasing NAD-concentration, which is the co-substrate for the deacetylase sirtuin 1 (SIRT1) [50]. With regard to dietary changes in methyl-donors, it is of particular interest that PGC1α activity can be enhanced additionally by its methylation dependent on protein arginine methyltransferase 1 [52]. In this context, dietary methyl-donor deficiency was recently described to impair fatty acid oxidation through PGC1α hypomethylation in rat liver [39] and myocardium [53]. Interestingly, PGC1α is also known to influence homocysteine homeostasis, likely via hepatic nuclear factor 4α mediated expression control of BHMT [54].
Together, one can speculate that in our study both AMPK and PGC1α may operate in the alleviation of TG accumulation in the livers of methyl-donor supplemented mice and that the increased BHMT protein expression observed in these mice may reflect enhanced PGC1α activity on its target genes. Considering the complex molecular network of metabolism regulation in living organisms, it will be very interesting for future experiments to determine whether MDS and HF diet impact the regulation of PGC1α activity by methylation and/or P-AMPK dependent phosphorylation associated with metabolic changes. In this context, the analysis of PGC1β, having similar metabolic targets, will be also of interest [50].
4.5. Increase of AMPKα phosphorylation in MDS mice
AMPKα phosphorylation mediated by upstream kinases is a prerequisite for AMPK activation [55,56]. AMPK senses the cellular energy state by AMP-dependent activation, antagonized by ATP [36]. AMPK is a heterotrimeric protein with a γ-subunit containing four tandem repeats of evolutionary conserved CBS-domains known to bind AMP or ATP [36]. However, CBS-domains can also bind SAM and recently a CBS-domain with bound SAM was crystallized [57]. As proposed by Finkelstein, AMPK could therefore also act as a SAM sensor [14]. Wang et al. reported an improved adipose tissue function by betaine supplementation in obese mice with improved insulin signaling and the prevention of endoplasmatic reticulum stress in adipose tissue [22]. Furthermore, betaine supplementation normalized plasma adiponectin concentrations which were decreased upon DIO [22]. Adiponectin is known to activate AMPK by an unknown mechanism [58], potentially by an endocrine mechanism. However, analysis of plasma adiponectin in our mice did not reveal a significant difference, suggesting that adiponectin effects on hepatic AMPK activation in MDS mice are negligible.
An enhanced supply of methyl-donors increases the hepatic biosynthesis and turnover of SAM which consumes ATP and the subsequent use of SAM for transmethylation produces cellular SAH and hence Hcy and adenosine. The thermodynamically favored SAH synthesis generates the necessity to constantly metabolize Hcy and adenosine in the methionine cycle [59,60]. Accumulating adenosine is primarily used by adenosine kinase in liver for AMP synthesis [15,61]. Therefore, it is conceivable that an enhanced SAM synthesis and turnover increases cellular AMP concentrations with an altered AMP/ATP ratio affecting AMPK activation. In addition, enhanced SAM turnover may be able to reduce the activity of AMPK specific protein phosphatases resulting in an increased AMPK phosphorylation status [62].
4.6. AMPK-mediated stimulation of l-carnitine acylation of fatty acids and increase of fatty acid β-oxidation
The increased phosphorylation state of AMPK observed in liver tissues of CMS and HFMS mice may translate into an increased activity of CPT1 and in turn increased fatty acid β-oxidation. The increased β-HAD activity supports the increased capacity for fatty acid oxidation, although this was not associated with enhanced 3-HB levels in MDS livers. Interestingly, Ogborn et al. described that a dietary betaine supplementation also did not alter hepatic 3-HB levels in a rat polycystic kidney disease model [63] suggesting that a methyl-donor supplementation not a priori also increases hepatic ketone body production. Our analysis of the various acyl-carnitines revealed that in the CMS and HFMS groups all short- and long-chain acyl-carnitines were reduced in levels compared to C tissues. Considering that acyl-carnitines are shuttled out of mitochondria via the CACT [64] in a one to one exchange process it is conceivable that the export of acyl-carnitines increases when the acyl-CoA flux through β-oxidation is limited by the availability of NAD and FAD and intermediate products. Acyl-carnitines are released into plasma and their analysis is used for diagnosing inherited diseases of β-oxidation represented for example by long-chain or medium-chain acyl-CoA dehydrogenase deficiencies [65]. In this context, the increased appearance of the acyl-carnitines in plasma may serve as a surrogate marker of a limited β-oxidation capacity. It is interesting to note that HF diets increased also all medium-chain acyl-carnitines (> C8 < C14) although MDS failed to affect their levels. Medium-chain fatty acids are known to be taken up into mitochondria independent of CPT1 and may be exported back into the cytosol by CACT as acyl-carnitines. As all other hepatic acyl-carnitines declined in concentration in MDS animal, it seems plausible to assume that this is a consequence of increased uptake of fatty acids into mitochondria through CPT1 that is no more inhibited by cytosolic malonyl-CoA when AMPK is activated. The importance of CPT1 in limiting fatty acid oxidation was recently elegantly shown in functional studies with genetically obese mice where increased CPT1 activity in the liver protected the mice against hepatic steatosis and insulin resistance [66]. Our data therefore argue for an AMPK-mediated stimulation of l-carnitine acylation of fatty acids by CPT1 that in turn increases fatty acid β-oxidation with a concomitant decline in short- and long-chain acyl-carnitines, NEFA and TG especially in livers from HFMS mice.
4.7. Conclusions
In summary, we have demonstrated that a MDS can prevent the progression of lipid accumulation in liver tissues of animals with NAFLD but it cannot reverse NAFLD. MDS leads to major changes in C1 metabolites and reduces hepatic free fatty acid and acyl-carnitine levels. These effects are associated with an increased activity state of AMPK and β-HAD enzyme activity that links C1-metabolism and β-oxidation. This is suggested by significant cross-correlations of hepatic SAH, Hcy and cystathionine concentrations with acyl-carnitine levels in liver tissues across all diets. AMPK may bind SAM directly via its CBS domains and thus acts as a direct SAM-sensor or alternatively responds with activation to increased AMP levels that arise from the enhanced methyl-group turnover. Considering the prime roles of PGC1α and AMPK in the complex molecular network of metabolism regulation and adaptations it will be of great interest to assess in future experiments whether the increase of AMPKα phosphorylation upon MDS leads to enhanced PGC1α methylation and activity.
Acknowledgments
The authors thank Johanna Welzhofer, Alexander Haag, Elmar Jocham and Ronny Scheundel for technical assistance.
Footnotes
Bernhard L. Bader and Hannelore Daniel contributed equally to this work.
Conflict of interest
HD is a consulting editor of Molecular Metabolism. There has been no significant financial support for this work that could have influenced its outcome.
Appendix A. Supporting information
There are two Supplemental Figures and seven Supplemental Tables associated with this manuscript.
References
- 1.Starley B.Q., Calcagno C.J., Harrison S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 2.Lazo M., Clark J.M. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Seminars in Liver Disease. 2008;28:339–350. doi: 10.1055/s-0028-1091978. [DOI] [PubMed] [Google Scholar]
- 3.Wree A., Kahraman A., Gerken G., Canbay A. Obesity affects the liver – the link between adipocytes and hepatocytes. Digestion. 2011;83:124–133. doi: 10.1159/000318741. [DOI] [PubMed] [Google Scholar]
- 4.Varela-Rey M., Embade N., Ariz U., Lu S.C., Mato J.M., Martinez-Chantar M.L. Non-alcoholic steatohepatitis and animal models: understanding the human disease. International Journal of Biochemistry & Cell Biology. 2009;41:969–976. doi: 10.1016/j.biocel.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 5.Angulo P. Nonalcoholic fatty liver disease. New England Journal of Medicine. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 6.Musso G., Gambino R., Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Progress in Lipid Research. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Rinella M.E., Elias M.S., Smolak R.R., Fu T., Borensztajn J., Green R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. Journal of Lipid Research. 2008;49:1068–1076. doi: 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu S.C., Alvarez L., Huang Z.Z., Chen L., An W., Corrales F.J. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Chantar M.L., Vazquez-Chantada M., Ariz U., Martinez N., Varela M., Luka Z. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walkey C.J., Yu L., Agellon L.B., Vance D.E. Biochemical and evolutionary significance of phospholipid methylation. Journal of Biological Chemistry. 1998;273:27043–27046. doi: 10.1074/jbc.273.42.27043. [DOI] [PubMed] [Google Scholar]
- 11.Noga A.A., Vance D.E. A gender-specific role for phosphatidylethanolamine N-methyltransferase-derived phosphatidylcholine in the regulation of plasma high density and very low density lipoproteins in mice. Journal of Biological Chemistry. 2003;278:21851–21859. doi: 10.1074/jbc.M301982200. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y., Su B., Jacobs R.L., Kennedy B., Francis G.A., Waddington E. Lack of phosphatidylethanolamine N-methyltransferase alters plasma VLDL phospholipids and attenuates atherosclerosis in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29:1349–1355. doi: 10.1161/ATVBAHA.109.188672. [DOI] [PubMed] [Google Scholar]
- 13.Williams K.T., Schalinske K.L. Homocysteine metabolism and its relation to health and disease. Biofactors. 2010;36:19–24. doi: 10.1002/biof.71. [DOI] [PubMed] [Google Scholar]
- 14.Finkelstein J.D. Metabolic regulatory properties of S-adenosylmethionine and S-adenosylhomocysteine. Clinical Chemistry and Laboratory Medicine. 2007;45:1694–1699. doi: 10.1515/CCLM.2007.341. [DOI] [PubMed] [Google Scholar]
- 15.Mato J.M., Martinez-Chantar M.L., Lu S.C. Methionine metabolism and liver disease. Annual Review of Nutrition. 2008;28:273–293. doi: 10.1146/annurev.nutr.28.061807.155438. [DOI] [PubMed] [Google Scholar]
- 16.Ball C.R. Actions of betaine, carnitine and choline on the pattern of hepatic liposis in mice fed a high-fat, low-protein diet. Anatomical Record. 1964;149:677–689. doi: 10.1002/ar.1091490411. [DOI] [PubMed] [Google Scholar]
- 17.Ji C., Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 18.Powell C.L., Bradford B.U., Craig C.P., Tsuchiya M., Uehara T., O'Connell T.M. Mechanism for prevention of alcohol-induced liver injury by dietary methyl donors. Toxicological Sciences. 2010;115:131–139. doi: 10.1093/toxsci/kfq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kharbanda K.K., Mailliard M.E., Baldwin C.R., Beckenhauer H.C., Sorrell M.F., Tuma D.J. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. Journal of Hepatology. 2007;46:314–321. doi: 10.1016/j.jhep.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 20.Song Z., Deaciuc I., Zhou Z., Song M., Chen T., Hill D. Involvement of AMP-activated protein kinase in beneficial effects of betaine on high-sucrose diet-induced hepatic steatosis. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2007;293:G894–G902. doi: 10.1152/ajpgi.00133.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwon D.Y., Jung Y.S., Kim S.J., Park H.K., Park J.H., Kim Y.C. Impaired sulfur-amino acid metabolism and oxidative stress in nonalcoholic fatty liver are alleviated by betaine supplementation in rats. Journal of Nutrition. 2009;139:63–68. doi: 10.3945/jn.108.094771. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z., Yao T., Pini M., Zhou Z., Fantuzzi G., Song Z. Betaine improved adipose tissue function in mice fed a high-fat diet: a mechanism for hepatoprotective effect of betaine in nonalcoholic fatty liver disease. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2010;298:G634–G642. doi: 10.1152/ajpgi.00249.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kathirvel E., Morgan K., Nandgiri G., Sandoval B.C., Caudill M.A., Bottiglieri T. Betaine improves nonalcoholic fatty liver and associated hepatic insulin resistance: a potential mechanism for hepatoprotection by betaine. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2010;299:G1068–G1077. doi: 10.1152/ajpgi.00249.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolff G.L., Kodell R.L., Moore S.R., Cooney C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB Journal. 1998;12:949–957. [PubMed] [Google Scholar]
- 25.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Pfaffl M.W., Tichopad A., Prgomet C., Neuvians T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnology Letters. 2004;26:509–515. doi: 10.1023/b:bile.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 27.Rubio-Aliaga I., Roos B., Sailer M., McLoughlin G.A., Boekschoten M.V., van Erk M. Alterations in hepatic one-carbon metabolism and related pathways following a high-fat dietary intervention. Physiological Genomics. 2011;43:408–416. doi: 10.1152/physiolgenomics.00179.2010. [DOI] [PubMed] [Google Scholar]
- 28.Kirsch S.H., Knapp J.P., Geisel J., Herrmann W., Obeid R. Simultaneous quantification of S-adenosyl methionine and S-adenosyl homocysteine in human plasma by stable-isotope dilution ultra performance liquid chromatography tandem mass spectrometry. Journal of Chromatography B – Analytical Technologies in the Biomedical and Life Sciences. 2009;877:3865–3870. doi: 10.1016/j.jchromb.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 29.Stabler S.P., Lindenbaum J., Savage D.G., Allen R.H. Elevation of serum cystathionine levels in patients with cobalamin and folate deficiency. Blood. 1993;81:3404–3413. [PubMed] [Google Scholar]
- 30.Römisch-Margl W., Prehn C., Bogumil R., Röhring C., Suhre C., Adamski J. Procedure for tissue sample preparation and metabolite extraction for high-throughput targeted metabolomics. Metabolomics. 2011;8:113–142. [Google Scholar]
- 31.Molero J.C., Waring S.G., Cooper A., Turner N., Laybutt R., Cooney G.J. Casitas b-lineage lymphoma-deficient mice are protected against high-fat diet-induced obesity and insulin resistance. Diabetes. 2006;55:708–715. doi: 10.2337/diabetes.55.03.06.db05-0312. [DOI] [PubMed] [Google Scholar]
- 32.R-Development-Core-Team . R Foundation for Statistical Computing; Vienna, Austria: 2009. R: a language and environment for statistical computing. [Google Scholar]
- 33.Smyth G.K. 2005. Limma: linear models for microarray data; pp. 397–420. (Bioinformatics and computational biology solutions using R and bioconductor). [Google Scholar]
- 34.Harrell F.J. 2009. Hmisc: Harrell Miscellaneous. R package version 3.7-0. [Google Scholar]
- 35.Browning J.D., Horton J.D. Molecular mediators of hepatic steatosis and liver injury. Journal of Clinical Investigation. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardie D.G. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochemical Society Transactions. 2011;39:1–13. doi: 10.1042/BST0390001. [DOI] [PubMed] [Google Scholar]
- 37.Ruderman N., Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nature Reviews Drug Discovery. 2004;3:340–351. doi: 10.1038/nrd1344. [DOI] [PubMed] [Google Scholar]
- 38.Weltman M.D., Farrell G.C., Liddle C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology. 1996;111:1645–1653. doi: 10.1016/s0016-5085(96)70028-8. [DOI] [PubMed] [Google Scholar]
- 39.Pooya S., Blaise S., Moreno Garcia M., Giudicelli J., Alberto J.M., Gueant-Rodriguez R.M. Methyl donor deficiency impairs fatty acid oxidation through PGC-1alpha hypomethylation and decreased ER-alpha, ERR-alpha, and HNF-4alpha in the rat liver. Journal of Hepatology. 2012;57:344–351. doi: 10.1016/j.jhep.2012.03.028. [DOI] [PubMed] [Google Scholar]
- 40.Desmarchelier C., Dahlhoff C., Keller S., Sailer M., Jahreis G., Daniel H. C57Bl/6N mice on a Western diet display reduced intestinal and hepatic cholesterol levels despite a plasma hypercholesterolemia. BMC Genomics. 2012;13:84. doi: 10.1186/1471-2164-13-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mihalik S.J., Goodpaster B.H., Kelley D.E., Chace D.H., Vockley J., Toledo F.G. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 2009;18:1695–1700. doi: 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muoio D.M., Newgard C.B. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nature Reviews Molecular Cell Biology. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 43.Vanni E., Bugianesi E., Kotronen A., De Minicis S., Yki-Jarvinen H., Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa? Digestive and Liver Disease. 2010;42:320–330. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 44.Park E.I., Garrow T.A. Interaction between dietary methionine and methyl donor intake on rat liver betaine-homocysteine methyltransferase gene expression and organization of the human gene. Journal of Biological Chemistry. 1999;274:7816–7824. doi: 10.1074/jbc.274.12.7816. [DOI] [PubMed] [Google Scholar]
- 45.Finkelstein J.D., Martin J.J., Harris B.J., Kyle W.E. Regulation of hepatic betaine-homocysteine methyltransferase by dietary betaine. Journal of Nutrition. 1983;113:519–521. doi: 10.1093/jn/113.3.519. [DOI] [PubMed] [Google Scholar]
- 46.Park E., Renduchintala M.S., Garrow T. Diet-induced changes in hepatic betaine-homocysteine methyltransferase activity are mediated by changes in the steady-state level of its mRNA. Nutritional Biochemistry. 1997;8:541–545. [Google Scholar]
- 47.Teng Y.W., Mehedint M.G., Garrow T.A., Zeisel S.H. Deletion of betaine-homocysteine S-methyltransferase in mice perturbs choline and 1-carbon metabolism, resulting in fatty liver and hepatocellular carcinomas. Journal of Biological Chemistry. 2011;286:36258–36267. doi: 10.1074/jbc.M111.265348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.ter Veld F., Primassin S., Hoffmann L., Mayatepek E., Spiekerkoetter U. Corresponding increase in long-chain acyl-CoA and acylcarnitine after exercise in muscle from VLCAD mice. Journal of Lipid Research. 2009;50:1556–1562. doi: 10.1194/jlr.M800221-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Faergeman N.J., Knudsen J. Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochemical Journal. 1997;323(Pt 1):1–12. doi: 10.1042/bj3230001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature Reviews Molecular Cell Biology. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jager S., Handschin C., St-Pierre J., Spiegelman B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teyssier C., Ma H., Emter R., Kralli A., Stallcup M.R. Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes & Development. 2005;19:1466–1473. doi: 10.1101/gad.1295005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia M.M., Gueant-Rodriguez R.M., Pooya S., Brachet P., Alberto J.M., Jeannesson E. Methyl donor deficiency induces cardiomyopathy through altered methylation/acetylation of PGC-1alpha by PRMT1 and SIRT1. Journal of Pathology. 2011;225:324–335. doi: 10.1002/path.2881. [DOI] [PubMed] [Google Scholar]
- 54.Li S., Arning E., Liu C., Vitvitsky V., Hernandez C., Banerjee R. Regulation of homocysteine homeostasis through the transcriptional coactivator PGC-1alpha. American Journal of Physiology – Endocrinology and Metabolism. 2009;296:E543–E548. doi: 10.1152/ajpendo.90719.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ingebritsen T.S., Lee H.S., Parker R.A., Gibson D.M. Reversible modulation of the activities of both liver microsomal hydroxymethylglutaryl coenzyme A reductase and its inactivating enzyme. Evidence for regulation by phosphorylation-dephosphorylation. Biochemical and Biophysical Research Communications. 1978;81:1268–1277. doi: 10.1016/0006-291x(78)91273-1. [DOI] [PubMed] [Google Scholar]
- 56.Hawley S.A., Davison M., Woods A., Davies S.P., Beri R.K., Carling D. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. Journal of Biological Chemistry. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 57.Lucas M., Encinar J.A., Arribas E.A., Oyenarte I., Garcia I.G., Kortazar D. Binding of S-methyl-5′-thioadenosine and S-adenosyl-L-methionine to protein MJ0100 triggers an open-to-closed conformational change in its CBS motif pair. Journal of Molecular Biology. 2010;396:800–820. doi: 10.1016/j.jmb.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 58.Yamauchi T., Kamon J., Minokoshi Y., Ito Y., Waki H., Uchida S. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nature Medicine. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 59.Finkelstein J.D. Methionine metabolism in mammals. Journal of Nutritional Biochemistry. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 60.De La Haba G., Cantoni G.L. The enzymatic synthesis of S-adenosyl-L-homocysteine from adenosine and homocysteine. Journal of Biological Chemistry. 1959;234:603–608. [PubMed] [Google Scholar]
- 61.Boison D., Scheurer L., Zumsteg V., Rulicke T., Litynski P., Fowler B. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:6985–6990. doi: 10.1073/pnas.092642899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinez-Chantar M.L., Vazquez-Chantada M., Garnacho M., Latasa M.U., Varela-Rey M., Dotor J. S-adenosylmethionine regulates cytoplasmic HuR via AMP-activated kinase. Gastroenterology. 2006;131:223–232. doi: 10.1053/j.gastro.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 63.Ogborn M.R., Nitschmann E., Bankovic-Calic N., Buist R., Peeling J. Dietary betaine modifies hepatic metabolism but not renal injury in rat polycystic kidney disease. American Journal of Physiology – Gastrointestinal and Liver Physiology. 2000;279:G1162–G1168. doi: 10.1152/ajpgi.2000.279.6.G1162. [DOI] [PubMed] [Google Scholar]
- 64.Vaz F.M., Wanders R.J. Carnitine biosynthesis in mammals. Biochemical Journal. 2002;361:417–429. doi: 10.1042/0264-6021:3610417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Hove J.L., Zhang W., Kahler S.G., Roe C.R., Chen Y.T., Terada N. Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: diagnosis by acylcarnitine analysis in blood. American Journal of Human Genetics. 1993;52:958–966. [PMC free article] [PubMed] [Google Scholar]
- 66.Orellana-Gavalda J.M., Herrero L., Malandrino M.I., Paneda A., Sol Rodriguez-Pena M., Petry H. Molecular therapy for obesity and diabetes based on a long-term increase in hepatic fatty-acid oxidation. Hepatology. 2011;53:821–832. doi: 10.1002/hep.24140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.