

Abstract
Opioid analgesics are commonly used in chronic pain management despite a potential risk of rewarding. However, it remains unclear whether opioid analgesia would enhance the opioid rewarding effect thereby contributing to opioid rewarding. Utilizing a rat paradigm of conditioned place preference (CPP) combined with ankle monoarthritis as a condition of persistent nociception, we showed that analgesia induced by either morphine or the nonsteroid anti-inflammatory drug ibuprofen increased CPP scores in arthritic rats, suggesting that analgesia itself had a rewarding effect. However, arthritic rats exhibited a significantly higher CPP score in response to morphine than ibuprofen. Thus, the rewarding effect of morphine was enhanced in the presence of persistent nociception, producing a phenomenon of analgesia-enhanced opioid reward. At the cellular level, administration of morphine activated a cascade of leptin expression, glial activation, and dopamine receptor upregulation in the nucleus accumbens (NAc), while administration of ibuprofen decreased glial activation with no effect on leptin expression in the NAc. Furthermore, the morphine rewarding effect was blocked in leptin deficient ob/ob mice or by neutralizing leptin or interleukin-1β in the NAc without diminishing morphine analgesia. The data indicate that systemic opioid can activate a leptin-mediated central mechanism in the NAc that led to the enhanced opioid rewarding effect. These findings provide evidence for an interaction between opioid analgesia and opioid rewarding, which may have implications in clinical opioid dose escalation in chronic pain management.
Keywords: conditioned place preference, leptin, nucleus accumbens, opioid, pain, reward
Introduction
Opioids are a class of pain reliever used to treat moderate to severe pain and have been increasingly prescribed over the last two decades (Compton and Volkow, 2006). Although opioid therapy is effective for the treatment of acute postoperative pain and pain related to terminal illness, recent reports have raised concerns over rewarding to and abuse of opioid analgesics in the setting of chronic nonmalignant pain management (Wesson et al., 1993; Eriksen et al., 2006; Ballantyne and LaForge, 2007). To date, it remains unclear whether opioid analgesia would enhance the opioid rewarding effect thereby contributing to opioid rewarding.
Recent studies have implicated glial activation in the cellular mechanisms of morphine tolerance and reward (Hutchinson et al., 2008). Exposure to morphine induced glial activation in the rat's brainstem, which was increased under a persistent nociceptive condition (Alonso et al., 2007), and exposure to morphine or methamphetamine increased the astrocyte expression in the nucleus accumbens (NAc) in association with the rewarding property of both morphine and methamphetamine in mice (Narita et al., 2006). It has also been shown that leptin affects hypothalamic astrocyte morphology and synaptic protein levels. (García-Cáceres et al., 2011). Leptin is an adipocytokine produced mainly by white adipose tissue and acts via neuronal receptors to regulate energy homeostasis (Ahima et al., 2006), memory processing (Farr et al., 2006), hippocampal synaptic plasticity (Shanley et al., 2001), and cytokine release from glial cells (Pinteaux et al., 2007). In rodents, intracerebroventricular leptin infusion enhanced the rewarding effect of d-amphetamine (Hao et al., 2006), whereas exogenous leptin attenuated acute food deprivation-induced relapse to heroin seeking (Shalev et al., 2001). Leptin also mediated nociceptive behavior induced by peripheral nerve injury (Lim et al., 2009) and induced pronociceptive mediators in rats with tactile allodynia (Maeda et al., 2009). Therefore, it would be of considerable interest to examine a cellular mechanism between leptin and glial expression in the NAc and its relationship with opioid analgesia and opioid rewarding.
We hypothesized that (1) the opioid rewarding effect would be enhanced in the presence of a persistent nociceptive condition and (2) this opioid rewarding effect would be mediated by a leptin-glia crosstalk in the NAc. To test these hypotheses, we used a rat paradigm of morphine conditioned place preference (mCPP) to model the cued association to a drug of abuse (Milekic et al., 2006) and a rat model of complete Freund's adjuvant (CFA)-induced ankle monoarthritis as a condition of persistent nociception (Hernstadt et al., 2009). These two models were combined in the same rats to examine a potential interaction between opioid analgesia and opioid rewarding at the behavioral and cellular level.
Materials and Methods
Animals
Male Sprague-Dawley rats (Charles River Laboratories) weighing 275–350 g (n = 260), and ob/ob (B6.V Lepob/J, n = 40) and wild-type (C57BL/6J, n = 40) mice at 8–10 weeks of age (The Jackson Laboratory) were used. Animals were housed in cages with water and food pellets available ad libitum and under controlled temperature (21 ± 2°C) and relative humidity (50 ± 10%). The animal room was artificially illuminated from 7:00 A.M. to 7:00 P.M. The animal protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and the experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Drugs
Ibuprofen was purchased from Sigma-Aldrich, leptin antagonist (rLA) from American Research Products, leptin from abcam, IL-1β antagonist (IL-1 ra) from R&D Systems, and sulpiride and SCH 39166 from Tocris Bioscience. Leptin antagonist was dissolved in 5% ethanol and 10% DMSO (Sigma-Aldrich; both diluted in normal saline and used as vehicle control). Leptin antagonist used in our experiments is a recombinant rat leptin (a polypeptide), resulting in L39A/D40A/F41A mutation, and was purified by chromatography. This leptin antagonist has been used to block the biological effect of leptin in previous in vivo and in vitro experiments (Gertler, 2006). The vehicle for leptin and IL-1β antagonist (IL-1 ra) was sterilzed PBS. The vehicle for sulpiride and SCH 39166 was DMSA (Sigma-Aldrich).
Surgical procedures
Ankle monoarthritis (CFA).
Monoarthritis was induced by injecting CFA (Sigma-Aldrich) into the rat's (or mouse) right tibio-tarsal joint (Butler et al., 1992) under brief anesthesia with 2% isoflurane. A 28 gauge needle was used and 50 μl (rats) or 20 μl (mice) CFA was injected. Control animals underwent the same procedure but were injected with a vehicle. Arthritic changes were confirmed during the experiments by assessing local redness and edema as well as spontaneous (posture and gait) and evoked (hyperalgesia and allodynia) nociceptive behaviors.
Intracranial cannula implantation.
For drug administration, an intracranial cannula was implanted. Under 2% isoflurane anesthesia, each rat was placed in a stereotaxic frame with both bregma and lambda lines at the horizontal level. A guide cannula (26 gauge, Plastics One) was implanted and secured by dental acrylic and a metal screw. The tip of a guide cannula was located just above each side of NAc (1.7 mm anterior to bregma, 2.2 mm lateral to the midline on either side and 6.7 mm ventral to the skull surface) according to a rat's brain atlas (Paxinos and Watson, 2006).
Injection procedures
For microinjection, a stainless steel needle (33 gauge, Plastics One) was inserted through the cannula into NAc extending 1.5 mm below the guide cannula and a total volume of 1 μl was slowly injected over 5 min using a Hamilton syringe by PE-10 tubing. After the infusions, animals were placed back into their home cages. The injection site was confirmed by the postmortem verification of the needle-tip location (see Fig. 6F). Data from rats (∼1–2 rats per group) with an incorrect needle-tip location was excluded from statistical analysis.
Figure 6.

Ibuprofen-CPP is independent of a leptin-glia crosstalk in NAc. A, Leptin (ob) levels in the NAc were not increased in sham or arthritic rats exposed to ibuprofen conditioning. B, Intra-NAc microinjection of LA (4 μg; once daily × 5 d) failed to reduce the CPP score in arthritic rats exposed to ibuprofen conditioning (CFA/Ibu/LA; n = 6). Intra-NAc LA alone did not change the baseline CPP score in sham rats exposed to ibuprofen conditioning (Ibu/LA; n = 6); *p < 0.05 compared with S/V. C–E, GFAP expression in the NAc (C) was downregulated, whereas Th (D), D1, and D2 (E) expression was upregulated, in CFA/Ibu; β-Actin: loading control; *p < 0.05, **p < 0.01, ***p < 0.001 compared with S/V. Data are shown as mean + SD. F, Location of microinjections into the NAc (AP: 1.7 mm from bregma; left: 2.2 mm; depth: 6.7 mm).
CPP test
The CPP protocol was modified from a previous study (Milekic et al., 2006). The CPP apparatus was a Plexiglas box (Med Associates) with two side chambers (dark vs light) of equal size (20.3 × 15.9 × 21.3 cm) separated by a middle chamber with a sliding door on each side.
Preconditioning (day 0).
Each animal was placed in the middle chamber with doors to each side chamber open and allowed to freely explore both side chambers for 10 min. The time spent in the light or dark chamber was recorded using a computerized program. Rats that spent most of their time (500 s of 10 min) in a single chamber (∼10% number of rats) were excluded from the experiment because of preconditioning bias. Animals were divided into four groups: (1) sham rats with saline conditioning (S/V), (2) arthritic rats with saline conditioning (CFA/V), (3) sham rats with morphine conditioning (S/MS), and (4) arthritic rats with morphine conditioning (CFA/MS).
Conditioning phase (days 1–5).
Each rodent received daily intraperitoneal injection of an assigned drug (morphine, ibuprofen) in the morning and placed in the assigned nonpreferred chamber for 30 min. Six hours later, the same rodent received saline and was again placed for 30 min in the preferred chamber. Control groups received saline twice a day with a 6 h interval.
Postconditioning.
each rodent was again placed in the middle chamber and allowed to freely explore both side chambers for 10 min. The difference in the time spent in the drug-paired chamber before and after conditioning was recorded, calculated using a computerized program, and designed as the CPP (difference) score, which is the time spent in the drug-paired compartment on the postconditioning test day minus the time spent in the same drug-paired compartment at baseline.
Nociceptive behavioral tests
The foot-withdrawal test was used to assess hyperalgesia to radiant heat (Hargreaves et al., 1988). Each animal underwent three trials and the results from these trials were averaged to yield mean withdrawal latencies. A set of von Frey filaments was used to examine mechanical allodynia using an up-and-down (in filament size) approach (Tal and Bennett, 1994). A single filament was applied perpendicularly to the plantar surface of a hindpaw five times and a positive response was defined as a clear withdrawal in at least one of five applications.
Experimental protocol for two cycles of conditioning
To examine whether the presence or absence of persistent nociception would influence CPP score in the same rats, we used an experimental protocol consisting of two cycles of conditioning separated by a period of 3 weeks that allowed rats to recover from ankle monoarthritis.
Cycle 1.
Rats first underwent a preconditioning test followed by the CFA or sham injection into a unilateral ankle joint. Beginning on day 1, each rat was subject to morphine (10 mg/kg), ibuprofen (90 mg/kg), or saline conditioning for 5 d. On day 6, each group of rats underwent a postconditioning test (Cycle 1).
Recovery period.
Upon completion of the first cycle, rats were allowed to recover from CFA-induced monoarthritis or sham condition for a period of 3 weeks. During this period, no drug treatment or intervention was given.
Cycle 2.
At the end of this recovery period, nociceptive behavioral tests and observation of the arthritic ankle were used to confirm the recovery from ankle monoarthritis. We retested baseline CPP score before the conditioning in Cycle 2. We found no differences in all groups as compared with their corresponding baseline CPP scores in Cycle 1 (p > 0.05; comparison between baseline CPP score in Cycles 1 and 2 in each corresponding group). The same morphine, ibuprofen, or saline conditioning used in Cycle 1 was repeated in the same rats (Cycle 2) and a postconditioning test was performed following 5 d of conditioning.
Primary astrocyte culture
Postnatal day 1 Sprague-Dawley rats were used for astrocyte culture. After removal of meninges, the cerebral cortices were digested with Trypsin/EDTA (Invitrogen) and samples were placed in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) culture medium. Cultures were maintained at 37°C in a 5% CO2 incubator for 4–6 weeks in 10% serum DMEM and the media were changed twice per week.
Immunohistochemistry
Under pentobarbital (50 mg/kg, i.p.) anesthesia, animals were perfused with saline through the ascending aorta followed by 4% paraformaldehyde containing 1.5% picric acid and 0.16 m PBS (pH 7.2–7.4, 4°C). Brain sections (30 μm) were blocked with 4% goat serum in 0.3% Triton X-100 for 1 h at room temperature and incubated for 24–48 h at 4°C with a primary antibody: iba-1 (abcam): 1:500, mouse monoclonal; tyrosine hydroxylase (Abcam): 1:1000, rabbit polyclonal; GFAP (BD Biosciences): 1:1000, mouse monoclonal; leptin (Santa Cruz Biotechnology): 1:200, rabbit polyclonal. Sections were then incubated for 1 h at room temperature with FITC- or cyanine 3-conjugated secondary antibody (1:300; Jackson ImmunoResearch). For control, primary antibodies were omitted in the process. Sections were examined with an Olympus fluorescence microscope, and images were captured with a digital camera (Olympus) and analyzed with Adobe Photoshop v7.
Western blot
Animals were killed by decapitation under pentobarbital anesthesia. Brain regions of interest were separately removed and homogenized in an SDS buffer containing a mixture of proteinase inhibitors (Sigma-Aldrich). Samples were separated on an SDS-PAGE gel (4–15% gradient gel; Bio-Rad) and transferred to PVDF membranes (Millipore). Membranes were blocked with 5% nonfat dried milk and incubated overnight (4°C) with a primary antibody: leptin (Santa Cruz Biotechnology): 1:200, rabbit polyclonal; D1 receptor (Millipore): 1:500, rabbit polyclonal; D2 receptor (Calbiochem): 1:800, rabbit polyclonal; GFAP (Abbiotec): 1:300, rabbit polyclonal; iba-1 (Wako): 1:300, rabbit polyclonal; tyrosine hydroxylase (Abcam): 1:5000, rabbit polyclonal. Membranes were then incubated for 1 h at room temperature with an HRP-conjugated secondary antibody (1:7000; Sigma-Aldrich). Blots were visualized in ECL solution (Millipore) for 5 min and exposed to hyperfilms (Kodak) for 1–10 min. Blots were again incubated in a stripping buffer (67.5 mm Tris, pH 6.8, 2% SDS, and 0.7% β-mercaptoethanol) for 30 min at 50°C and reprobed with an anti-β-actin antibody (1:12,000, mouse monoclonal; Abcam) as loading control. Western blots were made in triplicates. Band density was measured and normalized against a corresponding loading control band on the same blot using Quantity One analysis software (Bio-Rad).
ELISA
A standard curve was generated for each assay using the reference set supplied in the leptin ELISA kit (R&D Systems). Serum samples were read using a microplate reader (Bio-Tek Instruments) at wavelengths of 450–620 nm. The content of IL-1β in primary cell culture media was assayed using an immunoassay kit based on the manufacturer's instructions (R&D Systems). The sample concentration was calculated based on a standard curve.
Real-time RT-PCR
Total RNA was isolated from nucleus accumbens samples from animals using TRIzol reagent (Invitrogen). Two micrograms of total RNA was used to synthesize the first strand of cDNA using SuperScript III kit (Invitrogen) according to the manufacturer's protocol. Four microliters of cDNA was put into 20 μl vessel with 1 μl of 20× TaqMan Gene Expression Assay (Applied Biosystems). The genes examined were GFAP (Rn00566603 m1, NM 017009), TH (Rn00562500 m1, NM012740.3), D1 (Rn03062203s1, NM_012546) and D2 (Rn00561126m1, NM012547) for rats and D1 (Mm02620146s1, NM_010076) and D2 (Mm00438545m1, NM010077) for mice. The reaction was performed in duplicate with the following conditions: denaturing at 95°C for 30 s, annealing at 60°C for 2 min, extension at 68°C for 2 min for 35 using ABI Prism 7000 Sequence Detector System maintained by the MGH Neuroscience Center nucleic acid quantitation core facilities. GAPDH RNA gene expression was measured as control.
Statistics
All results are expressed as mean + SD. Two-way ANOVA, repeated across various time points, was used to detect overall differences among treatment groups. Whenever applicable, the data were also examined using two-way ANOVA repeated across treatment groups to examine overall differences among various time points. When a significant main effect was detected, the Waller–Duncan K-ratio t test was performed to determine between-group differences. This analysis was used to analyze the data from thermal hyperalgesia test and CPP test. The nonparametric Mann–Whitney U test was used to determine differences in threshold force in response to von Frey filament stimulation. For Western blot, RT-PCR, and ELISA analyses, differences were compared using one-way ANOVA followed by the Waller–Duncan K-ratio t tests. In all cases, differences were considered to be statistically significant at the level of α = 0.05.
Results
Morphine-CPP is enhanced in rats with monoarthritis
Utilizing a rat paradigm of mCPP, we first examined whether the morphine rewarding effect would be enhanced in rats with CFA-induced ankle monoarthritis. The rats were divided into four groups: (1) Sham rats with saline conditioning (S/V, n = 10), (2) arthritis rats with saline conditioning (CFA/V, n = 10), (3) sham rats with morphine conditioning (S/MS, n = 10), and (4) arthritis rats with morphine conditioning (CFA/MS, n = 10). Rats were exposed to morphine conditioning (10 mg/kg, i.p., once daily) for 5 consecutive days with or without CFA. The CPP score (difference in the time spent in conditioning chamber between days 0 and 6) was moderately increased in S/M, and substantially higher in arthritic rats, CFA/MS (Fig. 1A; ***p < 0.001 compared with S/V). But the CPP score was not changed in arthritic rats without morphing conditioning, CFA/V (Fig. 1A; p = 0.18 compared with S/V). The mCPP score nearly doubled in arthritic rats (CFA/MS) compared with sham rats (S/MS; Fig. 1A; ##p < 0.01 compared with S/MS). Next, we used the same CPP paradigm but replaced morphine with ibuprofen in both sham and arthritic rats. Ibuprofen conditioning (90 mg/kg, i.p.; Wilson et al., 2006) also produced only a moderate increase in CPP score in CFA/Ibu (n = 10) but not in S/Ibu (n = 10; Fig. 1B; ***p < 0.001 compared with S/V). Both morphine and ibuprofen treatment effectively reduced mechanical allodynia (Fig. 1D; ***p < 0.001 compared with S/V) and thermal hyperalgesia (Fig. 1E; ***p < 0.001 compared with S/V) in arthritic rats. These findings indicate that pain relief by itself was rewarding regardless of whether an opioid or nonopioid analgesic was applied. However, the morphine rewarding effect was significantly enhanced in rats with inflammatory pain compared with ibuprofen's rewarding effect.
Figure 1.

Morphine-CPP is enhanced in rats with monoarthritis. A, CPP score was increased in both sham (S/MS, n = 10) and arthritic (CFA/MS, n = 10) rats exposed to morphine conditioning. However, the CPP score was significantly higher in the CFA/MS than S/MS group; ***p < 0.001 compared with S/V; ## p < 0.01 compared with S/MS. B, CPP score was increased in arthritic (CFA/Ibu, n = 10) but not sham (S/Ibu, n = 10) rats exposed to ibuprofen (Ibu) conditioning; ***p < 0.001 compared with S/V C, The CFA/Ibu regimen increased the CPP score in Cycle 1 but not in Cycle 2. In contrast, the CPP score was also increased in Cycle 2 in the CFA/MS rats, similar to the CPP score in S/MS in Cycle 1(n = 8–10). MS/L-CFA: S/MS conditioning in Cycle 1 and CFA/MS conditioning in Cycle 2, n = 8. The CPP score in MS/L-CFA was much higher than CFA/MS; ***p < 0.001 compared with S/V; ## p < 0.01 compared with S/MS; $p < 0.05 compared with CFA/MS. D, E, Morphine or ibuprofen attenuated mechanical allodynia (D) and thermal hyperalgesia (E) when examined on day 6 in Cycle 1 (n = 8). Both allodynia and hyperalgesia were no longer detected after a 3 week recovery period (n = 8); ***p < 0.001, compared with S/V. F, CPP score was increased in arthritic rats, but not sham rats, exposed to 1.25 mg/kg morphine (i.p. once daily × 5 d, n = 8); ***p < 0.001 compared with S/V. G, H, Single administration of morphine (1.25 or 10 mg/kg, i.p.) attenuated both mechanical allodynia (G) and thermal hyperalgesia (H) at 15 and 30 min after the treatment, confirming that morphine produced a dose–response effect; *p < 0.05 compared with V/V, n = 8; S/V: sham rats with saline conditioning, n = 10; S/MS: sham rats with morphine conditioning, n = 10; CFA/V: arthritic rats with saline conditioning, n = 10; CFA/MS: arthritic rats with morphine conditioning, n = 10; S/Ibu: sham rats with ibuprofen conditioning, n = 10; CFA/Ibu: arthritic rats with ibuprofen conditioning, n = 10. RP: 3 week recovery period; C1D0: Cycle 1, Day 0; C1D6: Cycle 1, Day 6; C2D0: Cycle 2, Day 0; C2D6: Cycle2, Day 6; FWL, foot-withdrawal latency. Data are shown as mean + SD.
We then examined whether the intrinsic rewarding effect of morphine could be separated from the reward due to analgesia in arthritic rats. We allowed arthritic rats to recover from monoarthritis (3 weeks) after the first cycle of morphine, ibuprofen, or saline conditioning, as confirmed by recovery of allodynia and hyperalgesia (Fig. 1D,E) as well as signs of arthritis (redness, swelling). Rats recovered from arthritis (former arthritic rats), as well as rats without arthritis (sham rats), were re-exposed to a second cycle of morphine, ibuprofen, or saline conditioning that was identical to that in the first cycle. This second cycle of morphine conditioning only moderately increased CPP scores in these former CFA/MS and their CPP score was significantly lower than that in the first cycle (Fig. 1C; ##p < 0.01 compared with Cycle 1). There were no changes in CPP scores for ibuprofen (S/Ibu, CFA/Ibu) or saline conditioning (S/V, S/MS) in the second cycle. To examine whether previous morphine exposure would have an effect on subsequent pain-induced CPP modulation, we first exposed rats to morphine without CFA in the first cycle and then injected these same rats with CFA before morphine conditioning in the second cycle (Fig. 1C; MS/Late CFA, n = 8). In these rats, the mCPP score in the second cycle was also higher than that in the first cycle (##p < 0.01, compared with Cycle 1). In addition, the mCPP score for MS/L-CFA in Cycle 2 was higher than that of CFA/MS in Cycle 1 ($p < 0.05 compare with CFA/MS in Cycle 1). The data indicate that the intrinsic rewarding effect of morphine was compounded by the reward from pain relief in arthritic rats, producing a phenomenon of analgesia-enhanced opioid reward.
To examine whether both the intrinsic morphine rewarding effect and the reward due to morphine-induced analgesia would be dose-dependent, we used a low dose of morphine (1.25 mg/kg, i.p.; n = 8) to repeat the above experiment which used a high dose of morphine (10 mg/kg, i.p.) in rats with or without ankle monoarthritis. Conditioning with a low dose of morphine increased the CPP score only in CFA/MS but not in S/MS (Fig. 1F; ***p < 0.001 compared with S/V), indicating that the intrinsic morphine rewarding effect was a dose-dependent phenomenon. In arthritic rats, however, both the increase of CPP score (Fig. 1F) and the degree of analgesia (Fig. 1G,H) induced by 1.25 mg/kg morphine were lower than those induced by 10 mg/kg morphine (Fig. 1A), indicating that the reward due to morphine-induced analgesia in arthritic rats was dose-dependent as well.
Leptin level is elevated in NAc following mCPP
To examine the cellular mechanism of analgesia-enhanced opioid reward, we first determined whether the leptin level in serum and brain regions such as the NAc would be changed in association with mCPP. When examined on day 6 right after the behavioral test, the serum leptin level was substantially higher in arthritic rats exposed to morphine conditioning than in other groups (Fig. 2A; **p <0.01, *p < 0.05 compared with S/V). The leptin level was also increased in the bilateral NAc of S/MS rats and further increased in CFA/MS (Fig. 2B,C; **p < 0.01, ***p < 0.001 compared with S/V, #p < 0.05 compared with S/MS). To determine the role of leptin in analgesia-enhanced opioid reward, leptin-deficient ob/ob mice and age-matched wild-type mice first received CFA or sham injection to a hindpaw ankle, followed by exposure to either saline or morphine (5 mg/kg, i.p.) conditioning. In wild-type mice, the CPP score was increased in S/MS (n = 8) and further increased in CFA/MS (n = 8), but not in ob/ob mice, in response to morphine conditioning (n = 8; Fig. 2D; ***p < 0.001 compared with S/V, #p < 0.05 compared with S/MS). However, this absence of CPP score increase in ob/ob mice was restored by intraperitoneal administration of exogenous leptin (2 mg/kg, once daily; Niimi et al., 1999) in response to the same morphine conditioning (n = 8; Fig. 2D; ***p < 0.001 compared with S/V, %%%p < 0.001 compared with CFA/MS). Administration of exogenous leptin also increased the CPP score in wild-type mice. The CPP score in arthritic mice exposed to morphine with leptin (CFA/MS/Lep, n = 8) was more enhanced than that in wild-type mice exposed to CFA/MS without leptin (Fig. 2D; $p < 0.05 compared with CFA/MS).
Figure 2.
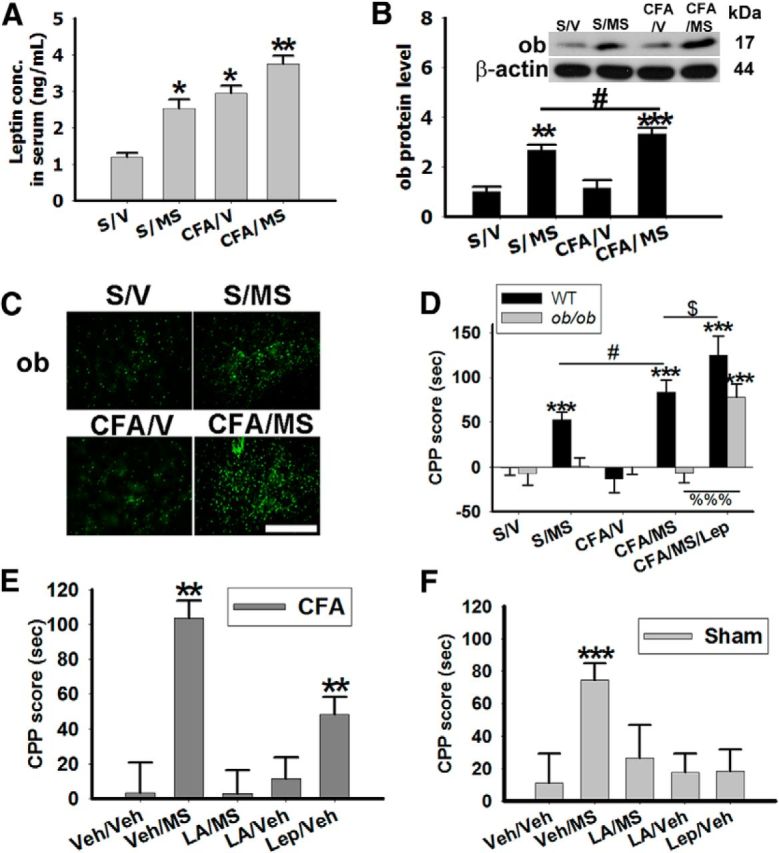
Leptin levels are elevated in the NAc following mCPP. A, The serum leptin concentration was increased in sham rats exposed to morphine conditioning (S/MS, n = 10) and in arthritic rats exposed to saline (CFA/V, n = 10) or morphine (CFA/MS, n = 10) conditioning; *p < 0.05, **p < 0.01 compared with S/V. B, Leptin (ob) levels were increased in the NAc of sham or arthritic rats exposed to morphine conditioning. β-Actin: loading control; **p < 0.01, ***p < 0.001 compared with S/V. C, Leptin immunoreactivity was increased in NAc. Scale bar, 100 μm. D, CPP score was not increased in ob/ob mice exposed to morphine conditioning (n = 8); ***p < 0.001 comparisons between ob/ob and wild-type (WT) mice. Exogenous leptin (2 mg/kg, i.p.; CFA/MS/Lep) increased the mCPP score in ob/ob mice (***p < 0.001 compared with ob/ob mice S/V, %%%p < 0.001 compared with ob/ob CFA/MS, n = 8) and in wild-type (***p < 0.001 compared with wild-type S/V, #p < 0.05 compared with wild-type S/M, $p < 0.01 compared with wild-type CFA/MS; n = 8). CFA/MS/Lep, Arthritic mice with morphine and leptin conditioning. E, F, CPP scores in arthritic rats (E) and sham rats (F). Intra-NAc LA (LA, leptin antagonist) (4 μg, LA/MS, n = 8) blocked the CPP scores in arthritic rats (E) and in sham rats (F). Intra-NAc microinjection of exogenous leptin (4 μg) enhanced the CPP score in CFA rats (Lep/V, n = 8) but failed to induce the CPP score in sham rats (Lep/V, n = 8); **p < 0.01 compared with Veh/Veh, ***p < 0.001 compared with Veh/Veh. Data are shown as mean + SD.
To examine the site of leptin actions, we microinjected a rat recombinant leptin antagonist (rLA, 4 μg, once daily × 5 d; Lim et al., 2009; n = 8) or vehicle (n = 8) into NAc of arthritic rats exposed to morphine conditioning. Intra-NAc administration of this leptin antagonist (LA/MS, n = 8) reduced the mCPP scores in both arthritic (Fig. 2E; p = 0.31, n = 6–8) and sham rats (Fig. 2F; p = 0.37), whereas intra-NAc injection of the leptin antagonist alone did not change the CPP score in arthritic rats exposed to saline conditioning (LA/V; Fig. 2E; n = 8). Conversely, administration of exogenous leptin (4 μg, once daily × 5 d; Sahu, 1998; n = 8) into NAc of CFA rats that were not exposed to morphine conditioning (Lep/V, n = 8) also increased CPP score (Fig. 2E; **p < 0.01 compared with V/V,), mimicking the effect of morphine conditioning. However, in sham rats, inta-NAc injection of exogenous leptin alone (Lep/V) did not change the CPP score (Fig. 2F; p = 0.27, n = 8). Collectively, these results indicate that leptin is likely to be critically involved in analgesia-enhanced opioid reward and the bilateral NAc is an important brain site of leptin action in this process.
Leptin induces glial activation: a leptin-glia crosstalk in NAc
Leptin has been suggested to induce cytokine release from glial cells and this action is related to the mechanism of morphine tolerance and reward (Pinteaux et al., 2007; Hutchinson et al., 2008). We therefore examined both astroglial (GFAP) and microglial (iba-1) expression in the NAc of sham and arthritic rats following saline or morphine conditioning (samples taken on day 6 right after behavioral tests). The GFAP and iba-1 expression in NAc was increased in S/MS and CFA/MS (Fig. 3A; **p < 0.01, ***p < 0.001 compared with S/V, #p < 0.05 compared with S/MS) but not in CFA/V (Fig. 3A). Moreover, we found that leptin was upstream to glial activation because: (1) upregulation of Gfap expression in NAc following S/MS and CFA/MS was detected in wild-type but not ob/ob mice. This effect was restored by administration of exogenous leptin (2 mg/kg, i.p. once daily × 5 d) in ob/ob mice, CFA/MS/Lep (Fig. 3C; #p < 0.05 compared with S/MS). The expression of Gfap without CFA (S/Lep) was enhanced by exogenous leptin in both wild-type and ob/ob mice (Fig. 3C; *p < 0.05 compared with S/V). (2) Upregulation of both GFAP and iba-1 in NAc following mCPP was blocked by bilateral administration of rLA (4 μg, once daily × 5 d, LA/MS) but not vehicle (V/MS) into NAc in arthritic rats (Fig. 3D; **p < 0.01 compared with V/V). (3) Exposure of a primary glial cell culture to exogenous leptin (1 μg/ml; Pinteaux et al., 2007) time-dependently increased interleukin-1β (IL-1β) release into the culture medium (Fig. 3E; **p < 0.01, *p < 0.05 compared with V, n = 12). Behaviorally, bilateral microinjection of the IL-1β receptor antagonist IL-1ra (100 μg, once daily × 5 d, n = 8; Konsman et al., 2008) into NAc in arthritic rats also prevented the mCPP score increase (Fig. 3F; **p < 0.01 compared with V/V, n = 6–8) without changing the intra-NAc leptin expression in these same rats (Fig. 3G; **p < 0.01 compared with V/V, n = 8). These results indicate that leptin induces both glial activation and release of IL-1β in the NAc, which contributed to the behavioral manifestation of analgesia-enhanced opioid reward.
Figure 3.

Leptin induces glial activation in NAc. A, Expression of GFAP and iba-1 in the NAc was upregulated in sham (S/MS) and arthritic (CFA/MS) rats exposed to morphine conditioning. β-Actin: loading control; ***p < 0.001 compared with S/V; #p < 0.05 compared with S/MS. B, GFAP and iba-1 immunoreactivity was increased in the NAc of these same rats. Scale bar, 100 μm. C, Gfap expression in the NAc was lower in ob/ob mice as compared with wild-type (WT) mice. Expression of Gfap in the NAc was not increased in ob/ob CFA/MS (n = 8), but exogenous leptin (2 mg/kg, i.p.) enhanced Gfap expression in ob/ob (CFA/MS/Lep, n = 8). Exogenous leptin also induced Gfap expression in sham ob/ob or sham wild-type mice (S/Lep, n = 8); *p < 0.05, ***p < 0.001 compared with wild-type S/V, $p < 0.05 compared with wild-type CFA/MS, #p < 0.05 compared with S/MS. D, Expression of GFAP and iba-1 was diminished by intra-NAc microinjection of a LA (4 μg) in arthritic rats exposed to morphine conditioning (LA/MS; n = 8); **p < 0.01 compared Veh/Veh. E, Leptin (1 μg/ml) stimulated the release of IL-1β in cultured astrocytes (n = 12); *p < 0.05, **p < 0.01 compared with vehicle 6 h. F, G, Intra-NAc microinjection of IL-1 ra (100 μg) prevented the increased CPP score in IL-1 ra/MS (F) but did not alter the leptin level in NAc (G; n = 8); **p < 0.01 compared with Veh/Veh. Data are shown as mean + SD.
Leptin and IL-1β regulate tyrosine hydroxylase expression in NAc
Leptin has been shown to modulate the mesolimbic dopaminergic system involved in energy regulation and food intake (Hommel et al., 2006). To determine whether a leptin-glia crosstalk in the NAc would stimulate dopaminergic neurons following morphine conditioning, we specifically examined the expression of tyrosine hydroxylase (TH), a marker of dopaminergic neurons (Perry et al., 2010). Expression of TH was significantly upregulated in S/M and CFA/MS than S/V (Fig. 4A,B; **p < 0.01). This increase in TH expression was prevented by the bilateral microinjection of either rLA (4 μg) or IL-1ra (100 μg) into NAc given once daily for 5 d (Fig. 4C). These findings indicate that the leptin-glia crosstalk in NAc stimulated dopaminergic neurons, shown as the upregulated TH expression, in rats with analgesia-enhanced opioid reward.
Figure 4.

Leptin and IL-1β regulate tyrosine hydroxylase expression in NAc. A, Expression of TH in the NAc was upregulated in S/MS and CFA/MS rats exposed to morphine conditioning; β-Actin: loading control; **p < 0.01 compared with S/V. B, TH immunoreactivity was expressed in the NAc. Scale bar, 50 μm. C, Intra-NAc microinjection of a leptin antagonist (LA) (4 μg) or an IL-1β antagonist (IL-1 ra, 100 μg), once daily for 5 d, prevented Th upregulation in LA/MS and IL-1 ra/MS; **p < 0.01 compared with Veh/Veh. Data are shown as mean + SD.
Leptin and IL-1β regulate the D1 and D2 receptor expression in NAc
The D1 and D2 dopaminergic receptor expression was upregulated in S/MS (Fig. 5A; **p < 0.01, *p < 0.05 compared with S/V) and further upregulated in CFA/MS (Fig. 5A; #p < 0.05 compared with S/MS). Once daily intra-NAc microinjection of IL-1ra (100 μg) for 5 d blocked upregulated D1 and D2 receptor expression (Fig. 5B). Moreover, the expression of both D1 and D2 receptors in NAc was not regulated by morphine conditioning in ob/ob mice, but was restored by exogenous leptin (2 mg/kg, i.p.; Fig. 5C,D; ***p < 0.001 compared with S/V). In wild-type, exogenous leptin enhanced the expression of D1 and D2 receptors compared with CFA/MS (Fig. 5C,D; $p < 0.05 compared with CFA/MS). At the behavioral level, intraperitoneal administration of the D2 receptor antagonist sulpiride (Zarrindast et al., 2007; 12 mg/kg, once daily × 5 d, n = 8) reduced the mCPP score in Sul/MS (n = 8) but did not alter baseline CPP score in S/Sul (n = 8; Fig. 5E; **p < 0.01). Administration of exogenous leptin failed to induce the mCPP score with Sul/MS conditioning (Fig. 5E). Sulpiride was used in this experiment because the D2 dopaminergic receptor has been shown to regulate behaviors related to drug abuse (Morutto and Phillips, 1998). To test the involvement of D1 receptors, SCH 39166 (a D1 receptor antagonist at 0.1 mg/kg, once daily × 5 d; Fenu et al., 2009) was used using the same design. The mCPP score in the SCH/MS group (n = 8) was not increased and administration of exogenous leptin also failed to enhance the mCPP score in rats with MS conditioning and SCH administration (Fig. 5F; **p < 0.01). These results indicate that leptin has both direct and indirect, via glial activation, modulatory effects on D1 and D2 receptor expression in the NAc. This leptin-mediated dopaminergic mechanism is critical to the behavioral manifestation of analgesia-enhanced opioid reward.
Figure 5.

Leptin and IL-1β regulate the D1 and D2 receptor expression in NAc. A, D1 and D2 receptor expression in the NAc was upregulated in S/MS and CFA/MS rats exposed to morphine conditioning; *p < 0.05 compared with S/V, **p < 0.01 compared with S/V, #P<0.05 compared with S/MS; β-Actin: loading control. B, Intra-NAc microinjection of the IL-1β antagonist IL-1 ra (100 μg), once daily for 5 d, prevented D1 and D2 receptor upregulation in the NAc of arthritic rats exposed to morphine conditioning (IL-1 ra/MS); ***p < 0.001 compared with Veh/Veh. D1 (C) and D2 (D) receptor mRNA expression in the NAc was not increased in ob/ob mice exposed to morphine conditioning (CFA/MS), but exogenous leptin (2 mg/kg, i.p., once daily × 5 d) restored D1 and D2 receptor upregulation (CFA/MS/Lep) in the NAc; *p < 0.05, **p < 0.01, ***p < 0.001 compared with S/V, #P<0.05 compared with S/MS, ##P<0.01 compared with S/MS, $P<0.05 compared with CFA/MS. E, F, The CPP scores decreased with an administration of the D2 antagonist sulpiride (Sul; 12 mg/kg, once daily × 5 d; E; n = 8) and D1 antagonist SCH39166 (SCH, 0.1 mg/kg, once daily × 5 d; F; n = 8) in rats exposed to morphine conditioning; **p < 0.01 compared with S/V. Data are shown as mean + SD.
Ibuprofen-CPP is independent of a leptin-glia crosstalk in NAc
To assess the role of leptin for preference to ibuprofen, we measured the expression of leptin. Compared with arthritic rats exposed to morphine conditioning (Fig. 2A), the leptin level was not increased in the NAc of either sham (S/Ibu) or arthritic (CFA/Ibu) rats exposed to ibuprofen conditioning (Fig. 6A; p = 0.67). In CFA/Ibu/LA rats, intra-NAc administration of rLA (4 μg, once daily × 5 d) failed to reduce the CPP score which was observed in arthritic rats exposed to ibuprofen conditioning in Fig. 1B (Fig. 6B). Ibuprofen conditioning in arthritic rats (CFA/Ibu) also did not increase the GFAP and iba-1 expression in NAc (Fig. 6C) although it upregulated the expression of Th, D1, and D2 receptors in the NAc (Fig. 6D,E; *p < 0.05, ***p < 0.001 compared with S/V). These results indicate that ibuprofen does not activate a leptin-mediated central reward mechanism, which is distinctively different from the mechanism of analgesia-enhanced opioid reward.
Discussion
We have demonstrated that: (1) the morphine rewarding effect was augmented in the presence of persistent nociception, producing a phenomenon of analgesia-enhanced opioid reward; (2) although ibuprofen-CPP was demonstrated in arthritic rats, this rewarding effect was much less robust than analgesia-enhanced opioid reward and disappeared after recovery from monoarthritis in the same rats; (3) morphine but not ibuprofen conditioning in arthritic rats activated a sequence of cellular responses in the NAc including an elevated leptin level, glial activation, release of IL-1β, and upregulation of D1/D2 dopaminergic receptors; and (4) the behavioral manifestation of analgesia-enhanced opioid reward was abolished in ob/ob mice and prevented by neutralizing leptin or IL-1β in the NAc.
Previous studies have shown that persistent nociception in rats may decrease the antinociceptive effect of morphine (Mao et al., 1995; Narita et al., 2005) and that the expression of morphine reward could be masked by the development of morphine tolerance (Suzuki et al., 1996). On the other hand, systemic morphine administration appears to increase CPP score in rats with nociception (Bardin et al., 2000) and arthritic rats self-administered more fentanyl (a potent opioid analgesic) than nonarthritic rats (Colpaert et al., 2001). More recently, the rewarding effect of a nonopioid analgesic was demonstrated in rats with nociception using a CPP paradigm (King et al., 2009). These previous findings suggest that (1) attenuation of nociception by an analgesic could be a rewarding experience in rodents and (2) opioid reward could be influenced by experimental procedures, such as opioid dose and experimental paradigm.
In the present study, we used (1) a counter-balanced design that included both sham and arthritic rats exposed to saline, morphine, or ibuprofen conditioning; (2) an experimental paradigm consisting of two identical cycles of saline, morphine, or ibuprofen conditioning separated by a period of recovery from monoarthritis in the same rats; (3) an analgesic dose of morphine (10 mg/kg) coupled with once daily conditioning in a CPP apparatus; and (4) concurrent validation of changes, or lack thereof, in nociceptive behavior in response to saline, morphine, or ibuprofen conditioning, in addition to performing CPP tests in the same rats. We showed that morphine conditioning produced a phenomenon of analgesia-enhanced opioid reward consisting of two distinguishable reward elements: (1) reward due to the intrinsic effect of morphine itself and (2) reward due to morphine-induced analgesia. We also showed that both reward elements were morphine dose-dependent. In contrast, changes in the CPP score in the ibuprofen group were likely due to the reward from ibuprofen-induced analgesia (antinociception). Of interest is that the degree of reward due to analgesia was nearly identical between arthritic rats exposed to morphine or ibuprofen conditioning, which is consistent with the finding that pain mediates a brain reward circuit (Gear et al., 1999; Becerra et al., 2001). Therefore, our findings suggest that pain relief by an analgesic, regardless of an opioid or a nonopioid, is likely to represent a “pleasure seeking” experience, which can be compounded by the intrinsic rewarding effect of an opioid but not a nonopioid analgesic. An alternative possibility is that rats exposed to vehicle (neither morphine nor ibuprofen) could express avoidance to the vehicle-paired chamber in the postconditioning test and this possibility was not explicitly examined in this study. Nonetheless, this experimental paradigm provides a unique approach to studying the interaction between opioid analgesia and opioid rewarding, an issue with significant clinical relevance.
As a result of the NAc being a critical site of a brain circuitry implicated in nicotine and opioid reward (Schmidt et al., 2001), we investigated a novel cellular mechanism of opioid reward through a leptin-glia crosstalk in the NAc: (1) an elevated leptin level, (2) leptin-induced glial activation, and (3) modulation of dopaminergic neurons. Leptin is a polypeptide hormone encoded by the obese (ob) gene (Zhang et al., 1994; Morash et al., 1999) and is known to modulate feeding behavior and nicotine craving primarily through the ob-Rb receptor expressed in broad brain regions (Cohen et al., 2001; Banks, 2004; von der Goltz et al., 2010). In this study, expression of leptin in the NAc was upregulated in arthritic rats exposed to morphine conditioning. Because the plasma leptin level was decreased following morphine administration in rats without nociception (Ferenczi et al., 2010) and in human subjects undergoing heroin withdrawal (Shi et al., 2009), this increased leptin level in the NAc of rats with analgesia-enhanced opioid reward may result from both CFA-induced inflammation and the intrinsic effect of morphine.
Recent studies have shown that minocycline (a microglial inhibitor) attenuated mCPP in rats without nociception (Hutchinson et al., 2008) and propentofylline (a glia modulator) suppressed methamphetamine- or morphine-induced CPP (Narita et al., 2006). Our data indicate that glial activation is downstream of the leptin increase in the NAc because (1) glial expression was increased in the NAc of rats with analgesia-enhanced opioid reward, (2) GFAP and iba-1 expression in the NAc was lower in ob/ob than wild-type mice, (3) ob/ob mice failed to show GFAP upregulation following morphine conditioning, which was reversed by exogenous leptin, and (4) leptin induced IL-1β release from cultured astrocytes. At the behavioral level, analgesia-enhanced opioid reward was (1) abolished in ob/ob mice but restored by exogenous leptin and (2) prevented by microinjection into NAc of the IL-1β antagonist IL-1 ra. In contrast, arthritic rats exposed to ibuprofen conditioning (a nonsteroidal anti-inflammatory drug) showed downregulation of GFAP expression in the NAc without an increase in the serum leptin level. It remains to be investigated as to why GFAP expression was reduced under our experimental condition. In a mouse model of Alzheimer's disease, ibuprofen suppressed plaque pathology, inflammation, and GFAP expression (Lim et al., 2000), and chronic administration of ibuprofen protected against behavioral manifestation in Alzheimer's disease (Richardson et al., 2002). Future studies may help explain how ibuprofen could play a role in “rewarding” but not “addiction.”
Dopaminergic receptors are important components of the cellular mechanisms of both opioid rewarding (Manzanedo et al., 2005) and nociception (Wood, 2006). Several brain regions including the NAc play an important role in cue-elicited reward-seeking behaviors including mCPP through regulation of the dopaminergic system (Fenu et al., 2006). Moreover, the D1 and D2 receptor mRNA is expressed in a primary astrocyte culture (Miyazaki et al., 2004) and leptin regulates the meso-accumbens dopamine pathway and signaling (Perry et al., 2010). In this study, we demonstrated that the leptin-glia crosstalk stimulated the intra-NAc dopaminergic system, which is an important mechanism of analgesia-enhanced opioid reward. First, the basal D1 and D2 receptor mRNA expression was lower in ob/ob mice than wild-type mice. Second, the D1 and D2 receptor upregulation was absent in ob/ob mice exposed to morphine conditioning but was restored by exogenous leptin. Third, arthritic rats with mCPP also showed TH upregulation in the NAc, which was blocked by either rLA or IL-1 ra. Fourth, exposure of primary cell cultures with leptin or IL-1β increased TH expression. Fifth, systemic administration of the D2 receptor antagonist sulpiride diminished analgesia-enhanced opioid reward. Given that both leptin and glial activation are also implicated in neuropathic pain (Lim et al., 2009), this leptin-mediated mechanism of analgesia-enhanced opioid reward could be relevant to other pain conditions, in addition to arthritic pain, requiring opioid analgesics.
In summary, analgesia-enhanced opioid award and ibuprofen-CPP are likely to be mediated through two distinct cellular mechanisms, although both mechanisms may involve the NAc dopaminergic system. For analgesia-enhanced opioid award, the dopaminergic system was activated by a leptin-glia crosstalk in NAc. For ibuprofen-CPP, activation of dopaminergic system was independent of a leptin-glia crosstalk in the NAc. Therefore, the present data may have important implications in clinical opioid therapy for chronic pain management. If analgesia-enhanced opioid reward were also demonstrated in chronic pain patients requiring opioid therapy, opioid requirement would be expected to increase causing prolonged opioid use, difficulty in weaning off opioid, and possible drug-seeking behaviors. Because modulating a leptin-mediated cellular mechanism can effectively prevent analgesia-enhanced opioid reward, several key elements of this mechanism as demonstrated in this study may serve as potential targets for developing therapeutic interventions to diminish the opioid rewarding effect and minimize the potential of prescription opioid rewarding in chronic pain management.
Footnotes
This work was supported by NIH Grants P20DA26002 and R01 DA036564.
The authors declare no competing financial interests.
References
- Ahima RS, Qi Y, Singhal NS, Jackson MB, Scherer PE. Brain adipocytokine action and metabolic regulation. Diabetes. 2006;55:S145–154. doi: 10.2337/db06-S018. [DOI] [PubMed] [Google Scholar]
- Alonso E, Garrido E, Díez-Fernández C, Pérez-García C, Herradón G, Ezquerra L, Deuel TF, Alguacil LF. Yohimbine prevents morphine-induced changes of glial fibrillary acidic protein in brainstem and alpha2-adrenoceptor gene expression in hippocampus. Neurosci Lett. 2007;412:163–167. doi: 10.1016/j.neulet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Ballantyne JC, LaForge KS. Opioid dependence and addiction during opioid treatment of chronic pain. Pain. 2007;129:235–255. doi: 10.1016/j.pain.2007.03.028. [DOI] [PubMed] [Google Scholar]
- Banks WA. The many lives of leptin. Peptides. 2004;25:331–338. doi: 10.1016/j.peptides.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Bardin L, Kim JA, Siegel S. The role of formalin-induced pain in morphine tolerance, withdrawal, and reward. Exp Clin Psychopharmacol. 2000;8:61–67. doi: 10.1037/1064-1297.8.1.61. [DOI] [PubMed] [Google Scholar]
- Becerra L, Breiter HC, Wise R, Gonzalez RG, Borsook D. Reward circuitry activation by noxious thermal stimuli. Neuron. 2001;32:927–946. doi: 10.1016/S0896-6273(01)00533-5. [DOI] [PubMed] [Google Scholar]
- Butler SH, Godefroy F, Besson JM, Weil-Fugazza J. A limited arthritic model for chronic pain studies in the rat. Pain. 1992;48:73–81. doi: 10.1016/0304-3959(92)90133-V. [DOI] [PubMed] [Google Scholar]
- Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI200113914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpaert FC, Tarayre JP, Alliaga M, Bruins Slot LA, Attal N, Koek W. Opiate self-administration as a measure of chronic nociceptive pain in arthritic rats. Pain. 2001;91:33–45. doi: 10.1016/S0304-3959(00)00413-9. [DOI] [PubMed] [Google Scholar]
- Compton WM, Volkow ND. Abuse of prescription drugs and the risk of addiction. Drug Alcohol Depend. 2006;83:S4–S7. doi: 10.1016/j.drugalcdep.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Eriksen J, Sjøgren P, Bruera E, Ekholm O, Rasmussen NK. Critical issues on opioids in chronic non-cancer pain: an epidemiological study. Pain. 2006;125:172–179. doi: 10.1016/j.pain.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Farr SA, Banks WA, Morley JE. Effects of leptin on memory processing. Peptides. 2006;27:1420–1425. doi: 10.1016/j.peptides.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Fenu S, Spina L, Rivas E, Longoni R, Di Chiara G. Morphine-conditioned single-trial place preference: role of nucleus accumbens shell dopamine receptors in acquisition, but not expression. Psychopharmacology (Berl) 2006;187:143–153. doi: 10.1007/s00213-006-0415-2. [DOI] [PubMed] [Google Scholar]
- Fenu S, Rivas E, Di Chiara G. Differential involvement of dopamine D1 receptors in morphine- and lithium-conditioned saccharin avoidance. Physiol Behav. 2009;96:73–77. doi: 10.1016/j.physbeh.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Ferenczi S, Núñez C, Pintér-Kübler B, Földes A, Martín F, Márkus VL, Milanés MV, Kovács KJ. Changes in metabolic-related variables during chronic morphine treatment. Neurochem Int. 2010;57:323–330. doi: 10.1016/j.neuint.2010.06.011. [DOI] [PubMed] [Google Scholar]
- García-Cáceres C, Fuente-Martín E, Burgos-Ramos E, Granado M, Frago LM, Barrios V, Horvath T, Argente J, Chowen JA. Differential acute and chronic effects of leptin on hypothalamic astrocyte morphology and synaptic protein levels. Endocrinology. 2011;152:1809–1818. doi: 10.1210/en.2010-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gear RW, Aley KO, Levine JD. Pain-induced analgesia mediated by mesolimbic reward circuits. J Neurosci. 1999;19:7175–7181. doi: 10.1523/JNEUROSCI.19-16-07175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler A. Development of leptin antagonists and their potential use in experimental biology and medicine. Trends Endocrinol Metab. 2006;17:372–378. doi: 10.1016/j.tem.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Hao J, Cabeza de Vaca S, Pan Y, Carr KD. Effects of central leptin infusion on the reward-potentiating effect of d-amphetamine. Brain Res. 2006;1087:123–133. doi: 10.1016/j.brainres.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hernstadt H, Wang S, Lim G, Mao J. Spinal translocator protein (TSPO) modulates pain behavior in rats with CFA-induced monoarthritis. Brain Res. 2009;1286:42–52. doi: 10.1016/j.brainres.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Chao LW, Kearney JJ, Zhang Y, Berkelhammer DL, Loram LC, Rozeske RR, Bland ST, Maier SF, Gleeson TT, Watkins LR. Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun. 2008;22:1248–1256. doi: 10.1016/j.bbi.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konsman JP, Veeneman J, Combe C, Poole S, Luheshi GN, Dantzer R. Central nervous action of interleukin-1 mediates activation of limbic structures and behavioural depression in response to peripheral administration of bacterial lipopolysaccharide. Eur J Neurosci. 2008;28:2499–2510. doi: 10.1111/j.1460-9568.2008.06549.x. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim G, Wang S, Zhang Y, Tian Y, Mao J. Spinal leptin contributes to the pathogenesis of neuropathic pain in rodents. J Clin Invest. 2009;119:295–304. doi: 10.1172/JCI36785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Kiguchi N, Kobayashi Y, Ikuta T, Ozaki M, Kishioka S. Leptin derived from adipocytes in injured peripheral nerves facilitates development of neuropathic pain via macrophage stimulation. Proc Natl Acad Sci U S A. 2009;106:13076–13081. doi: 10.1073/pnas.0903524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanedo C, Aguilar MA, Rodríguez-Arias M, Miñarro J. Sensitization to the rewarding effects of morphine depends on dopamine. Neuroreport. 2005;16:201–205. doi: 10.1097/00001756-200502080-00028. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995;61:353–364. doi: 10.1016/0304-3959(95)00022-K. [DOI] [PubMed] [Google Scholar]
- Milekic MH, Brown SD, Castellini C, Alberini CM. Persistent disruption of an established morphine conditioned place preference. J Neurosci. 2006;26:3010–3020. doi: 10.1523/JNEUROSCI.4818-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M, Diaz-Corrales FJ, Miyoshi K, Ogawa N. Direct evidence for expression of dopamine receptors in astrocytes from basal ganglia. Brain Res. 2004;1029:120–123. doi: 10.1016/j.brainres.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Morash B, Li A, Murphy PR, Wilkinson M, Ur E. Leptin gene expression in the brain and pituitary gland. Endocrinology. 1999;140:5995–5998. doi: 10.1210/endo.140.12.7288. [DOI] [PubMed] [Google Scholar]
- Morutto SL, Phillips GD. Interactions between sulpiride infusions within the perifornical region of the lateral hypothalamus and the nucleus accumbens on measures of locomotor activity and conditioned place preference. Behav Pharmacol. 1998;9:345–355. [PubMed] [Google Scholar]
- Narita M, Kishimoto Y, Ise Y, Yajima Y, Misawa K, Suzuki T. Direct evidence for the involvement of the mesolimbic kappa-opioid system in the morphine-induced rewarding effect under an inflammatory pain-like state. Neuropsychopharmacology. 2005;30:111–118. doi: 10.1038/sj.npp.1300527. [DOI] [PubMed] [Google Scholar]
- Narita M, Miyatake M, Narita M, Shibasaki M, Shindo K, Nakamura A, Kuzumaki N, Nagumo Y, Suzuki T. Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology. 2006;31:2476–2488. doi: 10.1038/sj.npp.1301007. [DOI] [PubMed] [Google Scholar]
- Niimi M, Sato M, Yokote R, Tada S, Takahara J. Effects of central and peripheral injection of leptin on food intake and on brain Fos expression in the Otsuka Long–Evans Tokushima Fatty rat with hyperleptinaemia. J Neuroendocrinol. 1999;11:605–611. doi: 10.1046/j.1365-2826.1999.00368.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. New York: Academic; 2006. [DOI] [PubMed] [Google Scholar]
- Perry ML, Leinninger GM, Chen R, Luderman KD, Yang H, Gnegy ME, Myers MG, Jr, Kennedy RT. Leptin promotes dopamine transporter and tyrosine hydroxylase activity in the nucleus accumbens of Sprague-Dawley rats. J Neurochem. 2010;114:666–674. doi: 10.1111/j.1471-4159.2010.06757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinteaux E, Inoue W, Schmidt L, Molina-Holgado F, Rothwell NJ, Luheshi GN. Leptin induces interleukin-1beta release from rat microglial cells through a caspase 1 independent mechanism. J Neurochem. 2007;102:826–833. doi: 10.1111/j.1471-4159.2007.04559.x. [DOI] [PubMed] [Google Scholar]
- Richardson RL, Kim EM, Shephard RA, Gardiner T, Cleary J, O'Hare E. Behavioural and histopathological analyses of ibuprofen treatment on the effect of aggregated Abeta(1–42) injections in the rat. Brain Res. 2002;954:1–10. doi: 10.1016/S0006-8993(02)03006-8. [DOI] [PubMed] [Google Scholar]
- Sahu A. Leptin decreases food intake induced by melanin-concentrating hormone (MCH), galanin (GAL) and neuropeptide Y (NPY) in the rat. Endocrinology. 1998;139:4739–4742. doi: 10.1210/endo.139.11.6432. [DOI] [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Gear RW, Levine JD. Nicotine withdrawal hyperalgesia and opioid-mediated analgesia depend on nicotine receptors in nucleus accumbens. Neuroscience. 2001;106:129–136. doi: 10.1016/S0306-4522(01)00264-0. [DOI] [PubMed] [Google Scholar]
- Shalev U, Yap J, Shaham Y. Leptin attenuates acute food deprivation-induced relapse to heroin seeking. J Neurosci. 2001;21:RC129. doi: 10.1523/JNEUROSCI.21-04-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci. 2001;21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Li SX, Zhang XL, Wang X, Le Foll B, Zhang XY, Kosten TR, Lu L. Time-dependent neuroendocrine alterations and drug craving during the first month of abstinence in heroin addicts. Am J Drug Alcohol Abuse. 2009;35:267–272. doi: 10.1080/00952990902933878. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kishimoto Y, Misawa M. Formalin- and carrageenan-induced inflammation attenuates place preferences produced by morphine, methamphetamine and cocaine. Life Sci. 1996;59:1667–1674. doi: 10.1016/0024-3205(96)00498-5. [DOI] [PubMed] [Google Scholar]
- Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:375–382. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- von der Goltz C, Koopmann A, Dinter C, Richter A, Rockenbach C, Grosshans M, Nakovics H, Wiedemann K, Mann K, Winterer G, Kiefer F. Orexin and leptin are associated with nicotine craving: a link between smoking, appetite and reward. Psychoneuroendocrinology. 2010;35:570–577. doi: 10.1016/j.psyneuen.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Wesson DR, Ling W, Smith DE. Prescription of opioids for treatment of pain in patients with addictive disease. J Pain Symptom Manage. 1993;8:289–296. doi: 10.1016/0885-3924(93)90157-Q. [DOI] [PubMed] [Google Scholar]
- Wilson AW, Medhurst SJ, Dixon CI, Bontoft NC, Winyard LA, Brackenborough KT, De Alba J, Clarke CJ, Gunthorpe MJ, Hicks GA, Bountra C, McQueen DS, Chessell IP. An animal model of chronic inflammatory pain: pharmacological and temporal differentiation from acute models. Eur J Pain. 2006;10:537–549. doi: 10.1016/j.ejpain.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Wood PB. Mesolimbic dopaminergic mechanisms and pain control. Pain. 2006;120:230–234. doi: 10.1016/j.pain.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Ebrahimi-Ghiri M, Rostami P, Rezayof A. Repeated pre-exposure to morphine into the ventral pallidum enhances morphine-induced place preference: involvement of dopaminergic and opioidergic mechanisms. Behav Brain Res. 2007;181:35–41. doi: 10.1016/j.bbr.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]