

Abstract
Neuronal nAChRs in the medial habenula (MHb) to the interpeduncular nucleus (IPN) pathway are key mediators of nicotine's aversive properties. In this paper, we report new details regarding nAChR anatomical localization and function in MHb and IPN. A new group of knock-in mice were created that each expresses a single nAChR subunit fused to GFP, allowing high-resolution mapping. We find that α3 and β4 nAChR subunit levels are strong throughout the ventral MHb (MHbV). In contrast, α6, β2, β3, and α4 subunits are selectively found in some, but not all, areas of MHbV. All subunits were found in both ChAT-positive and ChAT-negative cells in MHbV. Next, we examined functional properties of neurons in the lateral and central part of MHbV (MHbVL and MHbVC) using brain slice patch-clamp recordings. MHbVL neurons were more excitable than MHbVC neurons, and they also responded more strongly to puffs of nicotine. In addition, we studied firing responses of MHbVL and MHbVC neurons in response to bath-applied nicotine. Cells in MHbVL, but not those in MHbVC, increased their firing substantially in response to 1 μm nicotine. Additionally, MHbVL neurons from mice that underwent withdrawal from chronic nicotine were less responsive to nicotine application compared with mice withdrawn from chronic saline. Last, we characterized rostral and dorsomedial IPN neurons that receive input from MHbVL axons. Together, our data provide new details regarding neurophysiology and nAChR localization and function in cells within the MHbV.
Keywords: habenula, interpeduncular, nicotine, nicotinic, tobacco, withdrawal
Introduction
Recently, human genetic studies have uncovered a number of alleles that confer susceptibility to developing dependence to nicotine found in tobacco products (Bierut et al., 2007). The most striking risk alleles are located within a gene cluster on chromosome 15 containing the α3, β4, and α5 nAChR genes (Saccone et al., 2007, 2009; Bierut et al., 2008). These nAChR subunits, particularly α3 and β4, exhibit extremely high expression in both medial habenula (MHb) and interpeduncular nucleus (IPN) (Dineley-Miller and Patrick, 1992; Marks et al., 1992). Several rodent studies indicate a key role for α5- and/or β4-containing nAChRs in modulating nicotine intake. For example, α5 KO mice consume significantly more nicotine than their α5 WT counterparts, and reexpression of α5 subunits in MHb of α5 KO mice restores aversion to high doses of nicotine (Fowler et al., 2011). In a complementary study, Frahm et al. (2011) found that overexpression of β4-containing nAChRs in MHb enhances aversion to, and consequently reduces consumption of, nicotine. These studies, coupled with recent findings indicating the existence of distinct neuronal populations in ventral MHb (MHbV) (Aizawa et al., 2012), point to the need for a better understanding of nAChR function within specific cell populations in MHbV.
Several nAChR subunits exhibit their highest expression level in the MHb (Perry et al., 2002). In addition, most nAChR subunits found in heteromeric receptors are expressed in MHb (Dineley-Miller and Patrick, 1992; Marks et al., 1992), which theoretically increases the number of distinct nAChR subtypes found in MHb compared with other brain areas. Several studies have attempted to identify important nAChR subtypes in MHb neurons. Robin Lester and colleagues identified α3β4* (* = subunits in addition to those listed may be found in the indicated pentameric receptor) nAChRs as the predominant subtype in MHbV, with a minor contribution from β2* nAChRs in some cells (Quick et al., 1999). Studies in mice expressing gain-of-function α4 nAChR subunits demonstrated expression and function of α4* nAChRs in lateral MHbV neurons (Nashmi et al., 2007; Fonck et al., 2009).
Because of the small size of the MHb, prior studies using radioligand binding or mRNA in situ hybridization did not allow for high-resolution mapping of nAChR subunit localization in specific cell types within this brain area. Although basic electrophysiological features of MHb neurons are starting to be uncovered (Kim and Chang, 2005), we fail to understand how activation of MHb nAChRs changes excitability of the neurons in which they reside and the circuits into which they are integrated. To address these shortcomings in our understanding of MHb nAChR function, we combined immunohistochemistry with brain slice patch-clamp electrophysiology using mouse models. We generated knock-in mouse strains expressing α3, α4, β2, β3, and β4 nAChR subunits fused to GFP. Along with previously generated α6-GFP mice (Mackey et al., 2012), we localized six nAChR subunits in specific regions of MHb and IPN. Further, in recordings from MHb and IPN neurons in brain slices from C57BL/6 WT mice, we identified neuronal cell types that differ in their sensitivity to nicotine.
Materials and Methods
Materials.
CNQX and picrotoxin were purchased from Tocris Bioscience, and all other drugs and chemicals not indicated above were purchased from Sigma-Aldrich.
Mice.
C57BL/6 mice used for electrophysiology studies were purchased from The Jackson Laboratory. Bacterial artificial chromosome (BAC) transgenic mice expressing GFP-labeled α6 nAChR subunits were described previously (Mackey et al., 2012). Briefly, a BAC containing the α6 nAChR subunit gene was modified by recombineering to introduce a GFP sequence in-frame into the M3-M4 portion of the α6 open reading frame. This modified BAC was used to produce transgenic mice. In these mice, α6 nAChRs exhibit normal distribution in the brain, normal expression levels, and normal function (Mackey et al., 2012; Henderson et al., 2014). All groups of mice used in this study had approximately equal numbers of males and females. All mice were between 8 and 14 weeks old when experiments were conducted.
Construction and testing of GFP-labeled α3 and β4 nAChR subunits.
Mouse α3 and β4 nAChR cDNAs in pCDNA3.1 were a generous gift from Jerry Stitzel (Institute for Behavioral Genetics, University of Colorado, Boulder, CO). GFP was inserted in-frame into the M3-M4 loop region of the α3 and β4 nAChR subunit coding sequence. This insertion site yields normal, functional receptors when GFP (or similar fluorophore such as YFP or CFP) is inserted into α4 (Nashmi et al., 2003, 2007), α6 (Drenan et al., 2008a; Xiao et al., 2011; Mackey et al., 2012), β2 (Nashmi et al., 2003), and β3 (Drenan et al., 2008a) nAChR subunits. GFP insertion was executed using a modified, two-step QuikChange (Stratagene) reaction according to our previously published methods (Drenan et al., 2008a). Briefly, an Ala-Gly-Ala linker was inserted immediately 5′ and 3′ to the GFP coding sequence. AlaGlyAla-GFP-AlaGlyAla was inserted between residues Gly373 and Ala374 in mouse α3 and between residues Gly378 and Pro379 in mouse β4. All DNA constructs were fully sequenced before use.
To verify that GFP-labeled α3 and β4 nAChR subunits were functional, two-electrode voltage clamp in Xenopus laevis oocytes was conducted as previously described for other GFP-labeled nAChR subunits (Drenan et al., 2008a). Briefly, constructs were linearized and cRNA was produced using a T7 in vitro transcription kit (mMessage mMachine; Ambion). RNA yield was quantified with absorbance at 260 nm. High-quality WT or GFP α3:β4 cRNA was injected at a ratio of 2:3 by mass into stage V to VI X. laevis oocytes. After injection, oocytes were incubated for 1–4 d in ND-96 buffer (in mm: 96 NaCl, 2 KCl, 1 CaCl2, 1 MgCl2, and 5 HEPES/NaOH, pH 7.6) supplemented with 50 μg/ml gentamycin and 2.5 mm sodium pyruvate. Two-electrode voltage clamp was performed using an OpusXpress 6000A (Molecular Devices). Oocytes were clamped at a holding potential of −60 mV; 1 ml of ND-96 solution containing ACh (concentrations indicated in Fig. 1A) was applied to clamped oocytes for 12 s followed by a 2 min wash with drug-free ND-96. Data were sampled at 50 Hz and filtered at 20 Hz.
Figure 1.

Production of GFP-labeled nAChR subunits. A, Functional characterization of α3-GFP and β4-GFP subunits. mRNA encoding WT or GFP-fused mouse α3 and β4 nAChR subunits was transcribed and microinjected (ratio of 2 α subunits:3 β subunits) into X. laevis oocytes. Using two-electrode voltage clamp (holding potential = −60 mV), inward current responses were recorded during application of ACh at the indicated concentrations. Concentration-response curves were constructed, and data were fitted to the Hill equation. The number of oocytes analyzed was 11 (WT α3β4), 11 (α3GFP β4), and 7 (α3 β4GFP). B, The nAChR genes modified and/or reported on in this study are the following: α3, α4, α6, β2, β3, and β4. For each of these genes, the M3–M4 intracellular loop is the site for insertion of monomeric GFP (abbreviated simply as “GFP”) and is encoded by exon 5. Except for the α6-GFP strain, for each of these nAChR subunit genes, a targeting vector was built that contained a modified (GFP inserted in-frame) exon 5, a neomycin selection cassette flanked by loxP sites, several kilobases of homologous 5′ and 3′ sequence (homology arms), and a diphtheria toxin-negative selection cassette. Targeting vectors were linearized and electroporated into mouse embryonic stem cells. GFP insertion into exon 5 was verified as described in Materials and Methods, targeted ES cell clones were injected into blastocysts, and blastocysts were implanted into pseudopregnant surrogates. Chimeric mice were crossed to Cre-deleter mice to remove the neomycin selection cassette in vivo, and correct gene targeting in neo-excised, F1 progeny was analyzed and verified.
nAChR-GFP knock-in mice.
Five additional knock-in mouse strains were generated and studied in this project, as summarized in Figure 1. These nAChR-GFP subunits were designed to insert a monomeric enhanced GFP (called here simply GFP) in-frame into the M3-M4 intracellular loop. The α4-GFP knock-in strain was constructed, bred, and backcrossed to C57BL/6 at Caltech (4 generations) using procedures previously used to generate mice with yellow fluorescent protein (YFP)-tagged α4 subunits (Nashmi et al., 2007). Four strains were produced by genOway for the α3, β2, β3, and β4 nAChR subunits. The exact insertion site of GFP in the nAChR open reading frame of α4-GFP, β2-GFP, and β3-GFP mice was previously described (Nashmi et al., 2003, 2007; Drenan et al., 2008a; Srinivasan et al., 2011). nAChR exon/intron structure is relatively conserved for the genes under study in this paper, and the coding sequence for the M3-M4 loop is contained within exon 5 of the nAChR genes we manipulated. For each nAChR gene, a targeting vector was constructed containing exon 5 with GFP fused in-frame in the desired position in the M3-M4 loop coding sequence. Several kilobases of 5′ and 3′ sequence (relative to exon 5) were included to direct homologous recombination. A neomycin cassette flanked by loxP sites was inserted adjacent to the modified exon 5 in the targeting vector. A diphtheria toxin negative selection cassette was inserted adjacent to one of the homology arms (5′ or 3′).
Targeting vectors were linearized and electroporated into C57BL/6 embryonic stem cells. G418-resistant cell clones were isolated and amplified. First, clones were screened via PCR to identify those positive for both the 5′ and 3′ homologous recombination events. Next, clones positive in the 5′ and 3′ PCR assays were further evaluated for correct homologous recombination (and the absence of additional genomic rearrangements) via Southern blot with probes designed to detect both the 5′ and 3′ homologous recombination events. At least three positive ES cell clones were injected into 3.5-d-old embryos (blastocyst stage) derived from an albino C57BL/6 strain (C57BL/6J-Tyrc-2J/J), and reimplanted into pseudo-pregnant females and allowed to develop to term. Progeny males with evidence of high (>50%) chimerism (based on black vs white coat color) were selected for further breeding after reaching sexual maturity. To both (1) breed chimeras to the F1 generation and (2) excise the neomycin selection cassette from the genome of the F1 progeny, chimeric males were mated with female C57BL/6 Cre-deleter (proprietary to genOway) mice. To confirm germline transmission, neo-excision, and maintenance of the proper homologous recombination event, F1 mice were genotyped via both PCR and Southern blot. Summarizing, all nAChR subunit GFP mice are extensively backcrossed to C57BL/6 (α6-GFP, α4-GFP) or are fully isogenic with C57BL/6 (β3-GFP, β2-GFP, β3-GFP, and β4-GFP). DNA sequences for all targeting vectors, Southern blotting probes, and PCR primers are available upon request.
Chronic nicotine treatment.
To treat mice chronically with nicotine and to study nicotine withdrawal, mice were implanted with primed osmotic minipumps (model 2004; Alzet) filled with sterile saline or (−)-nicotine hydrogen-tartrate salt dissolved in sterile saline. Nicotine (free base) was delivered at a rate of 1 mg/kg/h for 14 d. On day 14, minipumps were surgically removed, and mice were killed 18–24 h later for electrophysiology experiments. Alternatively, on day 14 after implantation, some mice were injected intrapertoneally with mecamylamine (1 mg/kg) to precipitate withdrawal. Mice were transcardially perfused 90 min after mecamylamine injection, brains were removed, sectioned (50 μm thickness), and stained with anti-c-Fos antibodies as described below.
Immunohistochemistry.
For analysis of nAChR-GFP expression in mouse brain, mice under deep anesthesia were perfused transcardially with cold PBS followed by cold PBS containing 4% PFA. Brains were postfixed overnight in 4% PFA in PBS. Using MultiBrain Technology (NeuroScience Associates), nAChR-GFP brains were embedded in a gelatin matrix. After freezing the brain samples to −70°C with dry ice, they were sliced coronally into 35-μm-thick serial sections using a microtome. For anti-GFP staining, the following procedures were followed. Sections were washed 3 times for 5 min per wash in TBS, followed by incubation in peroxide solution (0.9% H2O2 in TBST, which is TBS containing 0.3% Triton X-100) for 30 min. Sections were rinsed 3 times for 5 min per wash in TBS, followed by incubation in blocking serum (normal horse serum diluted 80× into TBST) for 60 min. Sections were rinsed 3 times for 5 min per wash in TBS, followed by incubation in primary antibody solution (rabbit anti-GFP; 1:5000; Millipore) overnight at room temperature. Sections were washed 3 times for 5 min per wash in TBS, followed by incubation in solution containing biotinylated secondary antibodies (goat anti-rabbit biotinylated IgG; 1:240; Vector Laboratories) for 60 min. Sections were washed 3 times for 5 min per wash in TBS, followed by incubation in ABC solution (TBST containing Reagent A and B solutions, VECTASTAIN Elite ABC System; Vector Laboratories) for 120 min. Sections were washed 3 times for 5 min per wash, followed by incubation for 0–10 min in DAB-H2O2 solution (0.00017% DAB diluted into TBST containing 0.0015% H2O2) with continuous visual monitoring of DAB staining. Sections were washed 2 times for 5 min per wash, placed in fresh TBS, mounted onto glass slides, and coverslipped.
Some sections cut by NeuroScience Associates were stained at Purdue and imaged with confocal microscopy. For those, as well as c-Fos staining, the following procedures were followed. Sections were collected into 12-well tissue culture plates, washed 3 times for 10 min per wash in TBS, and blocked for 30 min in TBST containing 10% normal horse serum. Sections were washed 3 times for 10 min per wash and incubated in a solution containing TBST and primary antibodies (rabbit anti-GFP; 1:500; Invitrogen; rabbit anti-c-Fos; 1:400; Santa Cruz Biotechnology; and goat anti-ChAT; 1:100; Millipore) overnight at 4°C. Sections were washed 3 times for 10 min per wash in TBST and incubated in a solution containing secondary antibodies (chicken anti-rabbit Alexa-488; 1:500; Invitrogen; and/or donkey anti-goat Alexa-555; 1:500; Invitrogen) for 1–2 h at room temperature. Sections were then rinsed 3 times for 10 min per rinse, mounted onto glass slides, and coverslipped for confocal microscopy.
To test for colocalization of recorded neurons with ChAT, brain slices with neurobiotin-filled neurons were fixed overnight in 4% PFA in PBS and processed essentially as described above for confocal microscopy but with the following modifications. Sections were washed 2 times for 10 min per wash in PBS, blocked, and permeabilized in PBS containing 10% normal horse serum, 2% BSA and 0.3% Triton X-100 (PBST) for 60 min. Sections were incubated with PBST solutions containing primary antibody (goat anti-ChAT; 1:500; Millipore and streptavidin conjugated Alexa-488; 1:1000; Invitrogen) overnight at 4°C. Sections were washed 3 times for 5 min per wash with PBS, followed by incubation in PBST solutions containing secondary antibodies (Alexa-555 donkey-anti-goat; 1:500; Invitrogen) for 120 min at room temperature. Finally, sections were washed 3 times for 10 min per wash with PBS, mounted with anti-fade mounting medium (Vectashield; Vector Laboratories), and coverslipped.
Brain slice preparation for electrophysiology.
Two- to 3-month-old C57BL/6J mice (The Jackson Laboratory) and α4 knock-out (KO) mice of either sex were used in accordance with the guidelines of the National Institutes of Health Office of Laboratory Animal Welfare, and the protocols were approved by the Institutional Animal Care and Use Committee at Purdue University. Mice were anesthetized by intraperitoneal injection of a sodium pentobarbital (200 mg/kg), followed by transcardiac perfusion with oxygenated (95% O2/5% CO2) N-methyl-d-glucamine (NMDG) recovery solution (in mm: 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na+ ascorbate, 2 thiourea, 3 Na+ pyruvate, 10 MgSO4 · 7H2O, 0.5 CaCl2 · 2H2O, with pH adjusted to 7.3–7.4, 300–310 mOsm). The brain was quickly removed into ice-cold NMDG recovery solution for 1 min, and 200-μm-thick coronal slices containing MHb or IPN were cut with a vibrating microslicer (DTK-Zero 1; Ted Pella). Coordinates were as follows: (MHb: −1.9 anterior/posterior, 0.25 lateral, −2.5 dorsal/ventral; IPN: −3.5 anterior/posterior, 0.0 lateral, −4.5 dorsal/ventral). Slices were then incubated in this solution at 33°C for 12 min, after which they were kept at room temperature for at least 1 h in 95% O2/5% CO2-equilibrated HEPES-holding-solution containing the following (in mm): NaCl 92, KCl 2.5, NaH2PO4 1.2, NaHCO3 30, HEPES 20, glucose 25, Na+ ascorbate 5, thiourea 2, Na+ pyruvate 3, MgSO4 · 7H2O 2, and CaCl2 · 2H2O 2.
Patch-clamp electrophysiology.
Recordings were made under an upright microscope (Nikon Eclipse FN-1) equipped with infrared differential interference contrast optics for visualization. Slices were transferred to a recording chamber superfused with standard recording ACSF containing (in mm) NaCl 124, KCl 2.5, NaH2PO4 1.2, NaHCO3 24, glucose 12.5, MgSO4 · 7H2O 2, and CaCl2 · 2H2O 2, adjusted to pH 7.3–7.4, 300–310 mOsm. Recordings were performed at 32°C. Patch pipettes were made from borosilicate glass capillary tubing (1B150F-4; World Precision Instruments) using a micropipette puller (P-97; Sutter Instrument). The internal recording pipette solution contained the following (in mm): K+ gluconate 135, EGTA 5, HEPES 10, MgATP 2, NaGTP 0.1, CaCl2 0.5, MgCl2 2; pH adjusted to 7.3 with Tris base. In a subset of experiments, 2.5 mg/ml neurobiotin (Vector Laboratories) was added to the internal solution for intracellular labeling of recorded neurons, and the pH was readjusted. Currents were recorded using a Multiclamp 700B or an Axopatch 200B amplifier with a DigiData 1440A interface and Clampex 10.3 software (Molecular Devices) sampled at 5 kHz and low-pass filtered at 1 kHz. Series resistance was uncompensated.
For some drug applications, CNQX (10 μm), picrotoxin (100 μm), and atropine (1 μm) were added to the superfusion ACSF by dilution of a stock solution. Functional AMPAR and nAChR responses were examined as previously described (Engle et al., 2012, 2013). Briefly, a drug-filled glass pipette, identical to a typical recording pipette, was connected to a micropressure ejection system (PV-820 Pneumatic PicoPump; World Precision Instruments). Ejection pipettes were moved to within 20–40 μm of the recorded cell using a piezoelectric manipulator (PA-100/12; Piezosystem Jena), drug was applied for 250 ms (12 psi), and the ejection pipette was retracted.
Statistics and data analysis.
Summary data are presented in the text and figures as the mean ± SEM t tests (unpaired or paired, as indicated in figure legends) were used for all comparisons. All statistical tests were performed using Graphpad software (Prism). Concentration-response data were fitted to the Hill equation. Differences were considered significant at p < 0.05.
Results
We previously verified that XFP-tagged α4 (Nashmi et al., 2003, 2007), β2 (Nashmi et al., 2003), α6 (Drenan et al., 2008a; Xiao et al., 2009, 2011; Mackey et al., 2012), and β3 (Drenan et al., 2008a) subunits are functional in cell expression systems and in vivo in some cases, but we have not previously demonstrated the same for α3-GFP and β4-GFP subunits. To verify that these latter subunits are indeed functional, mRNA for α3-GFP and β4-GFP nAChR subunits was transcribed and microinjected into X. laevis oocytes. To mimic expression of these GFP-labeled subunits in the knock-in mice, α3-GFP was expressed with WT β4 subunits, and β4-GFP was expressed with WT α3 subunits. WT α3β4 receptor function was compared as a reference. ACh was applied to voltage-clamped oocytes for 1 s, and peak current responses were measured. Mean data (± SEM) were plotted and fitted to the Hill equation for WT α3β4, α3-GFPβ4, and α3β4-GFP nAChRs. Both α3-GFP and β4-GFP subunits supported relatively normal nAChR function compared with WT α3β4 nAChRs (Fig. 1A). α3-GFPβ4 and α3β4-GFP nAChRs had ACh EC50 values of 92 μm (95% confidence interval: 81–105 μm) and 262 μm (95% confidence interval: 246–279 μm) and Hill coefficients of 1.6 ± 0.1 and 1.8 ± 0.1, respectively. Reference WT α3β4 nAChRs had an ACh EC50 value of 125 μm (95% confidence interval: 113–137 μm) and a Hill coefficient of 1.6 ± 0.1, consistent with previous studies on this receptor (Fig. 1A) (Drenan et al., 2008a). We seek to make exon-replacement knock-in mice with a particular nAChR-GFP mutation only when the mutation is expected to result in a change in sensitivity of approximately twofold or less compared with the WT subunit. Although α3β4-GFP nAChRs did exhibit a slight reduction in sensitivity compared with WT α3β4 nAChRs in oocyte experiments (Fig. 1A), the change was approximately twofold in magnitude, so we proceeded with this GFP insertion to make β4-GFP mice.
For each of the α3, α4, β2, β3, and β4 nAChR genes, an exon replacement strategy in embryonic stem cells was used to insert an enhanced GFP sequence in-frame into exon 5 of the nAChR gene (Fig. 1B). This resulted in production of nAChR subunits with an intervening, functional GFP protein within the large, M3-M4 intracellular loop. In addition to these 5 new knock-in mouse strains, we also used BAC transgenic mice expressing GFP-fused α6 nAChR subunits, which have been reported previously (Mackey et al., 2012). nAChR-GFP mice were perfused and coronal sections were cut at 35 μm from bregma 2 mm to −4 mm (Fig. 2A). Sections were stained with anti-GFP antibodies and visualized with DAB histochemistry. Figure 2B–E shows representative images of GFP-stained coronal sections for all GFP mice (and C57BL/6 WT control mice; Fig. 2B) at 5 different anterior/posterior coordinates (bregma 2, 1, −1, −3, and −4 mm) corresponding to sites of known expression for these subunits. Subunits were grouped based on similar localization patterns in the brain: α4 and β2 (Fig. 2C), α3 and β4 (Fig. 2D), and α6 and β3 (Fig. 2E). Based on known localization patterns using methods, such as radioligand binding and mRNA in situ hybridization, no aberrant localization of GFP-labeled subunits was apparent in our staining results (Fig. 2B–E). α4 and β2 subunits were distributed throughout the brain, including but not limited to cerebral cortex, striatum, thalamus (including dorsal lateral geniculate nucleus), MHb, substantia nigra (SN), ventral tegmental area (VTA), and superior colliculus (SC) (Fig. 2C). α3 and β4 distribution was apparent in piriform cortex, MHb, fasciculus retroflexus (FR), and IPN, with additional α3 found apparent in SC (Fig. 2D). α6 was found at low levels in prefrontal cortex and striatum, MHb, SN, VTA, and SC (Fig. 2E). β3, which has been difficult to localize in the brain because it does not participate in a nAChR agonist binding site (Drenan et al., 2008a), has moderate levels in cortical areas, ST, MHb, FR, SN, VTA, and SC (Fig. 2E).
Figure 2.

Localization of GFP-fused nAChR subunits in mouse brain. A, Brain atlas diagram for five bregma coordinates (bregma 2 mm, 1 mm, −1 mm, −3 mm, −4 mm) corresponding to approximate bregma coordinates for data presented in B–E. B–E, Brains from six mouse strains expressing GFP-fused nAChR subunits were fixed, sectioned at 35 μm, and stained with anti-GFP antibodies. B, C57BL/6 WT brains were processed as a staining negative control. A representative (of n = 3) image of a coronal section is shown for each nAChR-GFP mouse strain at the indicated anterior/posterior coordinate. α4 and β2 (C), α3 and β4 (D), and α6 and β3 (E) were grouped together based on their coexpression in several brain areas.
To analyze nAChR localization in the MHb to IPN pathway, sections containing MHb (bregma −1 to −2 mm; Fig. 3A) and IPN (bregma −3.2 to −3.9 mm; Fig. 4A) were stained with anti-GFP primary antibodies and peroxidase-conjugated secondary antibodies, followed by visualization with a DAB reaction. Terminology for MHb subnuclei follows a recent study (Aizawa et al., 2012). α4, β2, and β4 nAChR subunits are restricted to the ventral two-thirds of the MHb (referred to as MHbV) (Fig. 3C,D). α3, α6, and β3 subunits are also largely restricted to the MHbV, but immunoreactivity for these subunits is also evident in a small number of cells in the dorsal one-third of MHb (MHbD) (Fig. 3D,E). α3 and β4 subunit localization is even and strong throughout the MHbV, with subunits also evident in efferent fibers that give rise to the FR (Fig. 3D). α4 subunits are exclusively found in the lateral portion of the MHbV (referred to as MHbVL) (Fig. 3C), consistent with previous reports (Nashmi et al., 2007; Fonck et al., 2009). In contrast to α4, α6 subunits are mainly found in the inferior portion of MHbV (MHbVI), with additional faint expression in the lateral portion of MHbD (Fig. 3E). β2 subunit immunoreactivity is seen throughout MHbV, with preference for central and inferior MHbV (Fig. 3C). β3 subunits are strong in MHbVI and MHbVC but very weak in MHbVL (Fig. 3E).
Figure 3.

Localization of GFP-fused nAChR subunits in MHb. A, Brain atlas diagram for three bregma coordinates (bregma −1 mm, −1.5 mm, and −2 mm) corresponding to anterior, medial, and posterior MHb (orange). B–E, Brains from six mouse strains expressing GFP-fused nAChR subunits were fixed, sectioned at 35 μm, and immunostained with anti-GFP antibodies. B, C57BL/6 WT brains were processed as a staining negative control. A representative (of n = 3) image of an MHb coronal section is shown for each nAChR-GFP mouse strain at the indicated anterior/posterior coordinate. α4 and β2 (C), α3 and β4 (D), and α6 and β3 (E) were grouped together based on coexpression in MHb and/or other brain areas.
Figure 4.
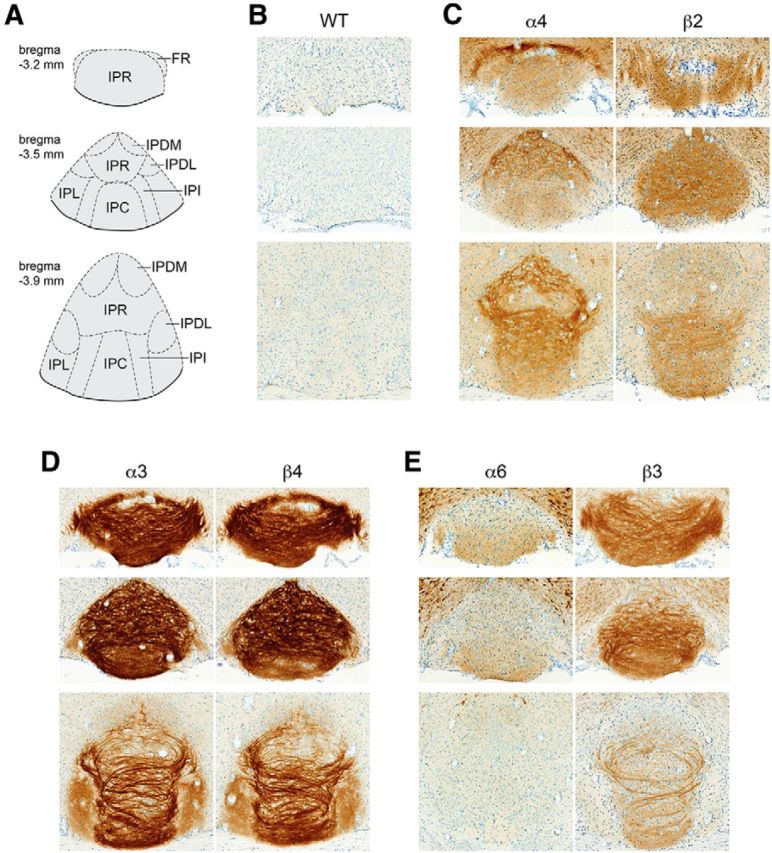
Localization of GFP-fused nAChR subunits in IPN. A, Brain atlas diagram for 3 bregma coordinates (bregma −3.2 mm, −3.5 mm, and −3.9 mm) corresponding to anterior, medial, and posterior interpeduncular nucleus. B–E, Brains from six mouse strains expressing GFP-fused nAChR subunits were fixed, sectioned at 35 μm, and immunostained with anti-GFP antibodies. B, C57BL/6 WT brains were processed as a staining negative control. A representative (of n = 3) image of an IPN coronal section is shown for each nAChR-GFP mouse strain at the indicated anterior/posterior coordinate. α4 and β2 (C), α3 and β4 (D), and α6 and β3 (E) were grouped together based on coexpression in MHb and/or other brain areas.
Next, nAChR subunit localization was analyzed in IPN from bregma −3.2 to −3.9 mm (Fig. 4A) using anti-GFP immunohistochemistry in C57BL/6 WT (Fig. 4B) and nAChR-GFP knock-in mice (Fig. 4C–E). Terminology used for IPN subnuclei is consistent with Wada et al. (1989) and the mouse brain atlas (Franklin and Paxinos, 2008): rostral subnucleus (IPR), dorsomedial subnucleus (IPDM), dorsolateral subnucleus (IPDL), caudal subnucleus (IPC), and intermediate subnucleus (IPI). Similar to MHb, α3 and β4 subunit levels are strong in IPR, IPDM, IPDL, IPC, and IPI, with moderate amounts of subunits also evident in IPL and IPDL (Fig. 4D). α4 subunits are strongest in IPR and IPDM, with lower levels of immunoreactivity evident in IPC and IPI (Fig. 4C). In contrast to α4, α6 subunits are found at low levels in ventral IPR and IPC with little to no α6 subunits found in dorsal IPR or IPDM (Fig. 4E). β2 subunit levels are modest throughout the rostral IPN but are restricted to IPC and IPI in caudal IPN (Fig. 4C). β3 levels are strong in most IPN subnuclei but were not found in dorsal IPR and/or IPDM (Fig. 4E).
The MHbV stains positive for cholinergic markers, such as ChAT and VAChT (Contestabile et al., 1987), and IPN nAChRs in presynaptic terminals from MHbV neurons are involved in ACh release (Grady et al., 2009). However, other reports indicate that MHbV neurons are glutamatergic and that nAChRs in presynaptic terminals in IPN are involved in regulating glutamate release (Ren et al., 2011; Hu et al., 2012). Using mouse strains expressing GFP-labeled nAChRs, we costained brain sections containing MHb with anti-GFP and anti-ChAT primary antibodies. The MHb was imaged at 60× magnification using confocal microscopy. Anti-GFP staining recapitulated our DAB staining results with respect to anatomical specificity; α3 and β4 subunits were found throughout the MHbV, α4 was restricted to MHbVL, α6 was largely restricted to MHbVI plus the MHbDC, and β2 and β3 subunits were found in MHbVI and MHbVC (Fig. 5A). All subunits were found in some/many ChAT(+) MHbV neurons, but we also found evidence for α3, α4, and α6 subunits localized to ChAT(−) cells (Fig. 5A). In contrast, nearly all cells that were immunoreactive for β2, β3, and β4 subunits were also ChAT(+) (Fig. 5B).
Figure 5.

nAChR MHb localization and function in ChAT(+) and ChAT(−) cells. A, For α3-GFP, α4-GFP, and α6-GFP mouse strains, coronal MHb sections were prepared, double-stained with anti-GFP and anti-ChAT antibodies, and imaged using confocal microscopy. Merged (green represents anti-GFP; red represents anti-ChAT; yellow represents colocalized pixels) micrographs of the MHb (top panels), and a high-resolution zoom of several cells in MHb (bottom panels) are shown. Single, diagonal arrowheads indicate cells expressing both ChAT and the nAChR-GFP subunit. Double, horizontal arrowheads indicate cells expressing nAChR-GFP subunits but not ChAT. Scale bars: top, 50 μm; bottom, 10 μm. B, For β2-GFP, β3-GFP, and β4-GFP mouse strains, coronal MHb sections were prepared, double-stained with anti-GFP and anti-ChAT antibodies, and imaged using confocal microscopy. Merged (green represents anti-GFP; red represents anti-ChAT; yellow represents colocalized pixels) micrographs of the MHb (top panels), and a high-resolution zoom of several cells in MHb (bottom panels) are shown. Single, diagonal arrowheads indicate cells expressing both ChAT and the nAChR-GFP subunit. Double, horizontal arrowheads indicate cells expressing nAChR-GFP subunits but not ChAT. Scale bars: top, 50 μm; bottom, 10 μm. C, Action potential firing patterns in MHb versus LHb neurons. Coronal habenular brain slices were prepared from C57BL/6 WT mice, and cell-attached recordings were made from MHb and LHb neurons. A representative trace is shown from a typical cell-attached recording from a MHb and LHb neuron. D, In whole-cell configuration, action potential firing in response to hyperpolarizing current (−50 pA) injections was recorded in MHb and LHb neurons. Representative traces show the latency to recovery of action potential firing, as well as the action potential firing frequency, after cessation of current injection. E, Responses to nicotine puffs in ChAT(+) and ChAT(−) MHb neurons. MHb neurons were voltage-clamped at −60 mV, and nicotine (30 μm, 250 ms) was puff-applied to the cell using a Picopump. Representative inward current deflections are shown for applications of nicotine at the indicated concentration. Responses from ChAT(+) and ChAT(−) neurons are shown. ChAT expression was later determined and assigned to each cell with a nicotine puff response as described in F. F, ChAT expression in MHb cells. For cells studied in E, neurobiotin was included in the patch pipette internal solution to mark the recorded cell. After successful nicotine puff experiments, neurobiotin-filled cells in slices were recovered and double-stained with streptavidin-Alexa-488 conjugates and anti-ChAT antibodies. A representative ChAT(+) and ChAT(−) MHb neuron that was studied with nicotine puffs is shown. The wide micrograph shows the location of the cell in the MHb. Inset shows whether the cell was ChAT (+) (yellow color) or ChAT (−) (green color). Scale bars: 50 μm; inset, 10 μm.
We were interested to further explore the possibility that ChAT(−) cells contained functional nAChRs, so we studied MHbV neurons and nAChR function using patch-clamp electrophysiology in brain slices from C57BL/6 mice. First, we developed the ability to distinguish MHb neurons from neurons in the adjacent lateral Hb (LHb). Whereas MHb neurons exhibited regular, tonic firing when action potentials were recorded in cell-attached recordings (Fig. 5C), LHb neurons were heterogeneous, frequently exhibiting phasic bursts of spontaneous action potentials followed by periods of inactivity (Fig. 5C). Further, “anode break” excitation differed between MHb and LHb neurons: in whole-cell mode, MHb neurons exhibited single action potentials following a brief hyperpolarizing current injection (anode break action potentials; Fig. 5D). In contrast, LHb neurons exhibited multiple low-amplitude anode break action potentials (Fig. 5D). The latency to action potential firing during anode break was often, but not always, shorter in LHb cells versus MHb cells (Fig. 5D).
Having established criteria to distinguish MHb from LHb neurons, we next recorded inward currents in response to nicotine in ChAT(+) and ChAT(−) neurons. For both ChAT(+) and ChAT(−) cells, we recorded robust responses to nicotine (30 μm) (Fig. 5E). Cells displaying nicotine-elicited inward currents were verified as ChAT(+) or ChAT(−) by filling with neurobiotin, double-staining with streptavidin-Alexa-488 conjugate and anti-ChAT antibodies, and confocal imaging. Representative ChAT(+) and ChAT(−) cells displaying nicotine-elicited inward currents are shown (Fig. 5F). These results suggest that nAChR function is not restricted to ChAT(+) cells in MHbV. Neurons studied in subsequent recordings reported below were not analyzed for ChAT expression status.
Based on differences in localization of nAChR subunits within regions of MHbV (Fig. 3), evidence for nAChRs in both ChAT(+) and ChAT(−) cells (Fig. 5), and emerging data showing specific gene expression in certain MHb to IPN circuits (Aizawa et al., 2012), we focused on distinguishing neurons within the MHbV region. In particular, we recorded from MHbVL and MHbVC neurons (Fig. 6A). In the cell-attached configuration, spontaneous action potential firing rates were similar in MHbVL and MHbVC neurons (MHbVL = 4.3 ± 0.5 Hz, MHbVC = 5.0 ± 0.6 Hz; unpaired t test, p = 0.208). In whole-cell configuration, MHbVL and MHbVC neurons had a similar resting membrane potential when pipettes were filled with a K+-gluconate-based internal pipette solution (MHbVL = −42.2 ± 1.7 mV, MHbVC = −42.6 ± 1.7 mV; unpaired t test, p = 0.429). In a measure of cellular excitability, we measured spike amplitude after a prolonged (1000 ms), depolarizing (50 pA) current injection (i.e., adaptation ratio) (Fig. 6F) (Bugaysen et al., 2010). MHbVL neurons exhibited significantly more decline in spike amplitude after depolarizing current injection compared with MHbVC neurons (Fig. 6G).
Figure 6.

Electrophysiological properties of MHbVL and MHbVC neurons. A, Diagram of approximate location of MHbVL and MHbVC. B, Hyperpolarization-activated cation currents (Ih) in MHbVC and MHbVL. MHb neurons were voltage-clamped at −60 mV, and the command voltage was pulsed to −120 mV for 1 s. The amplitude of the voltage-dependent inward current response was measured beginning with the current value at the end of the capacitive transient (indicated by the broken line), ending with the current value at the end of the voltage step. Representative Ih current traces are shown for MHbVL and MHbVC cells. Calibration: 100 pA, 200 ms. C, Quantification of Ih currents. A bar graph is shown for all recorded MHbVC (n = 13) and MHbVL (n = 18) cells. **p < 0.01 (unpaired t test). D, Measurement of rebound (anode break) delay in MHbVC and MHbVL neurons. MHb neurons recorded in whole-cell configuration were current-clamped (I = 0), followed by injection of hyperpolarizing (−75 pA) current. The latency to firing an action potential after the end of the pulse (from the broken line to the arrowhead) was measured for MHbVC and MHbVL neurons. Representative current traces are shown for MHbVL and MHbVC cells. Calibration: 50 mV, 200 ms. E, Quantification of rebound delay responses. A bar graph is shown for all responses from MHbVL (n = 18) and MHbVC (n = 13) neurons. **p < 0.01 (unpaired t test). F, Action potential amplitude adaptation in MHb neurons. MHb neurons were current-clamped (I = 0), and depolarizing current (50 pA) was injected. The amplitude of the first and last spike in the train was measured (between the two broken lines), and the adaptation ratio (last spike amplitude/first spike amplitude) was calculated for each cell. Calibration: 50 mV, 200 ms. G, Quantification of adaptation ratio measurements from F. A bar graph is shown for all adaptation ratio responses from MHbVL (n = 18) and MHbVC (n = 13) cells. **p < 0.01 (unpaired t test). H, J, For each recorded MHbVL (H; black open circles) and MHbVC (J; red open circles) neuron, Ih current amplitude was plotted on the x-axis, and rebound delay was plotted on the y-axis. Linear regression analysis was performed on the plotted data points, and r2 and p values are shown. I, K, For each recorded MHbVL (I; black open circles) and MHbVC (K; red open circles) neuron, Ih current amplitude was plotted on the x-axis, and adaptation ratio was plotted on the y-axis. Linear regression analysis was performed on the plotted data points, and r and p values are shown.
Tonic, spontaneous action potential firing (Fig. 5C) suggested that MHbV neurons may express hyperpolarization-activated inward cation current (Ih current). Both MHbVL and MHbVC neurons exhibited small but detectable Ih current in response to hyperpolarization (Fig. 6B), with MHbVC neurons exhibiting significantly more Ih current than MHbVL neurons (Fig. 6C). Differences in Ih current in MHbVL and MHbVC neurons suggested that these cells may respond differentially after hyperpolarizing current pulses. To test this idea, MHbVL and MHbVC neurons in current-clamp mode were briefly (100 ms) injected with hyperpolarizing (−75 pA) current pulses, and the delay from the end of the pulse to the first action potential (i.e., rebound delay) was measured (Fig. 6D). MHbVL neurons exhibited a significantly longer rebound delay compared with MHbVC neurons (Fig. 6E), consistent with the idea that Ih currents activated during the hyperpolarizing pulse help to depolarize the neuron, allowing it to reach threshold more quickly.
Next, we used linear regression analysis and tested whether two of the electrophysiological parameters we recorded were correlated with expression of Ih current. In both MHbVL and MHbVC neurons, Ih current amplitude (at Vm = −120 mV) was anti-correlated with rebound delay (Fig. 6H,J). Similarly, in both MHbVL and MHbVC neurons, Ih current was positively correlated with adaptation ratio (Fig. 6I,K) (Bugaysen et al., 2010). These results, in addition to revealing differences between MHbVL and MHbVC neurons, indicate that Ih current may play a general role in shaping the excitability of MHbV neurons.
Based on differences in nAChR subunit localization between MHbVL and MHbVC neurons (Fig. 3), and in part due to our results showing differences in excitability between these two regions of MHbV (Fig. 6), we next studied nAChR function in MHbVL and MHbVC cells using puff-application of nicotine. MHbVL and MHbVC neurons were recorded in whole-cell configuration. Neurons were voltage-clamped at −60 mV and nicotine was puff-applied using a second drug-filled pipette positioned 20–40 μm from the recorded cell. Interestingly, MHbVL neurons exhibited small but detectable responses to 1 μm nicotine (Fig. 7A,B). MHbVC neurons, on the other hand, did not respond appreciably to this concentration of nicotine (Fig. 7A,B,D). Both MHbVL and MHbVC neurons responded with inward currents following puff-application of 30 μm nicotine, but mean responses in MHbVL neurons were, again, larger than in MHbVC neurons (Fig. 7C,D).
Figure 7.

Nicotine-elicited inward currents in MHbV neurons. A, MHbVL or MHbVC neurons were voltage-clamped at −60 mV, and nicotine at the indicated concentration was puff-applied. Representative traces are shown for each nicotine concentration and cell type. Calibration: 1 μm nicotine, 25 pA, 5 s; 30 μm nicotine, 0.5 nA, 5 s. B, Mean nicotine (1 μm) responses from MHbVL (n = 7) and MHbVC (n = 8) neurons are shown. *p < 0.05 (unpaired t test). C, Mean nicotine (30 μm) responses from MHbVL (n = 7) and MHbVC (n = 7) neurons are shown. *p < 0.05 (unpaired t test). D, Data from B and C are plotted together on a logarithmic scale. *p < 0.05 (unpaired t test).
Although nicotine typically achieves concentrations of 50–200 nm in the CSF of smokers, concentrations of nicotine as high as 1 μm have been reported as well (Malkawi et al., 2009). Because nAChRs in MHbVL neurons are sensitive to 1 μm nicotine, we tested the idea that MHbVL neurons would respond to prolonged (∼10 min) exposure to 1 μm nicotine with altered action potential firing. Cell-attached recordings were made from MHbVL and MHbVC neurons, followed by bath application of 1 μm nicotine. Whereas MHbVC neurons did not significantly change their firing in response to 1 μm nicotine, MHbVL neurons exhibited a significant increase in firing rate relative to prenicotine baseline firing (Fig. 8A,D,E). Next, we sought to better understand the mechanism behind this difference in firing after nicotine application. Results from GFP-nAChR knock-in mice indicate that one defining factor of MHbVL neurons is their expression of α4 subunits (Fig. 3). Because α4* nAChRs are among the most sensitive to nicotine and ACh (Salminen et al., 2004, 2007), we tested the idea that MHbVL sensitivity to 1 μm nicotine (Fig. 8) required α4 subunits. We measured cell-attached firing in MHbVL neurons in slices from α4 knock-out mice, and we found that 1 μm nicotine did not significantly increase firing above baseline levels (Fig. 8B,D,E). Next, because β4 subunit levels are strong in MHbVL (Fig. 3D), we tested the hypothesis that β4 subunits play a role in the response to 1 μm nicotine in MHbVL cells. Pretreatment of cells with SR16584, a selective antagonist of α3β4* nAChRs, eliminated the increase in action potential firing rate elicited by 1 μm nicotine in MHbVL neurons (Fig. 8C–E). Together, these results point to a role for α4 and β4 subunits, and likely nAChR pentamers containing both of these subunits, in the response of the MHb/IPN pathway to nicotine.
Figure 8.

α3β4* and/or α4* nAChRs drive nicotine-elicited firing in MHbVL neurons. A, Cell-attached recordings from MHbVL (n = 8) and MHbVC (n = 8) neurons were established, and baseline firing was recorded for several minutes, followed by superfusion of nicotine (1 μm). Normalized firing rate is plotted, with baseline firing before nicotine application set to 1.0. B, Cell-attached recordings from α4 KO MHbVL neurons (n = 9) were conducted. As in A, baseline firing was recorded followed by firing in response to superfusion of 1 μm nicotine. Data from MHbVL WT neurons shown in A are replotted for reference using dashed lines and no symbols. C, Cell-attached recordings from WT MHbVL neurons (n = 6) were conducted in response to SR16584, a putative α3β4* nAChR antagonist. Baseline firing was recorded for several minutes, followed by superfusion of SR16584 (20 μm), then superfusion of nicotine (1 μm) plus SR16584. Data from MHbVL WT neurons shown in A (response to 1 μm nicotine alone) are replotted for reference using dashed lines and no symbols. D, Representative traces from cell-attached firing experiments described in A–C. A sample current trace from t = 2.5 min (baseline) and t = 10 min (in 1 μm nicotine) is shown for the indicated cell type, genotype, and/or pharmacological treatment. Calibration: 20 pA, 200 ms. E, Quantification of data shown in A–C. For each cell from each of 4 conditions described in A–C, the fold change in action potential firing was derived compared with predrug firing. A bar graph is shown for these 4 conditions. *p < 0.05 (paired t test comparing predrug firing and peak firing during drug application for each cell).
The MHb to IPN pathway is a key circuit mediating the somatic and affective signs of nicotine withdrawal in rodents, and presumably in humans. To determine whether MHbVL neurons play a role in nicotine withdrawal, we implanted C57BL/6 WT mice with osmotic minipumps delivering control saline or nicotine (1 mg/kg/h) for 14 d. To elicit withdrawal from nicotine, minipumps were removed, and brain slices were prepared for electrophysiology 18–24 h later (Fig. 9A). First, we measured nicotine-evoked currents using puff-application of 1 and 30 μm nicotine. Compared with saline-withdrawn mice, there was a trend toward a decrease in nicotine-evoked currents in MHbVL neurons from nicotine-withdrawn mice at 1 μm nicotine (Fig. 9B,C). Furthermore, there was a significant reduction in nicotine-evoked current amplitudes in nicotine-withdrawn MHbVL neurons at 30 μm nicotine (Fig. 9B,D). Next, we measured action potential firing in response to nicotine (1 μm) reexposure in MHbVL neurons from nicotine- and saline-withdrawn mice. Consistent with a reduction in nAChR responsiveness seen in puff-application experiments (Fig. 9D), MHbVL neurons from nicotine-withdrawn mice exhibited a blunted response to nicotine application in cell-attached firing experiments (Fig. 9E). Although raw baseline and nicotine-evoked firing rates in nicotine- and saline-withdrawn mice were similar (Fig. 9F), nicotine-withdrawn MHbVL neurons did not respond to 1 μm nicotine application with as large an increase in firing rate as saline-withdrawn MHbVL neurons (Fig. 9G). To verify that our minipump procedure did indeed deliver chronic nicotine to mice, we conducted a control experiment. Mice implanted with saline- or nicotine-containing minipumps for 14 d were given an intraperitoneal injection of mecamylamine (1 mg/kg) to precipitate withdrawal from chronic nicotine, and mice were perfused 90 min later for immunohistochemical analysis. Coronal sections containing the IPN were stained for c-Fos induction, and we noted a large increase in c-Fos-immunoreactive neurons in the IPN of mecamylamine-injected mice treated with chronic nicotine versus mecamylamine-injected mice treated with chronic saline (Fig. 9H). This result parallels a recent result obtained by Tapper and colleagues, who demonstrated that nicotine withdrawal results in activation of GABAergic neurons in the IPN (Zhao-Shea et al., 2013).
Figure 9.

nAChR function and nicotine-evoked action potential firing in MHbVL neurons after nicotine withdrawal. A, Nicotine withdrawal procedure. C57BL/6 WT mice were implanted with saline (SAL)- or nicotine (NIC)-containing osmotic minipumps. After 14 d, minipumps were removed to initiate withdrawal, and mice were killed 1 d later for slice preparation and electrophysiology. B, Representative traces from nicotine puff-application experiments. The indicated nicotine concentration was puff-applied to MHbVL cells in slices from animals that underwent withdrawal from chronic saline or chronic nicotine. Calibration: 1 μm nicotine, 10 pA, 5 s; 30 μm nicotine, 0.5 nA, 5 s. C, Mean nicotine (1 μm) responses from saline-withdrawn (n = 6) and nicotine-withdrawn (n = 9) MHbVL neurons are shown. D, Mean nicotine (30 μm) responses from saline-withdrawn (n = 6) and nicotine-withdrawn (n = 8) MHbVL neurons are shown. **p < 0.01 (unpaired t test). E, Time course for cell-attached firing in response to nicotine (1 μm) for saline-withdrawn (n = 4) and nicotine-withdrawn (n = 6) MHbVL neurons. MHbVL neurons were held in the cell-attached configuration and action potential firing was recorded before and after bath application of 1 μm nicotine. F, Raw firing frequencies from experiment described in E. Baseline and nicotine-elicited peak action potential firing frequency in MHbVL neurons from saline-withdrawn (n = 6) and nicotine-withdrawn (n = 6) mice is shown. G, Fold change in action potential firing in response to nicotine. Using raw firing frequency data from F, we calculated the fold change in action potential firing in response to nicotine application in MHbVL neurons from saline-withdrawn (n = 6) and nicotine-withdrawn (n = 6) mice. **p < 0.01 (unpaired t test). H, c-Fos induction in IPN neurons after mecamylamine-precipitated withdrawal. C57BL/6 WT mice were implanted with saline- or nicotine-containing osmotic minipumps as in A. On day 14, mice were given an injection of mecamylamine (1 mg/kg, i.p.) and were killed and perfused 90 min later. Coronal IPN sections were stained with anti c-Fos antibodies, and a representative image is shown. Scale bars, 30 μm.
To further study MHbVL to IPN circuitry, we recorded from IPN neurons in the IPR and IPDM. These areas, according to our immunohistochemistry data (Fig. 4), display high and specific expression of α4 nAChR subunits that largely originate in MHbVL. IPR/IPDM neurons were recorded in whole-cell configuration, first in current-clamp mode. Most IPR/IPDM neurons recorded from (19 of 25) did not fire spontaneously, and had an average resting membrane potential of −62 ± 1 mV. Interestingly, when positive current was injected during current-clamp recordings, two responses were evident. One class of cells was less sensitive to current injection, responding to positive current injection with moderate increases in firing (Fig. 10A, left). The other class of cells (Fig. 10A, right) was comparatively more sensitive to positive current injection and tended to exhibit rapid (>100 Hz) (Fig. 10D), phasic bursts of action potentials (Fig. 10A, right). For subsequent experiments (described below), we first recorded firing responses in current-clamp mode, and slower-firing and faster-firing neurons were classified as Type I and Type II IPR/IPDM neurons, respectively.
Figure 10.

IPR/IPDM neuron populations receiving input from MHbVL. A, IPR/IPDM neurons recorded in whole-cell configuration were held in current-clamp mode, and action potential firing was recording while the indicated amount of positive or negative current was injected. Based on the pattern of action potential firing, cells were classified as Type I or Type II. B, Type I or Type II IPR/IPDM neurons were voltage-clamped at −60 mV, and AMPA (100 μm) was puff-applied. Representative traces are shown for each cell type. Calibration: 300 pA, 2 s. C, Type I or Type II IPR/IPDM neurons were voltage-clamped at −60 mV, and nicotine (10 μm) was puff-applied. Representative traces are shown for each cell type. Calibration: 100 pA, 2 s. D, Quantification of action potential firing from A. Action potential firing after injection of the indicated amount of positive current was recorded, and the maximum instantaneous firing rate was derived and plotted for each current injection. The number of neurons analyzed was as follows: n = 16 (Type I), n = 9 (Type II). E, AMPA concentration response relation for Type I and Type II IPR/IPDM neurons. AMPA-evoked inward currents were recorded for a range of AMPA concentrations (in μm: 1, 3, 10, 30, 100, and 300). Data from Type I neurons were fitted to a single-component function, whereas data from Type II neurons were fitted to a biphasic function. The number of Type I neurons analyzed for each AMPA concentration was as follows: n = 3 (1 μm), n = 3 (3 μm), n = 7 (10 μm), n = 4 (30 μm), n = 16 (100 μm), n = 6 (300 μm). The number of Type II neurons analyzed for each AMPA concentration was as follows: n = 2 (1 μm), n = 2 (3 μm), n = 4 (10 μm), n = 3 (30 μm), n = 9 (100 μm), n = 3 (300 μm). F, Quantification of nicotine-evoked currents in Type I and Type II IPR/IPDM neurons. Cells were voltage-clamped at −60 mV, nicotine (1 and 10 μm) was puff-applied, and mean values are plotted. The number of Type I neurons analyzed for each nicotine concentration was as follows: n = 7 (1 μm), n = 7 (10 μm). The number of Type II neurons analyzed for each nicotine concentration was as follows: n = 7 (1 μm), n = 7 (10 μm). *p < 0.05 (unpaired t test).
MHb neurons express cholinergic and glutamatergic markers, and in optogenetic experiments, appear to use both ACh and glutamate to signal to IPN neurons (Ren et al., 2011). Thus, we reasoned that some IPN neurons likely express and use AMPA-type glutamate receptors (AMPARs). To study AMPARs, we puff-applied AMPA to IPR/IPDM neurons held in voltage-clamp mode similar to our previous study (Engle et al., 2013). As with action potential firing, we found two types of responses to AMPA that cosegregated with action potential firing in response to positive current injection. Type I IPR/IPDM neurons exhibited responses to puffed AMPA (100 μm) that were relatively small (−273.4 ± 30 pA; Fig. 10B, left), with a slower deactivation time constant of 16.7 ± 3 s. By contrast, Type II IPR/IPDM neurons were found to have comparatively larger (−1292.0 ± 116 pA; Fig. 10B, right) responses to AMPA with a much more rapid deactivation time constant of 5.7 ± 1 s. To determine whether these differences in response to AMPA were due to greater potency of AMPA or greater efficacy of AMPA, we studied AMPA concentration-response relations for Type I and Type II IPR/IPDM neurons. The AMPA concentration-response data from Type I neurons were fitted (r2 = 0.60) to a monophasic function (Fig. 10E), with an EC50 of 30.5 μm. Interestingly, AMPA concentration-response data from Type II neurons fit well (r2 = 0.87) to a biphasic function. The higher-sensitivity component had an EC50 of 3.3 μm, whereas the lower-sensitivity component had an EC50 of 53.7 μm. These differences in AMPAR function in Type I and Type II IPR/IPDM neurons suggest that AMPAR expression may be greater in Type II IPR/IPDM neurons or that different AMPAR subtypes are expressed in these two populations.
Our immunohistochemistry data (Fig. 4) suggest a prominent role for MHb-derived, presynaptic nAChRs in IPN. Our data do not, however, rule out a postsynaptic role for some nAChR subtypes in IPN. Several studies suggest a postsynaptic role for nAChRs in IPN (Wada et al., 1989; Hsu et al., 2013). To study the action of nicotine on IPR/IPDM neurons and to further characterize Type I and Type II IPR/IPDM neurons, we studied responses to puffed nicotine. Like firing responses and AMPA-evoked currents, nicotine-evoked currents exhibited a bimodal distribution: Type II neurons responded to 10 μm nicotine with larger-amplitude currents compared with Type I neurons (Fig. 10C,F). This difference was also evident in responses to 1 μm nicotine (Fig. 10F). Together, these recordings in two types of IPR/IPDM neurons reveal interesting differences between these cell types in excitability and coupling to glutamatergic and cholinergic afferents from MHbVL.
Discussion
nAChR expression in MHb to IPN pathway
In this study, we used newly developed nAChR-GFP knock-in mice to localize nAChR subunits in MHb neurons at subcellular resolution. Our data demonstrating localization of α3 and β4 subunits throughout the MHbV area are consistent with previous reports using antibody staining or Western blotting (Yeh et al., 2001; Gahring et al., 2004), radioligand binding (Salas et al., 2004), mRNA in situ hybridization (Wada et al., 1989; Winzer-Serhan and Leslie, 1997; Quik et al., 2000), and transgenic approaches (Frahm et al., 2011). Because most previous attempts to visualize nAChR subunits in MHb or IPN relied on radioligand binding, α3β4* nAChR binding failed to resolve cellular details of subunit location. Although α4 nAChR subunits were previously localized to MHbVL (Fonck et al., 2009), this is the first report demonstrating such a specific pattern for β2, β3, and α6 subunits.
We take particular note that α4, α6, β2, and β3 subunits exhibit a localization “gradient” across both the MHb and IPN. α4 subunits are confined to MHbVL and, in IPN, are localized preferentially in IPR and IPDM (Fig. 11C). Conversely, α6 subunits are confined to MHbVI and exhibit weak but detectable levels in the ventral IPR and in IPC (Fig. 11A). β2 MHbV localization is somewhat similar to α6, being stronger in MHbVI and weaker in MHbVC and MHbVL (Fig. 11A–C). β2 IPN localization is largely uniform from dorsal to ventral but is notably absent from IPR in caudal IPN sections. Last, β3 subunit levels are strong in MHbVI and MHbVC (Fig. 11A,B), but comparatively weaker in MHbVL. In a similar manner, IPN β3 levels are strong, except in IPDM and/or dorsal IPR. A gradient pattern for MHb molecular markers has been seen by other investigators. Aizawa et al. (2012) reported differential expression of markers in subdivisions of MHb, including tachykinin 1, vesicular glutamate transporter 1 and 2, the μ opioid receptor, and the orphan GPCR Gpr151. Because IPN receives dense innervations from MHb via the FR, the patterns seen for α4, α6, β2, and β3 suggest that specific subdivisions of MHb project to specific areas in IPN. Our data support previous observations that MHbVL projects largely to IPR/IPDM, whereas MHbVI projects preferentially to IPC (Contestabile and Flumerfelt, 1981). If nAChR activation in MHbVI and/or MHbVL can control neuronal excitability in these areas, then ligands specific for α4* or α6* nAChRs could specifically control the activity of cells in IPR/IPDM or IPC, respectively.
Figure 11.
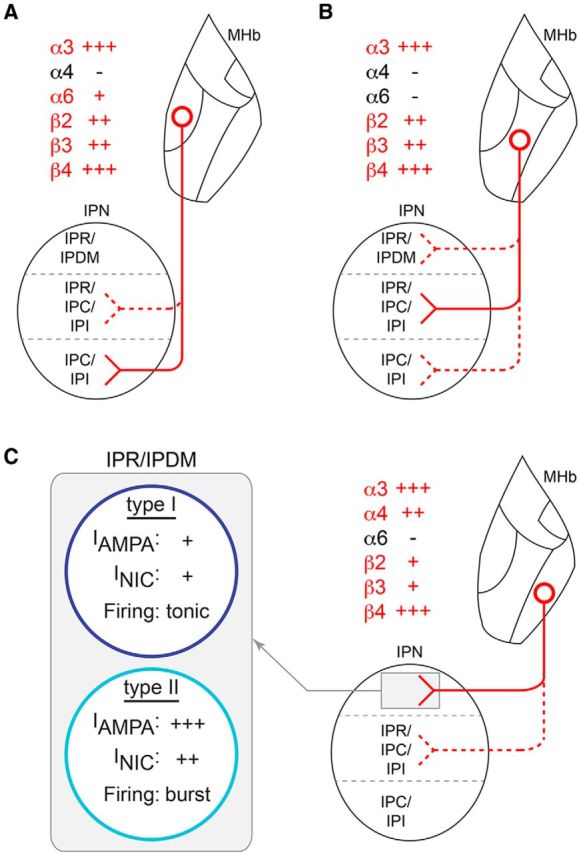
nAChRs and the MHbV to IPN pathway: summary schematic. A, nAChR-expressing cells of the MHbVI project preferentially to ventral IPN, and the indicated (red text) nAChR subunits are expressed in MHb somata and in presynaptic terminals in IPN. B, MHbVC projects preferentially to central regions of IPN. C, MHbVL projects preferentially to IPR/IPDM. At least two cell types exist in IPR/IPDM. Type I neurons exhibit relatively small inward currents in response to puffed nicotine (NIC) and AMPA and fire tonic action potentials in response to positive current injection. Type II neurons exhibit relatively (compared with Type I cells) large inward currents in response to puffed NIC or AMPA and fire bursts of action potentials following positive current injection.
The extremely high density of nAChRs in MHb axons allowed us to visualize the FR. Interestingly, MHb axons exit the MHb at two distinct sites, then merge to form the FR. Micrographs of α3 and β4 MHb localization clearly show axons exiting the MHb from the extreme ventral tip (either MHbVC or MHbVI) as well as in a more lateral direction from MHbVL (Fig. 3). We termed these exit sites the “ventral” and “lateral” exit sites. β2 subunit-containing axons entered the FR primarily via the ventral exit site, whereas α4-containing axons entered the FR from the lateral exit site. β3-containing exons entered the FR through both exit sites, and α6 subunit levels were too weak to clearly visualize any axons. The lateral and ventral exit sites that are defined by nAChR subunit content may correspond to the central and subependymal branches, respectively, of the medial root of the FR (Andres et al., 1999).
The FR undergoes putative excitotoxic degeneration after exposure to high concentrations of nicotine (Carlson et al., 2000, 2001), possibly due to activation of nAChRs that do not strongly desensitize (Labarca et al., 2001). α4β4* nAChRs appear to show reduced desensitization compared with β2* nAChRs (Lax et al., 2002), and in this study we have shown evidence for expression of this subtype in MHbVL neurons. nAChR-GFP mice will be useful in future experiments studying the relationship between nAChR activity and FR degeneration.
Surprisingly, nAChRs were not always seen within ChAT(+) neurons. Anti-ChAT staining has been used as a marker for the MHbV (Fowler et al., 2011; Frahm et al., 2011; Ren et al., 2011), and optogenetics experiments use the ChAT promoter to direct expression of channelrhodopsin and to therefore “control” MHbV activity (Ren et al., 2011; Hu et al., 2012). Most neurons expressing α3, β3, and β4 subunits were ChAT-immunopositive, consistent with their role in regulating ACh release in IPN (Grady et al., 2009). However, α6 and α4 subunits were often found in neurons that were ChAT(−). Future experiments will be required to determine what neurotransmitters these ChAT(−) neurons release and how nAChRs modulate their activity. Because recordings reported in Figures 6, 7, 8, and 9 are from neurons whose ChAT expression status was not studied, data from these studies may represent a pool of both ChAT(+) and ChAT(−) neurons.
nAChR function in MHbV neurons
Consistent with a previous (Kim and Chang, 2005) and a more recent (Görlich et al., 2013) report, we found that some MHb neurons exhibited spontaneous, tonic action potential firing in slice preparations. This tonic firing in MHb neurons is likely the result, at least in part, of expression of HCN channels and hyperpolarization-activated cation current (Ih) (Görlich et al., 2013). Although Ih current amplitudes in MHb neurons are smaller than those seen in SN pars compacta or VTA dopamine neurons (Engle et al., 2013), our analysis suggests an important role for Ih in shaping MHb neuron excitability. This is supported by a recent report, which demonstrated that inhibition of MHb HCN channel activity can elicit behaviorally relevant changes in IPN activity (Görlich et al., 2013). Ih current amplitudes in individual cells were positively correlated with that cell's ability to fire sustained and high-frequency trains of action potentials, and Ih current amplitude was negatively correlated with the latency to anode break firing (Fig. 6). These data suggest that MHb provides a tonic glutamatergic and/or cholinergic signal to IPN neurons (Grady et al., 2009; Ren et al., 2011; Hu et al., 2012).
Surprisingly, neurons in MHbVL, but not MHbVC, were capable of responding to 1 μm nicotine with responses similar in magnitude to those seen in reward-pathway VTA DA neurons at the same concentration of nicotine (Tapper et al., 2004; Drenan et al., 2008b). Furthermore, 1 μm nicotine also augmented MHbVL neuron action potential firing in a manner similar to that seen in VTA DA neurons (Liu et al., 2012), and loss of α4 subunits eliminated this effect (Fig. 8). MHbVC neurons, which are devoid of α4 subunits, did not express nAChRs that were sensitive enough to 1 μm nicotine to elicit inward currents (Fig. 7) or action potential firing (Fig. 8). Although SR16584 is generally considered a selective antagonist for α3β4* nAChRs (Zaveri et al., 2010), it has not been tested on α4β4* nAChRs, whose subunits are found in MHbVL and respond well to 1 μm nicotine (Luetje and Patrick, 1991; Puskar et al., 2011). Our data also suggest that α3β4* nAChRs play a role in the ability of MHbVL to respond to 1 μm nicotine (Fig. 8). Together, these recordings of action potential firing suggest at least two possibilities: (1) separable populations of α3β4* and α4* nAChRs may mediate nicotine's action in accelerating MHbVL neuron firing; or (2) α3α4β4* receptors are at least partially responsible for the increase.
In contrast to α4β2* nAChRs, α4β4* nAChRs are not expected to undergo marked upregulation in response to chronic nicotine (Srinivasan et al., 2011). In knock-in mice expressing YFP-labeled α4 subunits, α4 levels in MHb did not change significantly in response to chronic nicotine (Nashmi et al., 2007). Thus, if α4β4* and/or α3α4β4* nAChRs are expressed at high levels in MHbVL neurons, it would account for the blunted response to nicotine in these cells from nicotine-withdrawn mice (Fig. 9). A recent report corroborated our findings indicating that chronic nicotine reduces MHb neuronal excitability, although without determining which subregions of MHbV are involved (Dao et al., 2014). Together, these data suggest that reexposure of MHbVL neurons to nicotine after nicotine withdrawal may result in reduced firing of these neurons and reduced glutamate and/or ACh release onto IPR/IPDM target neurons. If IPR/IPDM cells receiving innervation from MHbVL neurons are GABAergic, as suggested recently (Hsu et al., 2013; Zhao-Shea et al., 2013), then reexposure to nicotine in nicotine-withdrawn animals could result in less activation of GABAergic IPR/IPDM neurons compared with nicotine exposure in control animals, which could ultimately contribute to relapse because of a reduction in aversive signals generated by the IPN (Fowler et al., 2011). Our withdrawal data highlight the importance of studying subregions of the MHb to IPN pathway, such as the MHbVL, as a recent study that did not restrict its analysis to specific MHbV areas found increased nicotine-evoked firing in neurons from nicotine-withdrawn mice (Görlich et al., 2013). Further, we recently found increased α6 subunit levels in MHbVI of mice treated chronically with nicotine (Henderson et al., 2014).
Our data reporting electrophysiologically distinct neuronal populations in IPR/IPDM (Fig. 10C) are novel and build upon recently published data suggesting the existence of distinct cellular populations in IPN (Hsu et al., 2013; Zhao-Shea et al., 2013). Compared with Type I IPR/IPDM neurons, Type II neurons may couple more strongly to cholinergic/glutamatergic input from MHbVL because of greater function of AMPA receptors (Fig. 10B,E) and a stronger response to nicotine (Fig. 10C,F). Overall, these studies provide new anatomical and mechanistic details regarding nAChR expression and function in the brain's key aversion pathway.
Footnotes
This work was supported by a Brain and Behavior Research Foundation (formerly National Alliance for Research on Schizophrenia and Depression) Young Investigator Award and National Institutes of Health Grant DA030396 to R.M.D., Grant DA028955 to H.A.L. and Michael J. Marks, and Grant NS034407 to Dennis A. Dougherty). We thank Elisha Mackey and Rahul Srinivasan for technical assistance.
The authors declare no competing financial interests.
References
- Aizawa H, Kobayashi M, Tanaka S, Fukai T, Okamoto H. Molecular characterization of the subnuclei in rat habenula. J Comp Neurol. 2012;520:4051–4066. doi: 10.1002/cne.23167. [DOI] [PubMed] [Google Scholar]
- Andres KH, von Düring M, Veh RW. Subnuclear organization of the rat habenular complexes. J Comp Neurol. 1999;407:130–150. doi: 10.1002/(SICI)1096-9861(19990428)407:1<130::AID-CNE10>3.0.CO%3B2-8. [DOI] [PubMed] [Google Scholar]
- Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, Swan GE, Rutter J, Bertelsen S, Fox L, Fugman D, Goate AM, Hinrichs AL, Konvicka K, Martin NG, Montgomery GW, Saccone NL, Saccone SF, Wang JC, Chase GA, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet. 2007;16:24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, et al. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008;165:1163–1171. doi: 10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugaysen J, Bronfeld M, Tischler H, Bar-Gad I, Korngreen A. Electrophysiological characteristics of globus pallidus neurons. PLoS One. 2010;5:e12001. doi: 10.1371/journal.pone.0012001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J, Armstrong B, Switzer RC, 3rd, Ellison G. Selective neurotoxic effects of nicotine on axons in fasciculus retroflexus further support evidence that this is a weak link in brain across multiple drugs of abuse. Neuropharmacology. 2000;39:2792–2798. doi: 10.1016/S0028-3908(00)00141-6. [DOI] [PubMed] [Google Scholar]
- Carlson J, Noguchi K, Ellison G. Nicotine produces selective degeneration in the medial habenula and fasciculus retroflexus. Brain Res. 2001;906:127–134. doi: 10.1016/S0006-8993(01)02570-7. [DOI] [PubMed] [Google Scholar]
- Contestabile A, Flumerfelt BA. Afferent connections of the interpeduncular nucleus and the topographic organization of the habenulo-interpeduncular pathway: an HRP study in the rat. J Comp Neurol. 1981;196:253–270. doi: 10.1002/cne.901960206. [DOI] [PubMed] [Google Scholar]
- Contestabile A, Villani L, Fasolo A, Franzoni MF, Gribaudo L, Oktedalen O, Fonnum F. Topography of cholinergic and substance P pathways in the habenulo-interpeduncular system of the rat: an immunocytochemical and microchemical approach. Neuroscience. 1987;21:253–270. doi: 10.1016/0306-4522(87)90337-X. [DOI] [PubMed] [Google Scholar]
- Dao DQ, Perez EE, Teng Y, Dani JA, De Biasi M. Nicotine enhances excitability of medial habenular neurons via facilitation of neurokinin signaling. J Neurosci. 2014;34:4273–4284. doi: 10.1523/JNEUROSCI.2736-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley-Miller K, Patrick J. Gene transcripts for the nicotinic acetylcholine receptor subunit, β4, are distributed in multiple areas of the rat central nervous system. Brain Res Mol Brain Res. 1992;16:339–344. doi: 10.1016/0169-328X(92)90244-6. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Nashmi R, Imoukhuede P, Just H, McKinney S, Lester HA. Subcellular trafficking, pentameric assembly, and subunit stoichiometry of neuronal nicotinic acetylcholine receptors containing fluorescently labeled α6 and β3 subunits. Mol Pharmacol. 2008a;73:27–41. doi: 10.1124/mol.107.039180. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, Bupp S, Heintz N, McIntosh JM, Bencherif M, Marks MJ, Lester HA. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity α6* nicotinic acetylcholine receptors. Neuron. 2008b;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SE, Broderick HJ, Drenan RM. Local application of drugs to study nicotinic acetylcholine receptor function in mouse brain slices. J Vis Exp. 2012;68:e50034. doi: 10.3791/50034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SE, Shih PY, McIntosh JM, Drenan RM. α4α6β2* nAChR activation on VTA DA neurons is sufficient to stimulate a depolarizing conductance and enhance surface AMPA receptor function. Mol Pharmacol. 2013;84:393–406. doi: 10.1124/mol.113.087346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonck C, Nashmi R, Salas R, Zhou C, Huang Q, De Biasi M, Lester RA, Lester HA. Demonstration of functional α4-containing nicotinic receptors in the medial habenula. Neuropharmacology. 2009;56:247–253. doi: 10.1016/j.neuropharm.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm S, Slimak MA, Ferrarese L, Santos-Torres J, Antolin-Fontes B, Auer S, Filkin S, Pons S, Fontaine JF, Tsetlin V, Maskos U, Ibañez-Tallon I. Aversion to nicotine is regulated by the balanced activity of β4 and α5 nicotinic receptor subunits in the medial habenula. Neuron. 2011;70:522–535. doi: 10.1016/j.neuron.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Ed 3. New York: Elsevier; 2008. [Google Scholar]
- Gahring LC, Persiyanov K, Rogers SW. Neuronal and astrocyte expression of nicotinic receptor subunit β4 in the adult mouse brain. J Comp Neurol. 2004;468:322–333. doi: 10.1002/cne.10942. [DOI] [PubMed] [Google Scholar]
- Görlich A, Antolin-Fontes B, Ables JL, Frahm S, Slimak MA, Dougherty JD, Ibañez-Tallon I. Reexposure to nicotine during withdrawal increases the pacemaking activity of cholinergic habenular neurons. Proc Natl Acad Sci U S A. 2013;110:17077–17082. doi: 10.1073/pnas.1313103110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L, Clementi F, Gotti C. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the α3β4* and α3β3β4* subtypes mediate acetylcholine release. J Neurosci. 2009;29:2272–2282. doi: 10.1523/JNEUROSCI.5121-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BJ, Srinivasan R, Nichols WA, Dilworth CN, Gutierrez DF, Mackey ED, McKinney S, Drenan RM, Richards CI, Lester HA. Nicotine exploits a COPI-mediated process for chaperone-mediated up-regulation of its receptors. J Gen Physiol. 2014;143:51–66. doi: 10.1085/jgp.201311102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YW, Tempest L, Quina LA, Wei AD, Zeng H, Turner EE. Medial habenula output circuit mediated by α5 nicotinic receptor-expressing GABAergic neurons in the interpeduncular nucleus. J Neurosci. 2013;33:18022–18035. doi: 10.1523/JNEUROSCI.2927-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Ren J, Zhang JE, Zhong W, Luo M. Natriuretic peptides block synaptic transmission by activating phosphodiesterase 2A and reducing presynaptic PKA activity. Proc Natl Acad Sci U S A. 2012;109:17681–17686. doi: 10.1073/pnas.1209185109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, Chang SY. Dendritic morphology, local circuitry, and intrinsic electrophysiology of neurons in the rat medial and lateral habenular nuclei of the epithalamus. J Comp Neurol. 2005;483:236–250. doi: 10.1002/cne.20410. [DOI] [PubMed] [Google Scholar]
- Labarca C, Schwarz J, Deshpande P, Schwarz S, Nowak MW, Fonck C, Nashmi R, Kofuji P, Dang H, Shi W, Fidan M, Khakh BS, Chen Z, Bowers BJ, Boulter J, Wehner JM, Lester HA. Point mutant mice with hypersensitive α4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci U S A. 2001;98:2786–2791. doi: 10.1073/pnas.041582598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax P, Fucile S, Eusebi F. Ca(2+) permeability of human heteromeric nAChRs expressed by transfection in human cells. Cell Calcium. 2002;32:53–58. doi: 10.1016/S0143-4160(02)00076-3. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhao-Shea R, McIntosh JM, Gardner PD, Tapper AR. Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing α4 and α6 subunits. Mol Pharmacol. 2012;81:541–548. doi: 10.1124/mol.111.076661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetje CW, Patrick J. Both α- and β-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci. 1991;11:837–845. doi: 10.1523/JNEUROSCI.11-03-00837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey ED, Engle SE, Kim MR, O'Neill HC, Wageman CR, Patzlaff NE, Wang Y, Grady SR, McIntosh JM, Marks MJ, Lester HA, Drenan RM. α6* nicotinic acetylcholine receptor expression and function in a visual salience circuit. J Neurosci. 2012;32:10226–10237. doi: 10.1523/JNEUROSCI.0007-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkawi AH, Al-Ghananeem AM, de Leon J, Crooks PA. Nicotine exposure can be detected in cerebrospinal fluid of active and passive smokers. J Pharm Biomed Anal. 2009;49:129–132. doi: 10.1016/j.jpba.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, Lester HA. Assembly of α4β2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci. 2003;23:11554–11567. doi: 10.1523/JNEUROSCI.23-37-11554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA. Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DC, Xiao Y, Nguyen HN, Musachio JL, Dávila-García MI, Kellar KJ. Measuring nicotinic receptors with characteristics of α4β2, α3β2 and α3β4 subtypes in rat tissues by autoradiography. J Neurochem. 2002;82:468–481. doi: 10.1046/j.1471-4159.2002.00951.x. [DOI] [PubMed] [Google Scholar]
- Puskar NL, Xiu X, Lester HA, Dougherty DA. Two neuronal nicotinic acetylcholine receptors, α4β4 and α7, show differential agonist binding modes. J Biol Chem. 2011;286:14618–14627. doi: 10.1074/jbc.M110.206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Ceballos RM, Kasten M, McIntosh JM, Lester RA. α3β4 subunit-containing nicotinic receptors dominate function in rat medial habenula neurons. Neuropharmacology. 1999;38:769–783. doi: 10.1016/S0028-3908(99)00024-6. [DOI] [PubMed] [Google Scholar]
- Quik M, Polonskaya Y, Gillespie A, Jakowec M, Lloyd GK, Langston JW. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J Comp Neurol. 2000;425:58–69. doi: 10.1002/1096-9861(20000911)425:1<58::AID-CNE6>3.0.CO%3B2-X. [DOI] [PubMed] [Google Scholar]
- Ren J, Qin C, Hu F, Tan J, Qiu L, Zhao S, Feng G, Luo M. Habenula “cholinergic” neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron. 2011;69:445–452. doi: 10.1016/j.neuron.2010.12.038. [DOI] [PubMed] [Google Scholar]
- Saccone NL, Saccone SF, Hinrichs AL, Stitzel JA, Duan W, Pergadia ML, Agrawal A, Breslau N, Grucza RA, Hatsukami D, Johnson EO, Madden PA, Swan GE, Wang JC, Goate AM, Rice JP, Bierut LJ. Multiple distinct risk loci for nicotine dependence identified by dense coverage of the complete family of nicotinic receptor subunit (CHRN) genes. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:453–466. doi: 10.1002/ajmg.b.30828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, Swan GE, Goate AM, Rutter J, Bertelsen S, Fox L, Fugman D, Martin NG, Montgomery GW, Wang JC, Ballinger DG, et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Pieri F, De Biasi M. Decreased signs of nicotine withdrawal in mice null for the β4 nicotinic acetylcholine receptor subunit. J Neurosci. 2004;24:10035–10039. doi: 10.1523/JNEUROSCI.1939-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Salminen O, Drapeau JA, McIntosh JM, Collins AC, Marks MJ, Grady SR. Pharmacology of α-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol Pharmacol. 2007;71:1563–1571. doi: 10.1124/mol.106.031492. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Pantoja R, Moss FJ, Mackey ED, Son CD, Miwa J, Lester HA. Nicotine up-regulates α4β2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J Gen Physiol. 2011;137:59–79. doi: 10.1085/jgp.201010532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW. Distribution of α2, α3, α4, and β2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol. 1989;284:314–335. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM. Codistribution of nicotinic acetylcholine receptor subunit α3 and β4 mRNAs during rat brain development. J Comp Neurol. 1997;386:540–554. doi: 10.1002/(SICI)1096-9861(19971006)386:4<540::AID-CNE2>3.0.CO%3B2-2. [DOI] [PubMed] [Google Scholar]
- Xiao C, Nashmi R, McKinney S, Cai H, McIntosh JM, Lester HA. Chronic nicotine selectively enhances α4β2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J Neurosci. 2009;29:12428–12439. doi: 10.1523/JNEUROSCI.2939-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Srinivasan R, Drenan RM, Mackey ED, McIntosh JM, Lester HA. Characterizing functional α6β2 nicotinic acetylcholine receptors in vitro: mutant β2 subunits improve membrane expression, and fluorescent proteins reveal responsive cells. Biochem Pharmacol. 2011;82:852–861. doi: 10.1016/j.bcp.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JJ, Yasuda RP, Dávila-García MI, Xiao Y, Ebert S, Gupta T, Kellar KJ, Wolfe BB. Neuronal nicotinic acetylcholine receptor α3 subunit protein in rat brain and sympathetic ganglion measured using a subunit-specific antibody: regional and ontogenic expression. J Neurochem. 2001;77:336–346. doi: 10.1046/j.1471-4159.2001.t01-1-00259.x. [DOI] [PubMed] [Google Scholar]
- Zaveri N, Jiang F, Olsen C, Polgar W, Toll L. Novel α3β4 nicotinic acetylcholine receptor-selective ligands: discovery, structure-activity studies, and pharmacological evaluation. J Med Chem. 2010;53:8187–8191. doi: 10.1021/jm1006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao-Shea R, Liu L, Pang X, Gardner PD, Tapper AR. Activation of GABAergic neurons in the interpeduncular nucleus triggers physical nicotine withdrawal symptoms. Curr Biol. 2013;23:2327–2335. doi: 10.1016/j.cub.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]