

Abstract
Objective
Candesartan cilexetil (CAN) is a poor aqueous soluble compound and a P-glycoprotein (P-gp) efflux pump substrate. These key factors are responsible for its incomplete intestinal absorption.
Methods
In this study, we investigated to enhance the absorption of CAN by improving its solubility and inhibiting intestinal P-gp activity. A phase solubility method was used to evaluate the aqueous solubility of CAN in PVP K30 (0.2–2%). Gibbs free energy values were all negative. Solubility was enhanced by the freeze drying technique. The in vitro dissolution was evaluated using the USP paddle method. The interaction between drug and carrier was evaluated by Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD) and Differential scanning calorimetry (DSC) studies. Naringin was selected as P-gp inhibitor. Absorption studies were performed using the everted gut sac model from rat jejunum. The drug analysis was performed by HPLC.
Results
FTIR spectra revealed no interaction between drug and PVP K30. From XRD and DSC data, CAN was in the amorphous form, which explains the cumulative release of drug from its prepared systems. We noticed an enhancement of CAN absorption by improving its solubility and inhibiting the P-gp activity. The significant results (p < 0.05) were obtained for freeze dried solid dispersions in the presence of P-gp inhibitor than without naringin (15 mg/kg) with an absorption enhancement of 8-fold.
Conclusion
Naringin, a natural flavonoid, has no undesirable side effects. Therefore, it could be employed as an excipient in the form of solid dispersions to increase CAN intestinal absorption and its oral bioavailability.
Keywords: Candesartan cilexetil, Naringin, Flavonoid, P-gp inhibitor, Intestinal absorption
1. Introduction
Candesartan cilexetil (CAN) is an antihypertensive angiotensin II receptor blocker, is rapidly and completely biaoactivated by ester hydrolysis at the ester link to form the active candesartan during absorption from the gastrointestinal (GI) tract McClellan and Goa, 1998. Oral administration of CAN shows low bioavailability, approximately 15% in humans, due to its low water (pKa 6.0) solubility (Vijaykumar et al., 2009) and efflux by drug resistance pumps in the gastrointestinal tract, limiting the oral absorption (Zhang et al., 2012, Lee et al., 2009, Kamiyama et al., 2010, Zhou et al., 2009). To overcome these problems, a solid dispersion approach can be utilized to increase the permeability (Shaikh and Avachat, 2011) and the dissolution rate of highly lipophilic drugs thereby improving their bioavailability (Arias et al., 1994, Zerrouk et al., 2001, Palmieri et al., 2002, Lee et al., 2005). Usually, solid dispersions are two-component systems in which the drug is incorporated in the hydrophilic carrier. The drug within the hydrophilic carrier may be dispersed molecularly or occur as amorphous components. The improved dissolution of the drug can be attributed to (i) enhanced solubility owing to its amorphous state or nano-particles (Kelvin’s law) Yonemochi et al., 1997, Yonemochi et al., 1999, Mura et al., 2002, Dai et al., 2007 (ii) increased surface area of nano particles of the drug for drug dissolution (Kubo and Mizobe, 1997, Purvis et al., 2006) and (iii) improved wetting caused by the hydrophilic carrier (Kawashima et al., 1975, Chow et al., 1995). Among all hydrophilic carriers, Polyvinylpyrrolidone (PVP) is the most often studied hydrophilic polymeric carriers (Janssens et al., 2008, Shah et al., 2009, Gupta et al., 2004).
Interaction of various sartans with PVP K30 was investigated in solution and in solid state. For example, solid dispersions with PVP K30 were found to improve the solubility and enhance the dissolution rate of valsartan, compared with β-CD and hydroxypropyl β-CD inclusion complexes. Freeze-dried valsartan/PVP K30 (1:5 M ratio) solid systems offer rapid dissolution profiles in comparison with the profiles of valsartan and its physical mixture with PVP K30 (Mahapatra et al., 2011). The effect of water-soluble polymers (PEG 4000 or PVP K-90) as a third component on the complexation of irbesartan with β-CD was investigated. The study revealed that the binding and the solubility were enhanced in the presence of the third component (PVP K-90 at 5%) in comparison with the binary system of irbesartan and β-CD (Rajashree et al., 2009).
Intestinal absorption is another key factor for the bioavailability of oral dosage forms (Chillistone and Hardman, 2011). It is well-known that influx and efflux transporters such as P-glycoprotein (P-gp) expressed on the membrane of epithelial intestinal cells have a substantial impact on drug absorption. Influx transporters facilitate drug absorption, whereas efflux transporters prevent the drug absorption (Scherrmann, 2009).
P-gp alters intestinal permeation of hydrophobic compounds by averting the influx into cells and facilitates drug efflux from intestinal cells back into the lumen. Inhibiting P-gp may improve drug absorption across intestinal barriers (Ishikawa et al., 2004, Linardi and Natalini, 2006, Kim, 2002). P-gp inhibitors act as high avidity substrates (e.g. verapamil, quinidine) or block its function by binding to it (e.g. sulfhydryl-substituted purine) (Föger, 2009).
These two key factors (poor water solubility and P-gp efflux pumps) are well known for incomplete absorption of orally administered drugs and thus limit their bioavailability (Streubel et al., 2006, Dahan and Amidon, 2009).
Therefore; still it is a real problem to improve the intestinal absorption and the oral bioavailability of candesartan cilexetil though Nekkanti et al. and Gao et al. reported that the oral bioavailability of candesartan was improved about 1.5-fold and 10-fold by nanosuspension (Nekkanti et al., 2009) and nanoemulsion (Gao et al., 2011) respectively until today. However, no reports showed the use of naringin as a pharmaceutical excipient in the presence of candesartan solid dispersions to increase candesartan intestinal absorption and therefore its oral bioavailability till today.
This study was intended to investigate the influence of the manufacturing process used to prepare glassy materials on the physicochemical properties of the products. Parameters of particular importance were product homogeneity, dissolution, physical stability, and drug/polymer interactions. The following study constraints were applied to allow comparisons to be made between manufacturing techniques. Only one polymer, poly(vinylpyrrolidone) K30 (PVP), was selected for use with all three manufacturing techniques. PVP was selected as it has high aqueous solubility, a high glass transition temperature, is miscible with a range of poorly soluble drugs, and forms hydrogen bonds with certain drugs (Forster et al., 2001). Hydrogen bonding is widely recognized as an important mechanism to increase amorphous stability (Matsumoto and Zografi, 1999). All products were prepared at a drug/PVP ratio of 1:2 (w/w) as this was suitable for processing using all three manufacturing techniques. The poorly soluble compound selected was candesartan cilexetil (Patterson et al., 2005). Therefore, in this study an attempt was made to enhance candesartan cilexetil intestinal absorption by improving its solubility as solid dispersions with hydrophilic carrier (PVP K30) using different pharmaceutical interventions and inhibiting the intestinal efflux transporters, P-gp using naringin. Permeability studies were conducted using the in vitro everted sac technique.
2. Materials and methods
2.1. Materials
Candesartan cilexetil (purity more than 99%, melting temperature = 163 ± 0.5 °C) was kindly donated as a generous gift by Dr. Reddy’s Labs, (Hyderabad, India). Polyvinyl pyrrolidone K30 (PVP-K30) was supplied by SD Fine Chem. Ltd., Mumbai, India. Naringin, Na2HPO4, KH2PO4, NaOH, KCl, citric acid NaCl, CaCl2, MgCl2 and NaHCO3 were obtained from Sigma Aldrich (Hyderabad, India). Other chemicals used were of analytical grade. All drug solutions were freshly prepared before use.
2.2. Phase solubility study
Phase solubility studies were performed as described by Higuchi and Connors (Higuchi and Connors, 1965). Pure drug (CAN), in amounts that exceeded its solubility, was added to a sequence of stoppered conical flasks with 25 ml of aqueous PVP-K30 solutions of various concentrations (0.2–2%) in double distilled water and shaken for 48 h on a rotary flask shaker. After reaching equilibrium, the samples were filtered through a 0.45 μm filter paper, suitably diluted with double distilled water and assayed for the drug content at λmax of 255 nm using a UV spectrophotometer (Shimazdu-1601, Shimazdu Corp, Kyoto, Japan) against a blank prepared using the same concentration of PVP-K30 in double distilled water. All assays were carried out at room temperature in triplicate.
2.3. Buffer Solution phosphate pH 6.8
77.3 ml of disodium phosphate R (71.5 g/l) was mixed with 22.7 ml of citric acid solution R (21 g/l).
2.4. Buffer solution phosphate pH 7.4
250 ml of potassium phosphate was added to 393.4 ml of NaOH (0.1 M).
2.5. Preparation of physical mixture and solid dispersion
2.5.1. Physical mixtures
Physical mixtures of candesartan cilexetil (1:2 w/w) were prepared by pulverization using a sample mill (SK-M2 model, Tokyo, Japan) with polyvinyl pyrrolidone K30 as a carrier and screened through a 60 mesh sieve. The physical mixtures containing drug to carrier proportions (1:2 w/w) were mixed manually with the appropriate amount of pure drug and carrier in a ceramic bowl using a polymeric spatula until a homogeneous mixture was obtained. The samples were screened through a 60 mesh sieve and then stored in a desiccator until further study. Optical microscopy and sieve analysis indicated that all the physical mixtures had similar particle sizes (mean particle size 150 and 250 μm diameter, n = 50).
2.5.2. Kneading method
The mixture of candesartan cilexetil (4 g) and PVP-K30 (8 g) was wetted (in portions) with water (4 ml) and kneaded thoroughly for 30 min in a glass mortar. The homogeneous paste, so formed was dried in an oven at 40 °C vacuum for 24 h. Dried powder was screened through a 60 mesh sieve and stored in a desiccator until further study. Optical microscopy and sieve analysis indicated that all the solid dispersions had similar particle sizes (mean particle size was between 150 and 250 μm diameter, n = 50).
2.5.3. Spray drying method (SPD)
The solid dispersions of CAN were prepared by evaporation of a hydro-alcoholic solution of CAN and PVP K30 in optimized ratio (1:2 w/w) using a spray dryer (Buchi B-191, India). The solution was prepared by dissolving 1 g of CAN in 50 ml of methanol and 2 g of PVP K30 in 100 ml of distilled water and mixing both solutions, which produce a clear solution. The solvent was evaporated at the following sets of conditions: air flow rate at 500 l/h, under an atomization pressure of 2 kg/cm2 with a solution flow rate of 4 mL/min. The inlet temperature: 120 °C and outlet temperature 90 °C. The spray-dried products were dried in a vacuum oven over a desiccant at 40 °C for 24 h. The product thus obtained was screened through a 60 mesh sieve, doubly wrapped in aluminum foil and stored in a desiccator until further use. Optical microscopy and sieve analysis indicated that all the solid dispersions had similar particle sizes (mean particle size was between 150 and 250 μm diameter, n = 50).
2.5.4. Freeze-drying method (FD)
Physical mixtures of drug (1:2 w/w, 20 g) and PVP-K30 (40 g) were added to 500 ml of double distilled water and placed on a mechanical shaker for 5 days. The resultant solution was prepared by dissolving 1 g of CAN in 50 ml of methanol and 2 g of PVP K30 in 100 ml of distilled water and mixing both solutions, which produce a clear solution. The solvent was then evaporated in vacuum at 50 °C with a rotary evaporator. The samples thus obtained were ground and sieved through a (<38 μm) mesh. Solid samples (0.5 g) were introduced in 20 ml vials and loaded on a freeze-dryer (SMART LYO® Freeze Dryers). After thermal equilibration, the shelf temperature was lowered to −40 °C. The product was maintained at this temperature for 2 h. Then, the system was evacuated to a pressure of 30 torr and the shelf temperature was adjusted to −35 °C and held for 4 h. Afterward, the shelf temperature was raised successively to −20 °C (8 h), then 0 °C (8 h) and finally 20 °C (2 h). After drying the samples (5–6 mg), the vials were capped within 5 min and stored at room temperature (22–24 °C) in a desiccator containing silica gel. The sample was in the form of amorphous flakes prepared by freeze-drying and hence the particle size was not determined.
2.6. Evaluation of solid dispersions and physical mixtures
2.6.1. Drug content determination
Drug: PVP-K30 solid dispersions and physical mixtures equivalent to 10 mg of drug (CAN) in 10 ml of methanol, were filtered using a 0.45 μm filter paper, suitably diluted with phosphate buffer (pH 6.8) and the drug content determined using the calibration curve of pure drug in methanol by a UV spectrophotometer (V-530, JASCO, Japan) at 255 nm against phosphate buffer as blank. Experiments were done in triplicate.
2.6.2. Saturation solubility studies
A saturation solubility study was carried out to determine the increase in the solubility of pure drug compared with the physical mixture and solid dispersions. Excess amount of pure drug (CAN) physical mixtures and solid dispersions were added to 10 ml of phosphate buffer (pH 6.8) in a 50 ml volumetric flask. Samples were stirred at 100 rpm temperature (37 °C) in a controlled water bath for 48 h. Samples were then filtered through 0.45 μm filters, suitably diluted and analyzed spectrophotometrically at 255 nm respectively. Experiments were done in triplicate.
2.7. Characterization of CAN: PVP-K30 (1:2) solid dispersions
2.7.1. Fourier Transform Infrared (FT-IR)
FT-IR is a useful tool to identify drug excipient interaction. The spectra were obtained using a Fourier Transform Infrared spectrometer (FTIR-8400, Shimadzu Corporation, Japan) with a data station. About 2–3 mg of the sample was mixed with dry potassium bromide (KBr) and the samples were scanned from 4000 to 400 cm–1 with the resolution of 4 cm–1.
2.7.2. X-ray powder diffraction (XRPD)
XRPD analyses were performed for PVP-K30–pure drug (CAN) physical mixtures and PVP-K30–pure drug (CAN) solid dispersions using an X-ray diffractometer (PW 1729, Philips, Netherlands). The samples were irradiated with monochromatized Cu Kα radiation (1.5406 Å) and analyzed between 5° and 50° 2θ. Voltage and current used were 40 kV and 40 mA respectively. The range and the chart speed were 2 × 103 CPS and 10 mm/degree 2θ, respectively.
2.7.3. Differential scanning calorimetry (DSC)
Differential scanning calorimetry (DSC) has been a widely used calorimetric tool to study the solid state interaction of drug with PVP-K30. Modulated temperature DSC equipped with an intracooler (DSC 60 Shimazdu, Japan) was used. Indium/zinc standards were used to calibrate the temperature and enthalpy scale. The samples were heated in pierced aluminum pans at a rate of 10 °C/min over a temperature range of 30–300 °C. Inert atmosphere was maintained by purging nitrogen gas at a flow rate of 40 ml/min.
2.7.4. Dissolution study
Dissolution studies were performed using a USP II Paddle dissolution apparatus (TDT-06P, Electrolab, India). To avoid aggregation of powder in contact with the dissolution medium, samples equivalent to 10 mg of pure drug (CAN) were mixed with 10 g glass beads, dispersed in 5 ml dissolution medium and then were placed in 900 ml phosphate buffer (pH 6.8) maintained at 37 ± 0.5 °C and stirred at 50 rpm. Samples were collected periodically at 2, 4, 6, 8, 10, 15, 20, 25, and 30 min, filtered through a 0.45 μm filter paper and replaced with a fresh dissolution medium. The concentrations were analyzed spectrophotometrically at 255 nm. Drug release studies were carried out in triplicate.
2.7.5. Preparation of the everted gut sacs
Freeze-dried solid dispersions of candesartan cilexetil (1:2 w/w) were selected for the intestinal absorption study. The experiments were carried out on male Sprague–Dawley rats (250–270 g; 10–15-week-old) purchased from the National Institute of Nutrition (NIN), Hyderabad. They were housed in large polypropylene cages with a 12-h light/dark cycle with free access to water and maintained on commercial feed obtained locally. After overnight fasting, the rats were anesthetized with ether and the small intestine was isolated and rinsed with Tyrode buffer solution (containing in mM: 15 glucose, 11.90 NaHCO3, 136.9 NaCl, 4.2 NaH2PO4, 2.7 KCl, 1.2 CaCl2 and 0.5 MgCl2) at room temperature and immediately placed at 37 °C in an oxygenated (95% O2 and 5% CO2) Tyrode solution.
Briefly, the intestinal segment was stripped off the adhering tissue, washed with Tyrode solution and cut into small sacs (3–4 cm in length). Each segment was gently everted over a glass rod and filled with 0.5 ml of Tyrode buffer solution (pH 7.4) using a 1-ml plastic syringe and tied with a silk suture at one end. The other end was connected to a small cannula using silk suture. The rats were finally sacrificed with overexposure to ether. The sacs were placed in 15 ml degassed oxygenated Tyrode solution in an oscillating water bath at 37 °C in 20 ml capacity test tubes. One ml each of four samples (each equivalent to 1 mg/ml of CAN in Tyrode solution) – control (pure drug), pure drug with naringin (15 mg/kg), and freeze-dried binary systems (1:2 w/w) with and without naringin (15 mg/kg) were placed in respective test tubes to ensure full exposure of the whole sac to the drug and maintained at 37 °C using a shaking water bath. At the end of each time point i.e. 15, 30, 45, 60, 75, 90, 105 and 120 min, the corresponding everted sacs were removed from the test tube, rinsed with Tyrode solution three times, blotted dry and weighed. Then, the contents of the sacs were collected and the sacs were weighed again to adjust for the content volume. The experiments were repeated three times (n = 3) and drug absorption was expressed as a percentage.
All the animal experiments were performed in accordance with the guidelines established by the Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA, New Delhi) and approved by the Institutional Animal Ethics Committee of Vaagdevi Institute of Pharmaceutical Sciences, Warangal with registration number 1533/PO/a/11/CPCSEA dated 7th April, 2012.
2.8. Analytical method validation
RP-HPLC method was used to analyze the CAN samples in phosphate buffer (pH 7.4). Validation was carried out for precision, linearity, specificity, accuracy, limit of quantification and ruggedness according to US Pharmacopoeia (FDA, 1997) and International Conference on Harmonization (ICH) guideline ICH (Q2R1), 2007.
2.8.1. HPLC sample analysis
The concentrations of CAN inside and outside the everted gut sac were assayed using a Shimadzu HPLC system (LC – 20A series) equipped with a UV detector, micro-volume double plunger pump (10 μl/stroke) and manual injector type model 7725-port sample injection valve. The system was controlled through SCL-10Avp or SCL-10A software. Samples from inside or outside the everted gut sac media were withdrawn, filtered through a 0.45μ filter and 20 μl of samples were injected into the HPLC system. HPLC analysis of samples was performed under the following conditions: a Shim-pack VP-ODS or equivalent ODS column (Particle size 5 μm, column dimension: I.D 4.6 mm × length 150 mm) maintained at room temperature; an isocratic mobile phase which consisted of acetonitrile:methanol:water:glacial acetic acid (40:35:25:0.1 v/v/v/v) at pH 6.8 respectively; and a flow rate of 1 ml/min at ambient temperature. The detective wavelength was set at 255 nm. The retention time was 4 min. Quantification of candesartan was made by measuring the peak areas to that of standard peaks under the standard conditions. The analysis was repeated three times (n = 3) to validate the obtained readings.
3. Data analysis
Data analysis was computed using SPSS version 19. The results were represented as mean ± standard deviation (±SD) for all the experiments. The apparent permeability average values (Papp) were compared for each sample using one way analysis of variance (ANOVA) test. Student’s t-test was used to compare drug diffusion data sets. The difference was considered significant at p ⩽ 0.05.
3.1. Phase-solubility studies
The apparent stability constant, Ks was computed from the phase-solubility profiles for drug–carrier ratio as described below:
The values of Gibbs free energy of transfer, , of candesartan cilexetil from aqueous solution of the carrier (PVP-K30) were calculated according to the relationship:
where, So and Ss are the molar solubilities of pure drug in the aqueous solution and the carrier respectively.
3.2. Dissolution efficiency (DE) studies
The drug release profiles were characterized by calculating the dissolution efficiency (DE) of the samples. The dissolution efficiency of a pharmaceutical dosage form is defined as the area under the dissolution curve up to a time, t, expressed as a percentage of the area of the rectangle described by 100% dissolution at the same time. DE can be calculated from the following equation.
where, y is the percent of drug released at time, t.
3.3. Calculation of the apparent permeability coefficients
Apparent permeability coefficient (Papp) was determined according to Eq. (1): (Lassoued et al., 2011)
| (1) |
where Papp (cm/s) is the apparent permeability coefficient, dQ/dt (mg/s) is the amount of drug transported across the membrane per unit time, A (cm2) is the surface area available for permeation and C0 (mg/ml) represents the initial concentration of the drug outside the everted gut sacs. The mean and the standard deviation values of Papp were calculated and expressed in 10−6 cm/s unit.
3.4. Calculation of the percentage of drug recovery and drug retained (Lassoued et al., 2011)
The percentage of drug recovery (R %) was calculated according to Eq. (2):
| (2) |
where Cr,end and Cd,end (mg/ml) are the drug concentrations measured at the end of the experiment inside and outside the sacs respectively; C0 (mg/ml) is the initial concentration of the drug outside the everted gut sacs; Vr and Vd (ml) are the volumes of the mucosal and the serosal media respectively.
The percentage of drug retained (Ad %) on the intestinal tissues was determined according to Eq. (3):
| (3) |
3.5. Water flux measurement
The water flux, resulting from both water absorption and efflux in the jejunum segment was determined according to the following equation: (Park et al., 2011, Khemiss et al., 2009)
where P1 is the weight of the empty and blotted sac, P2 is the weight of the sac + 500 μl Tyrode solution (pH 7.4) and P3 is the weight of the sac at the end of experiment. The water flux was determined after 2 h of experiment and the results were expressed in g water/g fresh intestine. Assays for the evaluation of water flux were repeated six times (n = 6).
4. Results and discussion
4.1. Phase solubility analysis
The phase solubility of candesartan cilexetil in the presence of PVP-K30 is shown in Fig. 1. The apparent stability constant (Ks) was found to be 179.1 m−1. An increase in solubility of the drug was linear (r2 > 0.9) with an increase in concentration of polymer (0.2–2 μg/ml) in water showing an AL type solubility curve. Gibbs free energy change, values were all negative at various PVP-K30 concentrations (0.2–2 μg/ml) as shown in Table 1.
Figure 1.
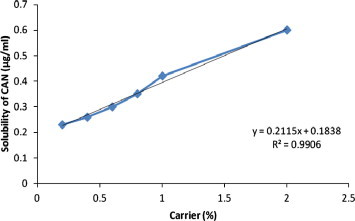
Phase solubility analysis plot.
Table 1.
Gibbs free energy of transfer for solubilization process of CAN in aqueous solutions of PVPK30 at 37 °C.
| Polymer concentration (%w/v) | (kJ/mole) |
|---|---|
| 0.2 | −0.58 |
| 0.4 | −0.91 |
| 0.6 | −1.25 |
| 0.8 | −1.63 |
| 1 | −2.0 |
| 2 | −2.9 |
4.2. Evaluation of CAN:PVP-K30 (1:2) physical mixtures and solid dispersions
4.2.1. Drug content
Preliminary experiments by preparing physical mixtures and solid dispersions using CAN and PVP-K30 in different drug-to-carrier ratios showed maximum solubility with 1:2 drug to polymer ratio. Hence, the ratio of CAN:PVP-K30 (1:2) was selected for the study.
Percentage drug content of physical mixtures and solid dispersions was found within the range of 83–94% as shown in Table 2. The maximum percent of drug content was found to be 93.30 % in the freeze-dried complex.
Table 2.
Percentage drug content and saturation solubility of drug: PVPK30 (1:2).
| Methods of preparation | Drug content (%) | Saturation solubility (μg/ml) | % Increase in solubility |
|---|---|---|---|
| Pure drug | – | 4.56 ± 0.23 | – |
| Physical mixture | 72 ± 0.6 | 17.92 ± 1.05 | 392.98 |
| Kneading | 82 ± 0.53 | 22.3 ± 1.45 | 489.03 |
| Spray drying | 90.2 ± 0.73 | 33.2 ± 1.89 | 728.07 |
| Freeze drying | 93.3 ± 0.65 | 42.5 ± 2.31 | 932.01 |
(Mean ± SD, n = 3).
4.2.2. Saturation solubility study
The saturation solubility of physical mixtures and solid dispersions is presented in Table 2. The freeze-dried solid dispersion showed maximum saturation solubility (42.15 μg/ml).
4.3. Characterization of CAN:PVP-K30 (1:2) solid dispersions and physical mixtures
4.3.1. FT-IR
Fig. 2 illustrates the FT-IR spectra of the samples under study. The spectrum of candesartan cilexetil shows prominent peaks at 2940, 2931, 1715, 1612, 1600, 1347 and 1281 cm−1 corresponding to C–H aromatic stretching, C–H aliphatic stretching, C O stretching in ester, C O stretching in acid, C N aromatic stretching, C C aromatic, C–N and –C–O–ester. The prominent peaks of PVP-K30 are at 2950, 1651, 1512 and 1270 cm−1 corresponding to C–H aliphatic stretching, C O stretching in acid, C stretching and C–N stretching respectively. Physical mixtures of CAN/PVP (Fig. 2C) exhibited spectra corresponding to a superposition of their parent components and a broad peak at 3390 cm−1 (broad band). In the spectra of all other prepared solid dispersion systems (Fig. 2D and F), the characteristic peaks of PVP were present at almost the same positions along with a broad peak at 3390 cm−1. Moreover, all the spectra showed no peaks other than those assigned to CAN and PVP.
Figure 2.

FT-IR spectra of single component and binary systems of CAN and PVPK30, physical mixture, kneading, spray dried and freeze dried. (A) CAN, (B) PVPK30, (C) physical mixture, (D) kneading, (E) spray drying, and (F) freeze drying (top to bottom).
4.3.2. Powder X-ray diffractometry [PXRD]
PXRD patterns of pure drug and CAN:PVP-K30 (1:2) physical mixtures and solid dispersions are shown in Fig. 3. The powder X-ray diffractrograms of pure CAN exhibited characteristic diffraction peaks at a diffraction angle of 2θ at 9.94, 11.48, 14.42, 16.97, 18.93, 21.80, 22.82, 24.73, 27.48, and 29.17. The spectrum of PVP exhibited complete absence of any diffraction peak, which is characteristic of an amorphous compound. The diffractrograms of all prepared systems showed peaks similar to PVP and absence of major diffraction peaks corresponding to CAN. However, the diffractrograms of CAN:PVP-K30 (1:2) freeze dried solid dispersions were found to be more diffuse than other prepared systems showing no characteristic peaks of pure drug.
Figure 3.
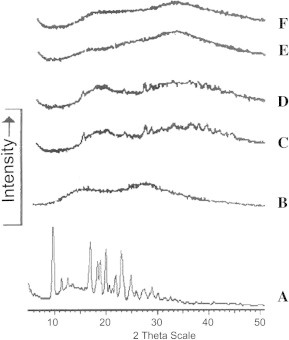
PXRD diffractrograms of single components and binary systems of CAN and PVPK30, physical mixture, kneading, spray dried and freeze dried. (A) CAN, (B) PVPK30, (C) physical mixture, (D) kneading, (E) spray drying, and (F) freeze drying.
4.3.3. Differential scanning calorimetry [DSC]
Thermograms of pure drug and CAN:PVP-K30 (1:2) physical mixtures and solid dispersions are shown in Fig. 4. DSC studies showed the melting endothermic peak for pure CAN at 164.9 °C, whereas the scan of PVP-K30 showed a broad endotherm ranging from 100 to 140 °C due to the presence of residual moisture. The physical mixture thermogram showed an exothermic peak at 210 °C (due to recrystallization) and an endothermic peak corresponding to superposition of their parent components between 100 and 140 °C (Fig. 4C). In the curves of all other prepared systems of solid dispersions with PVP (Fig. 4D and E), a very broad endotherm characteristic of amorphous PVP between 100 and 150 °C was noticed. The thermogram of CAN:PVP-K30 (1:2) freeze dried solid dispersions showed a drastic shift in the endothermic peak of PVP-K30 and complete disappearance of peaks of the pure drug (Fig. 4F).
Figure 4.
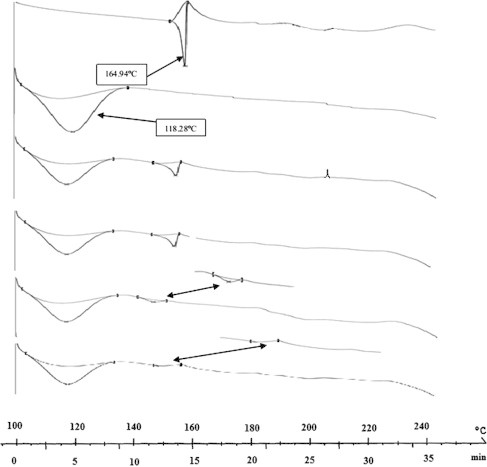
DSC thermograms of single component and binary systems of CAN and PVPK30, physical mixture, kneading, spray dried and freeze dried. (A) CAN, (B) PVPK30, (C) physical mixture, (D) kneading, (E) spray drying, and (F) freeze drying (top to bottom).
4.3.4. In vitro release profile of complexes
Dissolution profiles of pure drug, and all other prepared systems (SDs and PMs) were carried out in phosphate buffer (pH 6.8) as shown in Fig. 5. DP30 min values (percent drug dissolved within 30 min) and t50 % (time to dissolve 50% drug) values for different samples are reported in Table 3. From data presented in Table 3 and Fig. 5, it is evident that the dissolution rate of pure drug is very low. Solid dispersions of CAN with PVPK30 (1:2) by the freeze-drying method significantly enhanced the dissolution rate of CAN (80–85%) as compared to pure drug and all other prepared systems. Percent of drug dissolved at 10 and 30 min was highest in magnitude for solid dispersions prepared by the freeze drying technique than all other prepared systems. Freeze dried binary systems showed maximum enhancement in dissolution rate (DE30 = 80.4%).
Figure 5.
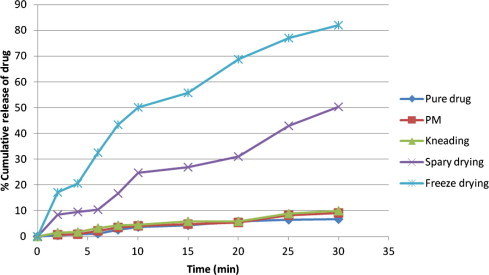
Cumulative percent of drug release from pure drug and prepared systems.
Table 3.
Percent drug dissolved within 30 min (DP30 min), time to dissolve 50% drug (T50%) for pure CAN and its prepared systems.
| Methods of preparation | DP30 min | T50 (min) |
|---|---|---|
| CAN | 6.57 | >30 |
| Physical mixture | 9.09 | >30 |
| Kneading method | 9.72 | >30 |
| Spray drying | 49.5 | 30 |
| Freeze drying | 80.4 | 10 |
4.3.5. Permeability studies
Preliminary experiments of the everted gut sac apparent permeability studies showed the maximum permeability coefficient, Papp (cm/s) for naringin at 15 mg/kg over other concentrations i.e. 5 and 10 mg/kg, respectively. Hence, the highest concentration of naringin (15 mg/kg) was selected for the study. The everted gut sac apparent permeability coefficients, Papp (cm/s) of CAN alone; CAN + naringin (15 mg/kg); CAN–PVP-K30 binary systems (freeze dried solid dispersion) at drug: polymer weight ratio (1:2 w/w) with and without naringin (15 mg/kg) across the everted gut sac are shown in Table 4. Data are expressed as mean ± SD and results are expressed in cm/s.
Table 4.
Apparent permeability coefficients Papp (cm/s) and percentages of drug recovery and drug retention for CAN in the presence of naringin across the everted gut sac model.
| In-vitro absorption model | Sample | Percentage of drug recovery ± S.Da (%) | Percentage of drug retention ± S.Da (%) | Papp (×10−6 cm/s) |
|---|---|---|---|---|
| Everted Sac technique | CAN (control) | 91.35 ± 1.84 | 8.65 ± 1.84 | 0.75 ± 0.08 |
| CAN + Naringin (15 mg/kg) | 91.65 ± 1.63 | 8.35 ± 1.63 | 3.40 ± 0.14 | |
| CAN freeze dried solid dispersions (1:2 w/w) | 93.35 ± 1.72 | 6.65 ± 1.72 | 3.99 ± 2.01 | |
| CAN freeze dried solid dispersions (1:2 w/w) + Naringin (15 mg/kg) | 96.05 ± 1.61 | 3.95 ± 1.61 | 12.40 ± 2.12 |
Data are presented as mean ± SD; (n = 3).
The apparent permeability value (Papp) of CAN was 0.76 ± 0.08 × 10−6 cm/s. Although CAN–PVP-K30 binary systems (Freeze dried solid dispersions) with and without naringin (15 mg/kg) showed a significant increase in apparent permeability coefficients (P < 0.05) when compared with controls (CAN and CAN + naringin). The highest Papp values (12.40 ± 2.12 × 10−6 cm/s) were found with freeze dried solid dispersions in the presence of naringin (15 mg/kg). However, the Papp coefficient of freeze dried solid dispersion (1:2; w/w) in the absence of P-gp inhibitors (naringin) was 3.99 ± 2.01 × 10−6 cm/s (Fig. 6).
Figure 6.
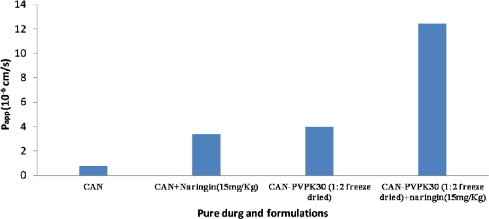
Apparent permeability coefficients Papp (cm/s) of pure drug and prepared systems in the presence of naringin.
4.3.6. Percentage of drug recovery and drug retained determination
The percentages of drug recovery (R %) and drug retention (Ad %) of CAN; CAN + naringin (15 mg/kg); CAN–PVP-K30 binary systems (Freeze dried) at drug: polymer weight ratio 1:2 (w/w) with or without naringin (15 mg/kg) across the everted gut sac model is shown in Table 4. The percentages of drug retention on the intestinal barrier were below 10% for all experiments.
4.3.7. Water flux studies
During all the experiments a transfer of water molecules from outside (mucosal side) to inside (serosal side) the everted gut sacs was noticed, demonstrating absorption expressed in g water/g fresh intestine. A significant increase in water influx (P < 0.05) was aggravated after the addition of naringin (15 mg/kg) in the mucosal medium. Water flux results for CAN–PVP-K30 binary systems were 1.35 ± 0.21 g water/g fresh intestine for freeze dried solid dispersion with naringin as shown in Fig. 7.
Figure 7.
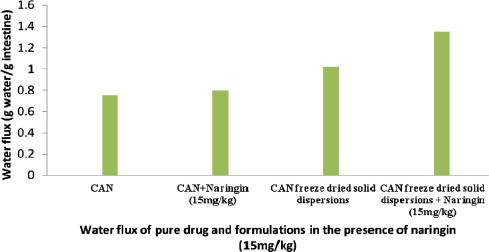
Water flux of pure drug and prepared systems in the presence of naringin.
5. Discussion
In this study, we attempted to enhance the intestinal absorption of candesartan cilexetil by improving its solubility, modulating the physicochemical nature into the amorphous form and inhibiting P-gp efflux transporters. The solubility of candesartan cilexetil increased owing to dispersion of the drug in PVP-K30. A Gibbs free energy change indicated the spontaneous nature of CAN solubilization demonstrating that the reaction was favorable with increase in the concentration of PVP-K30. This may probably be because of soluble complexes between water-soluble polymeric carrier and poorly soluble drug. The improved solubility is attributed to the wettability of PVP-K30 forming an intermolecular hydrogen bond between CAN and PVP-K30 and a decrease of the interfacial tension between the drug and the aqueous medium. The freeze-dried solid dispersion showed maximum amount of drug content and saturation solubility than other formulations due to amorphous nature of the drug in PVP-K30. Thus, enhancement of solubility for CAN in 1:2 ratio was 10.1-fold at 37 °C by PVPK30. The chemical interaction between drug and PVP-K30 offers insights into the changes in the infrared profiles of dispersions. It was noted that the acid carbonyl stretching bend of CAN shifted from 1715 cm−1 toward lower frequencies (1713 cm−1) which might be attributed to intermolecular hydrogen bonding between CAN and >N– and C O of PVP-K30. Moreover, the absorption band at 3390 cm−1 was broadened which attributed to the presence of water confirming the broad endotherm detected in the DSC experiments. Thus, IR spectra did not reveal dramatic changes in characteristic peaks of CAN molecule dispersed into the matrices of PVP-K30 suggesting the absence of well defined chemical interactions between the drug and the polymer. The assessment of degree of crystallinity of the sample is done by Powder X-ray diffraction spectroscopy. The complete dispersion of drug and PVP-K30 reduces the number of crystalline structures or appearance of halo at the baseline indicating amorphousness of the drug in the given sample. The diffractrograms were found to be more diffuse compared to the pure drug without characteristic peaks indicating the formation of amorphous solid state. Thus, the final product sample (freeze dried) demonstrated fewer and diffuse peaks, signifying an increase in solubility and drug release. DSC enables quantitative endothermic or exothermic phase transformations of drug in the given sample demonstrating complete dispersion of pure drug within the matrices of PVP-K30. The characteristic features of CAN peak were lost. This indicated that CAN was no longer present as a crystalline material, but was converted into the amorphous state. DSC thermogram of solid dispersions (Fig. 4) presented a straight line with absence of any thermodynamic transitions. In DSC thermogram of a freed dried prepared system, no distinct glass transition (Tg) was evident till 290 °C (Fig. 4F). In the amorphous form, there was no indication of Tg in DSC suggesting that CAN is a good glass former and requires more sensitive techniques to determine its Tg. The absence of melting peak clearly indicated the existence of the amorphous state of drug, which was also confirmed by XRPD (Fig. 3) showing a halo, characteristic of amorphous form. The DSC scan of all formulations shows a broad melting endotherm between 100 and 140 °C indicating loss of water from PVP-K30 followed by diminishing or absence of a drug peak. Thus, DSC studies confirmed that the drug is present as a solid solution inside the PVP-K30 matrix of freeze dried product. Extent of solubility and dissolution was found to show a relationship with particle size of binary prepared systems. It was seen that the particle size range (150 and 250 μm) for physical mixtures and solid dispersions prepared by kneading method and spray drying were responsible for lower dissolution. However, powder dissolution of spray and freeze dried prepared systems shows improved dissolution over physical mixtures and kneading method, because the highly water soluble PVP-dispersion of the drug may have resulted in rapid dissolution of spray dried solid dispersions. Nevertheless, freeze dried prepared samples contain a lower particle size <38 μm diameter and amorphous flakes and hence shows faster dissolution. The increase in drug release in freeze dried solid dispersions is due to the dispersion effect of the hydrophilic carrier and lowering of surface tension of the dissolution medium (Galia et al., 1999). Freeze dried solid dispersion presented dramatic improvements in the rate as well as the extent of drug release (Fig. 5) and presented highest drug release (80% drug released in 30 min). In the early 5 min period, there was about 5-fold increase of drug release from freeze dried solid dispersion compared to pure CAN. This improved drug release attributed to the presence of amorphous form of CAN, as confirmed by DSC and PXRD studies. Other factors such as increased wettability, solubilization of the drug by the carrier at the diffusion layer, and the reduction or absence of aggregation and agglomeration increased the rate of dissolution.
Based on all of the above findings, the possible mechanisms for an increased release rate from solid dispersions have been postulated and are summarized as follows: (i) reduction of crystallinity (ii) increased water absorption by the hydrophilic carrier which leads to increased wettability (iii) solubilization effect by the carrier and (iv) phase transition from crystalline to amorphous nature.
Candesartan cilexetil absorption assessment was performed with the in vitro everted gut sac model prepared from rat jejunum. (Bohets et al., 2001, Wang et al., 2007) The technique is a simple and economical method. It is an efficient tool to assess P-gp’s role in the intestinal absorption of drug compounds. The validity of this technique can be explained by histological studies. Histological slides observed under light microscopy after 120 min of experiment showed that the intestine remained intact morphologically with microvillosities and intact brush border (Lassoued et al., 2011). We investigated the effect of naringin, a natural flavonoid in grape fruit on candesartan absorption at 15 mg/kg. Permeability studies were undertaken for pure CAN; CAN + naringin and CAN–PVP-K30 binary systems (Freeze dried product) in the absence or presence of P-gp inhibitors (naringin). We noted a significant (P < 0.05) enhancement of CAN permeability compared with pure drug alone (13.87-fold). This enormous increase may have been attributed to the solubility improvement of CAN in the amorphous form as solid dispersions (Weuts et al., 2003). The addition of naringin in the mucosal medium significantly (P < 0.05) increased the permeability values by 14.99-fold of the drug in freeze dried solid dispersions compared to the permeability values of the drug in freeze dried solid dispersions without naringin in the mucosal medium. This could be attributed to the inhibitory effect of naringin on the P-gp efflux transporter indicating that candesartan may be a substrate for P-gp5 and the intestinal cytochrome P450 (CYP) 3A4 isoenzyme activity (Choi and Han, 2005).
In this study, it is indicated that the intestinal absorption of freed dried solid dispersion was better than that of pure drug alone. There was significant difference in intestinal absorption between solid dispersion and pure drug (p < 0.05) and insignificant difference between pure drug and pure drug + naringin (p > 0.05). Therefore, it is inferred that CAN is dispersed in the carrier greatly and was ready to absorb from the intestinal medium. The improvement of intestinal absorption was attributed to the physical characteristics of CAN in solid dispersion, but not because of the simple effect of excipients (PVP K30) Cheng et al., 2010.
In this study, we also demonstrated the water flux for CAN alone; CAN + naringin and CAN–PVP-K30 binary systems in the presence and absence of P-gp inhibitors (naringin). We found that there was a transfer of water from the outside to the inside of the sacs in the presence of P-gp inhibitor, which translated as complete absorption than in the absence of naringin. The comparison at the end of the assays (after 120 min) showed that the water flux was greater for CAN–PVP-K30 freeze dried solid dispersions (P < 0.05) with P-gp inhibitor than without P-gp inhibitor. This could be explained by the fact that the absorption of CAN might be resulted by the inhibition of intestinal efflux transporter P-gp as previously reported for intestinal absorption of digoxin concerning the role of P-gp where the water absorption increased significantly in the duodenum and the jejunum with verapamil, a P-gp inhibitor by Sababi et al. (2001). During permeation experiments undertaken with the everted gut sac model, it was important to assess the percentages of CAN recovery (R %) outside and inside the sacs and the percentage of CAN retained (Ad %) on the intestinal tissue. The results showed that the values for R% were above 90%, indicating that the amount of drug retained by the everted gut sac tissues was very less. This phenomenon may have been due to the accumulation of drug into the muscular layer as reported previously (Park et al., 2011) during the passage from mucosal to serosal side on the everted gut sac.
6. Conclusion
Solubility, dissolution and permeability are the fundamental triad governing the oral drug absorption and their enhancement remains the most challenging job for pharmaceutical scientists. Low drug solubility manifests many in vivo limitations like incomplete release from the dosage form, poor bioavailability, increased food effect and higher inter-patient variability. CAN being a lipophilic molecule, practically insoluble in water and a substrate of P-glycoprotein, could potentially demonstrate solubility and permeability limited bioavailability. Therefore, high energy amorphous forms as freeze dried solid dispersions may be useful for such lipophilic drugs where solubility is dependent on hydrophilic forces of a carrier, and disruption of crystal lattice may result in enhanced solubility. Therefore, the current study suggests that on enhancement of aqueous solubility with high energy amorphous forms and inhibition of efflux transporters using naringin, which are capable of mediating the absorption of CAN, the oral bioavailability can be enhanced. Therefore, naringin could be used as a pharmaceutical excipient in candesartan cilexetil solid dispersions to increase its intestinal absorption and oral bioavailability.
7. Declaration of interest
The authors report no declaration of interest.
Footnotes
Peer review under responsibility of King Saud University.

Contributor Information
S. Gurunath, Email: s.gurunath1979@gmail.com.
Baswaraj K. Nanjwade, Email: bknanjwade@yahoo.co.in.
References
- Arias M.J., Gines J.M., Moyano J.R., Rabasco A.M. The application of solid dispersion technique with D-mannitol to the improvement in oral absorption of triamterene. J. Drug Target. 1994;2:45–51. doi: 10.3109/10611869409015892. [DOI] [PubMed] [Google Scholar]
- Bohets H., et al. Strategies for absorption screening in drug discovery and development. Curr. Top. Med. Chem. 2001;1:367–383. doi: 10.2174/1568026013394886. [DOI] [PubMed] [Google Scholar]
- Cheng Juan, Hong Wu Zheng, Neng Ping Qineng, Wang Bogang, Lu Junjun. The absorption characteristics of bifendate solid dispersion in rat intestinal tissue. Drug Dev. Ind. Pharm. 2010;36(3):283–291. doi: 10.1080/03639040903140571. [DOI] [PubMed] [Google Scholar]
- Chillistone S., Hardman J. Factors affecting drug absorption and distribution. Anaesth. Intensive Care Med. 2011;12:151–155. [Google Scholar]
- Choi J.S., Han H.K. Enhanced oral exposure of diltiazem by the concomitant use of naringin in rats. Int. J. Pharm. 2005;305:122–128. doi: 10.1016/j.ijpharm.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Chow A.H.L., Hsia C.K., Gordon J.D., Young J.W.M., Vargha-Butler E.I. Assessment of wettability and its relationship to the intrinsic dissolution rate of doped phenytoin crystals. Int. J. Pharm. 1995;126:21–28. [Google Scholar]
- Dahan A., Amidon G.L. Small intestinal efflux mediated by MRP2 and BCRP shifts sulfasalazine intestinal permeability from high to low, enabling its colonic targeting. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;297:G371–G377. doi: 10.1152/ajpgi.00102.2009. [DOI] [PubMed] [Google Scholar]
- Dai W.G., Dong L.C., Song Y.Q. Nanosizing of a drug/carrageenan complex to increase solubility and dissolution rate. Int. J. Pharm. 2007;342:201–207. doi: 10.1016/j.ijpharm.2007.04.032. [DOI] [PubMed] [Google Scholar]
- FDA Guidance for industry: dissolution testing of immediate release solid oral dosage forms. US Department of Health and Human Services, Centre for Drug Evaluation and Research CDER. 1997:1–11. [Google Scholar]
- Föger F. In: Oral Delivery of Macromolecular Drugs: Barriers, Strategies and Future Trends. Bernkop-Schnürch A., editor. Springer Science and Business Media LLC; New York: 2009. Strategies to overcome efflux pumps; pp. 123–136. [Google Scholar]
- Forster A., Hempenstall J., Tucker I., Rades T. Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis. Int. J. Pharm. 2001;226:147–161. doi: 10.1016/s0378-5173(01)00801-8. [DOI] [PubMed] [Google Scholar]
- Galia E., Horton J., Dressman J.B. Albendazole generics—a comparative in vitro study. Pharm. Res. 1999;16:1871–1875. doi: 10.1023/a:1018907527253. [DOI] [PubMed] [Google Scholar]
- Gao Fang, Zhang Zhiwen, Bu Huihui, Huang Yan, Gao Zhiwei, Shen Jianan, Zhao Chunjie, Li Yaping. Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: performance and mechanism. J. Control. Release. 2011;149:168–174. doi: 10.1016/j.jconrel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Gupta P., Kakumanu V.K., Bansal A.K. Stability and solubility of celecoxib-PVP amorphous dispersions: a molecular perspective. Pharm. Res. 2004;21:1762–1769. doi: 10.1023/b:pham.0000045226.42859.b8. [DOI] [PubMed] [Google Scholar]
- Higuchi T., Connors K.A. Phase solubility techniques. Adv. Anal. Chem. Instrum. 1965;4:117–121. [Google Scholar]
- ICH (Q2R1), 2007 (4). Guideline on validation of analytical procedures: text and methodology. In: Proceedings of the International Conference on Harmonization of technical requirements for registration of pharmaceuticals for human use, Geneva.
- Ishikawa T., et al. Functional evaluation of ABCB1 (P-glycoprotein) polymorphisms: high-speed screening and structure–activity relationship analyses. Drug Metab. Pharmacokinet. 2004;19:1–14. doi: 10.2133/dmpk.19.1. [DOI] [PubMed] [Google Scholar]
- Janssens S., et al. Formulation and characterization of ternary solid dispersions made up of Itraconazole and two excipients, TPGS 1000 and PVPVA 64, that were selected based on a supersaturation screening study. Eur. J. Pharm. Biopharm. 2008;69:158–166. doi: 10.1016/j.ejpb.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Kamiyama E., Nakai D., Mikkaichi T., Okudaira N., Okazaki O. Interaction of angiotensin II type 1 receptor blockers with P-gp substrates in Caco-2 cells and hMDR1-expressing membranes. Life Sci. 2010;86(1–2):52–58. doi: 10.1016/j.lfs.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Kawashima Y., Saito M., Takenaka H. Improvement of solubility and dissolution rate of poorly water-soluble salicylic acid by a spray-drying technique. J. Pharm. Pharmacol. 1975;27:1–5. doi: 10.1111/j.2042-7158.1975.tb09369.x. [DOI] [PubMed] [Google Scholar]
- Khemiss F., et al. Effect of in vitro exposure to Vibrio vulnificus on hydro-electrolytic transport and structural changes of sea bream (Sparus aurata L.) intestine. Fish Physiol. Biochem. 2009;35:541–549. doi: 10.1007/s10695-008-9265-7. [DOI] [PubMed] [Google Scholar]
- Kim R.B. Drugs as P-glycoprotein substrates, inhibitors and inducers. Drug Metab. Rev. 2002;34:47–54. doi: 10.1081/dmr-120001389. [DOI] [PubMed] [Google Scholar]
- Kubo H., Mizobe M. Improvement of dissolution rate and oral bioavailability of a sparingly water-soluble drug, (+/−)-5-[[2-(2-naphthalenylmethyl)-5- benzoxazolyl]-methyl] – 2, 4-thiazolidinedione, in co-ground mixture with D-mannitol. Biol. Pharm. Bull. 1997;20:460–463. doi: 10.1248/bpb.20.460. [DOI] [PubMed] [Google Scholar]
- Lassoued M.A., et al. Comparative study of two in vitro methods for assessing drug absorption: sartorius SM 16750 apparatus versus everted gut sac. J. Pharm. Pharm. Sci. 2011;14:117–127. doi: 10.18433/j3gc7r. [DOI] [PubMed] [Google Scholar]
- Lee S., Nam K., Kim M.S., Jun S.W., Park J.S., Woo J.S., Hwang S.J. Preparation and characterization of solid dispersions of itraconazole by using aerosol solvent extraction system for improvement in drug solubility and bioavailability. Arch. Pharm. Res. 2005;28:866–874. doi: 10.1007/BF02977355. [DOI] [PubMed] [Google Scholar]
- Lee B.S., Kang M.J., Choi W.S., Choi Y.B., Kim H.S., Lee S.K., Lee J., Choi Y.W. Solubilized formulation of olmesartan medoxomil for enhancing oral bioavailability. Arch. Pharm. Res. 2009;32(11):1629–1635. doi: 10.1007/s12272-009-2117-x. [DOI] [PubMed] [Google Scholar]
- Linardi R.L., Natalini C.C. Multi-drug resistance (MDR1) gene and P-glycoprotein influence on pharmacokinetic and pharmacodynamic of therapeutic drugs. Cien. Rural. 2006;36:336–341. [Google Scholar]
- Mahapatra Anjan K., Murthy P.N., Biswal Sudarsan, Mahapatra Abikesh P.K., Pradhan Siba P. Dissolution enhancement and physicochemical characterization of valsartan in solid dispersions with β-CD, HP β-CD, and PVP K-30. Dissolution Technol. 2011;18(1):39–45. [Google Scholar]
- Matsumoto T., Zografi G. Physical properties of solid molecular dispersions of indomethacin with poly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-vinyl-acetate) in relation to indomethacin crystallization. Pharm. Res. 1999;16:1722–1728. doi: 10.1023/a:1018906132279. [DOI] [PubMed] [Google Scholar]
- McClellan K.J., Goa K.L. Candesartan cilexetil: a review of its use in essential hypertension. Drugs. 1998;56(5):847–869. doi: 10.2165/00003495-199856050-00013. [DOI] [PubMed] [Google Scholar]
- Mura P., Cirri M., Faucci M.T., Gines-Dorado J.M., Bettinetti G.P. Investigation of the effects of grinding and co-grinding on physicochemical properties of glisentide. J. Pharm. Biomed. Anal. 2002;30:227–237. doi: 10.1016/s0731-7085(02)00252-2. [DOI] [PubMed] [Google Scholar]
- Nekkanti V., Pillai R., Venkateshwarlu V., Harisudhan T. Development and characterization of solid oral dosage form incorporating candesartan nanoparticles. Pharm. Dev. Technol. 2009;14(3):290–298. doi: 10.1080/10837450802585278. [DOI] [PubMed] [Google Scholar]
- Palmieri G.F., Cantalamessa F., Di Martino P., Nasuti C., Martelli S., Lonidamine Solid dispersions: in vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2002;28:1241–1250. doi: 10.1081/ddc-120015357. [DOI] [PubMed] [Google Scholar]
- Park K.J., et al. Clinical, pharmacokinetic and pharmacogenetic determinants of clopidogrel resistance in Korean patients with acute coronary syndrome. Korean J. Lab. Med. 2011;31:91–94. doi: 10.3343/kjlm.2011.31.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson J., James M., Forster A., Lancaster R., Butler J., Rades T. The influence of thermal and mechanical preparative techniques on the amorphous state of four poorly soluble compounds. J. Pharm. Sci. 2005;94:1998–2012. doi: 10.1002/jps.20424. [DOI] [PubMed] [Google Scholar]
- Purvis T., Vaughn J.M., Rogers T.L., Chen X., Overhoff K.A., Sinswat P., Hu J., McConville J.T., Johnston K.P., Williams R.O. Cryogenic liquids, nanoparticles, and microencapsulation. Int. J. Pharm. 2006;324:43–50. doi: 10.1016/j.ijpharm.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Rajashree S.H., Suneeta N.S., Vilasrao J.K. Studies on the effect of water-soluble polymers on drug-beta-cyclodextrin complex solubility. AAPS PharmSciTech. 2009;10:858–863. doi: 10.1208/s12249-009-9274-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sababi M., et al. The role of P-glycoprotein in limiting intestinal regional absorption of digoxin in rats. Eur. J. Pharm. Sci. 2001;14:21–27. doi: 10.1016/s0928-0987(01)00161-0. [DOI] [PubMed] [Google Scholar]
- Scherrmann J.M. Transporters in absorption, distribution and elimination. Chem. Biodivers. 2009;6:1933–1942. doi: 10.1002/cbdv.200900171. [DOI] [PubMed] [Google Scholar]
- Shah J., Vasanti S., Anroop B., Vyas H. Enhancement of dissolution rate of valdecoxib by solid dispersions technique with PVP K 30 & PEG 4000: preparation and in vitro evaluation. J. Incl. Phenom. Macrocycl. Chem. 2009;63:69–75. [Google Scholar]
- Shaikh S.M., Avachat A.M. Enhancement of solubility and permeability of candesartan cilexetil by using different pharmaceutical interventions. Curr. Drug Deliv. 2011;8(4):346–353. doi: 10.2174/156720111795767997. [DOI] [PubMed] [Google Scholar]
- Streubel A., et al. Drug delivery to the upper small intestine window using gastroretentive technologies. Curr. Opin. Pharmacol. 2006;6:501–508. doi: 10.1016/j.coph.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Vijaykumar N., Raviraj P., Venkateshwarlu V., Harisudhan T. Development and characterization of solid oral dosage form incorporating candesartan cilexetil. Pharm. Dev. Technol. 2009;14(3):290–298. doi: 10.1080/10837450802585278. [DOI] [PubMed] [Google Scholar]
- Wang J., et al. Maximising use of in vitro ADMET tools to predict in vivo bio-availability and safety. Expert Opin. Drug Metab. Toxicol. 2007;3:641–665. doi: 10.1517/17425255.3.5.641. [DOI] [PubMed] [Google Scholar]
- Weuts I., Kempen D., Six K., Peeter J., Verreck G., Brewster M., Van den Mooter G. Evaluation of different calorimetric methods to determine the glass transition temperature and molecular mobility below Tg for amorphous drugs. Int. J. Pharm. 2003;259:17–25. doi: 10.1016/s0378-5173(03)00233-3. [DOI] [PubMed] [Google Scholar]
- Yonemochi E., Ueno Y., Ohmae T., Oguchi T., Nakajima S., Yamamoto K. Evaluation of amorphous ursodeoxycholic acid by thermal methods. Pharm. Res. 1997;14:798–803. doi: 10.1023/a:1012114825513. [DOI] [PubMed] [Google Scholar]
- Yonemochi E., Kitahara S., Maeda S., Yamamura S., Oguchi T., Yamamoto K. Physicochemical properties of amorphous clarithromycin obtained by grinding and spray drying. Eur. J. Pharm. Sci. 1999;7:331–338. doi: 10.1016/s0928-0987(98)00040-2. [DOI] [PubMed] [Google Scholar]
- Zerrouk N., Chemtob C., Arnaud P., Toscani S., Dugue J. In vitro and in vivo evaluation of carbamazepine-PEG 6000 solid dispersions. Int. J. Pharm. 2001;225:49–62. doi: 10.1016/s0378-5173(01)00741-4. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Gao F., Bu H., Xiao J., Li Y. Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: in vitro characteristics and absorption mechanism in rats. Nanomedicine. 2012;8(5):740–747. doi: 10.1016/j.nano.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Zhou L., Chen X., Gu Y., Liang J. Transport characteristics of candesartan in human intestinal Caco-2 cell line. Biopharm. Drug Dispos. 2009;30(5):259–264. doi: 10.1002/bdd.664. [DOI] [PubMed] [Google Scholar]