

Abstract
Non-communicable diseases such as cancer, atherosclerosis and diabetes are responsible for major social and health burden as millions of people are dying every year. Out of which, atherosclerosis is the leading cause of deaths worldwide. The lipid abnormality is one of the major modifiable risk factors for atherosclerosis. Both genetic and environmental components are associated with the development of atherosclerotic plaques. Immune and inflammatory mediators have a complex role in the initiation and progression of atherosclerosis. Understanding of all these processes will help to invent a range of new biomarkers and novel treatment modalities targeting various cellular events in acute and chronic inflammation that are accountable for atherosclerosis. Several biochemical pathways, receptors and enzymes are involved in the development of atherosclerosis that would be possible targets for improving strategies for disease diagnosis and management. Earlier anti-inflammatory or lipid-lowering treatments could be useful for alleviating morbidity and mortality of atherosclerotic cardiovascular diseases. However, novel drug targets like endoglin receptor, PPARα, squalene synthase, thyroid hormone analogues, scavenger receptor and thyroid hormone analogues are more powerful to control the process of atherosclerosis. Therefore, the review briefly focuses on different novel targets that act at the starting stage of the plaque form to the thrombus formation in the atherosclerosis.
Keywords: Atherosclerosis, Hyperlipidemia, Drug targets, Endoglin, Heat shock protein
1. Introduction
Cancer, diabetes, obesity, myocardial infarction, hyperlipidemia, chronic respiratory diseases and cardiovascular disease (CVD) like atherosclerosis are the major non-communicable diseases (NCD). These NCD are the leading causes of death worldwide in not only high-income population but in the middle and low group (Margaret, 2010). Atherosclerosis is the chronic inflammatory disease which overall increases the morbidity and the mortality rate (John, 2000).The development of the level of atherosclerosis from an early fatty streak lesion to a highly hazardous rupture-prone plaque is because of the many cellular and molecular events at each level hence, it is the inflammatory event (Robert, 2005). These fatty streaks are one of the signs of atherosclerosis, which is first observed by Russell Holman and this fatty streak develops, thrombosis or hemorrhage (Brian et al., 2012).
A persistent increase in circulating low-density lipoprotein (LDL) levels in the body is one of the most important causes for the initiation and progression of atherosclerosis. In this respect, macrophages play a very important role by increasing accumulation of lipids in blood vessels, leading to inflammation and plaque formation. There are two different theories proposed to describe the events involved in atherogenesis. The first hypothesis is “response to injury,” in which endothelial lining’s injury initiates events of deposition of LDL in the intimal space, followed by recruitments and migration of monocyte-derived macrophages, which take up modified LDL and become foam cells (John, 2000; Ross, 1993; Ross et al., 1977). The second hypothesis is “response to retention”; LDL is deposited in the intimal space and undergoes modification. Modified LDL serves as a chemoattractant for monocytes and macrophages. Macrophages remove modified LDL via scavenger receptors and become foamy (Williams and Tabas, 1995). In brief, LDL accumulates within the intimal space and subsequently undergoes modification such as oxidation, converting LDL into oxidized LDL, which acts as a major immunogen (Wilensky and Hamamdzic, 2007). Oxidation of LDL in the intimal space is still not known exactly; however, lipoxygenases, myeloperoxidase, inducible nitric oxide synthase and NADPH oxidases have LDL oxidation capacity (Li and Glass, 2002).The activated endothelial cells (EC) enhance the expression of adhesion molecules, such as intracellular adhesion molecule, vascular cell adhesion molecule, E- and P-selectin. Adhesion molecules have a major role in monocyte recruitment on the arterial endothelium. P-selectin binds to P-selectin glycoprotein ligand-1, which is present on monocytes (John, 2005; Elstad et al., 1995). P-selectin glycoprotein ligand-1 allows the capturing, rolling and activation of monocytes (Weyrich et al., 1995). P and E-selectin has similar functions in atherosclerosis progression (Chandak et al., 2011). VCAM-1 is another important molecule in monocyte recruitment and firm adhesion to the endothelial surface. Very late antigen-4 expressed by monocytes is a ligand for vascular cell adhesion molecule -1. Intracellular adhesion molecule has a role in monocyte adhesion, spreading and migration into the sub-endothelial space (Chandak et al., 2011). Monocytes enter the endothelial space and differentiate into macrophages by macrophage colony-stimulating factor secreted by EC and smooth muscle cells (SMCs). Activated macrophages in the sub-endothelial space secrete macrophage-chemoattractant protein-1. EC induces the expression of macrophage-chemoattractant protein-1 in the presence of oxidized phospholipids, which are components of oxidized LDL (Li and Glass, 2002). Macrophages remove modified LDL via scavenger receptor A, scavenger receptor B1 and cluster of differentiation 36. After internalization, endosomes are transferring LDL to lysosomes. After lysosomal degradation of LDL, free cholesterol is produced, which is re-esterified into cholesteryl esters via acyl-CoA: cholesterol acyltransferase1 (ACAT1). Accumulation of lipid-laden macrophages (foam cells) in the arterial wall is the hallmark of atherosclerosis. The atherosclerosis is the major complication of diabetes mellitus because many studies found that the diabetic patients have 2–5 times the death rate than non-diabetic patients (Gerrity and Antonoy, 1997). Atherosclerosis progression is characterized by development of plaque on the insides of arteries, which later hardens and narrows the arteries, leads to reduced supply of oxygen-rich blood to organs and other parts of the body. This can lead to various serious cardiovascular complications like heart attack, stroke, or even death (Paolo et al., 1995). Fig. 1 shows the development of atherosclerotic-plaque in the coronary artery. Management of adverse cardiovascular outcomes of atherosclerosis through risk factor identification and modification has been an active area of research over the past few decades. As a result, several large scale randomized clinical trials and observational studies have shown intensive risk reduction therapy to be very effective and critical in reducing adverse cardiovascular outcomes in patients with atherosclerosis (Mervi et al., 2012).
Figure 1.
Shows the atherosclerosis-plaque develops in the coronary arteries.
2. Epidemiology
In 1997, the latest year for which statistics are available, CVD claimed 953,110 lives in the United States presenting 41% of all deaths. CVD is the foremost source of morbidity and mortality in the western world. The risk of cardiovascular diseases is wicked because these diseases are derived per year than the later seven primary cause of death combined. Under the age of 65–70 approximately 1/6 population was killed by CVD of which 30% were men and 24% were women and 41% were men, and 40% were women in the black population. The World Health Organization predicts that continued global economic prosperity will likely lead to an epidemic of cardiovascular disease as developing countries are continuously acquiring Western habits which are the principal problem over the world. Hence, CVD is the major health problem that will only have wider effects over the resultant decades (John, 2000).
The class of cardiovascular diseases mainly includes NCD and is the cause of 57 million global deaths in 2008, 36 million or 63% happened, and predominantly NCD includes, atherosclerosis, diabetes, cancers and chronic respiratory diseases (Anthony, 2005). Low and middle-income regions show nearly 80% of the burden of diseases like cardiovascular disease, cancer, diabetes and chronic respiratory diseases (Margaret, 2010). Hypertension is one of the few recognized determinants of atherosclerosis. Various expressions of the atherosclerotic vascular disease are a leading cause of death and disability in industrialized countries. Arterial hypertension is found in 65 million Americans and over 1 billion individuals worldwide (John, 2005). Based on epidemiological studies of atherosclerosis using autopsy material in five European cities, which were carried out with an interval of 25 years in the 1960s and 1980s, respectively, it is revealed that atherosclerosis in young so-called practically healthy people, 20–39 years of age, who died from accidental causes, closely reflects the level of atherosclerosis in the population as a whole (Valentin and Nils, 2004).
Recently, China reports that the more effect of LDL and triglycerides (TG) shows age related specific value. The major risk of atherosclerosis increased 16–36% from 1950 to 1987 due to the increased dietary fat in Taiwan (Wen-Harn and Benjam, 1995).The self- regulating risk factor for atherosclerosis is hyperhomocysteinemia. It is found that the updated epidemic impact of cardiovascular disease in India is very high that is the incidence rate is almost 80–120 per 1000 populations. European population shows more levels of homocysteine as compared to Asian Indians which was believed to cause twice as many coronary heart disease (CHD) deaths. This indicates that homocysteine was an independent risk factor in Asian Indians, which probably contributed to the increased CHD risk (Sainani et al., 2008). In 1990–1991 Chin-Shan Community Cardiovascular Cohort (CCCC) study reported that the postmenopausal women showed higher atherogenic lipids than in premenopausal women. The postmenopausal increases were particularly significant in total cholesterol (TC) and low density lipoprotein cholesterol (LDL-C) among women aged 45–49 years, and in TG and Apo B among women aged 50–54 years (Pao-Ling et al., 2002).
Each year in Australia approximately 48,000 ‘major coronary events’ occur, equivalent to about 130 events per day of these events, about 40% are fatal. With medical advances worldwide, the number of people surviving an acute cardiac event and therefore, living with established CHD, has been greater than before (Barbara et al., 2011).The primary mortality and the disability are adjusted in Europe and are raised in developing countries due to atherosclerosis of the arterial vessel wall and to thrombosis formation. Indirect and direct healthcare cost in the European Union, the economic cost of CVD represents annually 192 billion. In the United States, it is established as 5.4 million, individuals have been diagnosed with diabetes and around 2.4 million are without diagnosis. Death rates among adults, atherosclerosis with diabetes are 2–4 times higher than for those without diabetes. It is observed that approximately 14,000,000 people in the United States have atherosclerotic coronary disease as a major complication of diabetes mellitus (John, 2000; Jonathan et al., 2012).
3. Risk factors
The aspect of the risk related to a specific disease in a behavior that is sturdily and constantly associated with that disease is defined as a risk factor. Atherogenesis is a multifactorial and complex progression which makes it difficult to clearly define the risk attributable to any one risk factor process. For clear understanding of a risk factor, a trait must be causally linked to a particular disease in a manner that is strongly and consistently associated with that disease. Obesity is clearly associated with an increased risk of atherosclerosis; however, this association seems to be mediated in large part by abnormalities in diabetes, hypertension, and lipoproteins which are all known risk factors for atherosclerosis (Sandra and Frank, 2011).The purpose of discussion is to focus on established risk factors and their contribution in the development of atherosclerosis.
3.1. Cigarette smoking
The relation of the cigarette smoking and heart disease was observed from 1940s which shows the relationship between smoking and heart diseases. Surgeon General update report showed that smoking increases atherosclerotic disease by more than 50% and doubles the incidence of coronary heart disease (John, 2000). Smoking increases arterial wall stiffness (resulting in reduced distensibility and compliance), increases fibrinogen levels, increases platelet aggregation, decreases high-density lipoprotein cholesterol levels, and increases hematocrit (John, 2000; Sarah and Bruce, 2009; David, 2007). Smoking causes an inflammatory state with high concentrations of C-reactive protein and fibrinogen, and it has long been recognized as a risk factor for cardiovascular disease (John, 2000). Genetic factors may influence whether smokers develop carotid, coronary and peripheral vascular atherosclerosis. Different genetic variants of the paraoxonase-1, which codes for an enzyme with antioxidant properties, are associated with greater risk for atherosclerosis in smokers than in nonsmokers (Bradford and Thomas, 2002). Exposure to cigarette smoke significantly up-regulates EPHX2 gene and protein expression in the vasculature, suggesting a possible role of this gene for its deleterious effects (Qi et al., 2007). The cessation of smoking declines stroke risk associated with former smoking to almost that of risk level of nonsmokers at 2–5 years from cessation (John, 2000; Sarah and Bruce, 2009; Bradford and Thomas, 2002; Steve and Laleh, 2004).
3.2. Hypertension
Atherosclerosis is one of the major contributing factors which may influence the course of the atherosclerosis (Sarah and Bruce, 2009; Steve and Laleh, 2004). Hypertension and atherosclerosis are intimately linked by the EC which forms at the interior surface of the vascular system. This lumen-vascular wall boundary function as a semi permeable membrane, maintains nonthrombogenic blood-tissue interface (by regulating thrombosis, thrombolysis, and platelet adherence), modulates vascular tone and blood flow, and metabolizes hormones. Also it regulates immune and inflammatory reactions, modify lipoproteins in the arterial wall, and regulate the growth of other cell types, particularly smooth muscle cell reactions. Shearing forces associated with wide arterial pulse pressures serve to activate the endothelium to induce genetic expression like procoagulant, inflammatory, and other responses that ultimately prove harmful to the endothelium and vessel wall. Chronic and repetitive endothelial injury promotes intimal thickening and a shift from the contractile phenotype to the proliferative-synthetic phenotype. Alteration of shear stress during hypertension, such as elevated pulse pressures, and stimuli from additional risk factors combine to increase endothelial permeability. Lipids accumulate within the intima at sites of increased endothelial permeability and endure various changes like oxidative modification of LDL by free radicals, formation of foam cells, increased monocyte adhesion. Oxidized LDL further stimulates the release of growth factors and cytokines, which is cytotoxic to EC and smooth muscle cells, and is immunogenic, inducing antibodies to oxidized lipoproteins. These events cumulate in thin-walled atherosclerotic plaques containing foam cells and activated macrophages. The hemodynamic shearing force of hypertension ruptures the vulnerable plaque. A ruptured plaque leads to atherothrombosis, occlusion of the lumen (John, 2005).
3.3. Diabetes mellitus
Type 2 diabetes patients are frequently prone to atherosclerotic complications, which account for more than 80% of all diabetic mortality (Sainani et al., 2008). Dyslipidemia characterized by elevated small dense LDL levels, lowered high density lipoproteins (HDL) levels, and elevated TG levels is associated with coronary heart diseases (Sarah and Bruce, 2009). Type 2 diabetes also promotes formation of coronary calcification, which is a potential marker for subclinical atherosclerosis (Bradford and Thomas, 2002). Thus, diabetes represents a major causative risk factor to atherosclerosis in the developed world (Anthony et al., 2005).
3.4. Serum cholesterol
The relation between cholesterol and atherosclerosis is explicit naturally. Elevated serum cholesterol levels are associated with higher atherosclerotic stroke risk (Sarah and Bruce, 2009). Cholesterol is an essential compound for the synthesis of bile acid, many hormones such as testosterone, estrogen, dihydroepiandrosterone, progesterone, cortisol and required to produce vitamin D. Plaques are formed by LDL cholesterol and oxidized LDL deposits in arterial walls, which later grows and rupture, leading to artery-blocking by formation of small blood clots (Anthony, 2005). It has been identified that multiple loci, polymorphisms of the paraoxonase-1, mutations of the endothelial constitutive nitric oxide synthase influence serum LDL cholesterol level (Bradford and Thomas, 2002).
The hypercholesterolemia is a disorder which affects approximately one in 500 persons from the general population. Heterozygote hypercholesterolemia disorder contains apparently 2–3-fold elevated plasma cholesterol whereas homozygotes have 4 ± 6-fold elevation in plasma cholesterol due to functional defects in the LDL receptor that interfere with LDL clearance. This leads to myocardial infarction by the age of 60 in heterozygotes and this age is reduced to 15 in patients who are homozygous (John, 2000).
Fig. 2 outlines the contribution of several etiological factors such as hypertension, elevated oxidized low density lipoprotein level, hyperglycemia, cigarette smoking, and homocysteine in the endothelial injury which further leads to initiation of atherosclerosis because of upregulation of genes and activation of macrophages and T-lymphocytes and increases lipid deposition. The reduced lumen diameter is the outcome of events occurred in intraluminal space, subendothelial matrix and smooth muscle cells during the progress of atherosclerotic-plaque.
Figure 2.
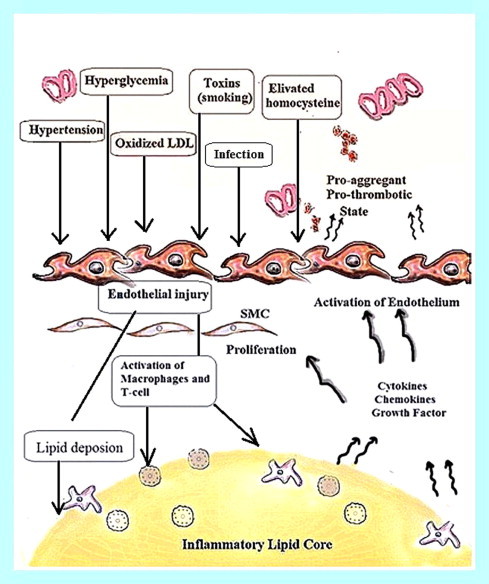
Shows endothelial injury progression for atherosclerosis initiation model. Several risk factors, such as hypertension, elevated oxidized low density lipoprotein level, hyperglycemia, cigarette smoking, and homocysteine promote atherogenesis through upregulation of genes and activation of macrophages and T-lymphocytes and increase lipid deposition. The top of the figure represents the intraluminal space, and the bottom represents the subendothelial matrix, and SMC.
3.5. Newer risk factors
Homocysteine is a highly reactive amino acid exhibiting toxicity for vascular endothelium and enhances auto-oxidation of LDL. Hyperhomocysteinemia is a potential risk factor for atherosclerosis, which interacts with other vascular risk factors and has demonstrable genetic and dietary determinants. Polymorphisms of 5,10-methylene tetrahydrofolate reductase is associated with atherosclerosis (Steve and Laleh, 2004). Augmented serum uric acid (SUA) (Markus et al., 2012) has been associated with coronary artery diseases (CAD), coronary calcification, carotid atherosclerotic plaques, increased carotid intima-media thickness, arterial stiffness and with the metabolic syndrome (Mervi et al., 2012). Modified malondialdehyde and oxidized LDL have been playing an increasing role in atherosclerotic lesions. Elevated titers of autoimmune antibodies specific for malondialdehyde (MDA)-modified LDL predicted the progression of carotid atherosclerosis (Paul and Desire, 1998). Patients with urinary stone disease should be at higher risk for other vascular complications and atherosclerosis is one of them (Alexander et al., 2011). Genetic risk factors related to the angiotensin converting enzyme (ACE), are also identified as increasing the risk of atherosclerosis (Pieter et al., 2001). Porphyromonas gingivalis, a major periodonto-pathogen, is associated with progression of atherosclerosis. P. gingivalis can accelerate atheroma deposition by inflammatory response, which exacerbates vascular inflammation via secreted cytokines and/or chemokines that ultimately modulate early atherogenesis that spontaneously develop atherosclerosis. Pathogens can upregulate the expression of innate immune receptors on the surfaces of both endothelial cells and macrophages (Qian et al., 2009). The recent update found that the intracellular bacterial pathogen Chlamydia pneumoniae causes respiratory tract infection and has been associated with atherosclerosis and coronary artery disease. C. pneumoniae is involved in all stages of atherosclerosis including initiation, inflammation, fibrous plaque formation, plaque rupture and thrombosis (James and Brian, 2001). Low level of ascorbic acid is a major risk factor and indicator of atherosclerotic CVD, including hypertension and elevated concentrations of LDL, acute phase proteins, and hemostatic factors (Sagar et al., 2012). Increased platelet action in the body can increase the atherosclerosis risk and plaque calcification also increases the risk of atherosclerosis (Amala et al., 2012). Leptin may contribute to the development of classic risk factors of atherosclerosis such as arterial hypertension and diabetes mellitus (Jerzy, 2006).
4. New drug targets of atherosclerosis
4.1. Activators of peroxisome proliferator-activated receptor
Peroxisome proliferator-activated receptor (PPARs) agonists are considered as a therapeutic target for diabetes, dyslipidemia, and atherosclerosis because they are the members of the nuclear receptor major family that characterizes as fatty acid-activated transcription factors; they are the regulator of numerous metabolic pathways. Three different PPAR genes (α, β/δ, and γ) have been identified. Kidney, liver, skeletal and cardiac muscle, and adrenal gland tissue include in lipid oxidation mainly characterized by the PPARα. Activated PPARα increases expression of lipoprotein lipase and apolipoprotein A-V and decreases hepatic apolipoprotein C-III. These actions lower plasma TGs in chylomicrons and very low density lipoprotein (VLDL) particles, thus liberating fatty acids (FAs), which are taken up and stored as fat in adipocytes or metabolized in skeletal muscle. Also PPARα activation increases hepatic apolipoprotein A -I and –II expressions, which raise HDL cholesterol levels, and promote HDL-mediated cholesterol efflux from macrophages by inducing ATP-binding cassette A1 transporter. PPARα is also associated with regulation of genes involved in FAs uptake, β-oxidation and down-regulates a protein apolipoprotein C-III that inhibits TG hydrolysis by lipoprotein lipase, and it also interferes with genes involved in reverse cholesterol transport like apolipoprotein A-I and A-II. PPARγ is present in adipose tissue, macrophages, and vascular smooth muscles, while PPARγ is mainly expressed in skeletal muscle and adipose tissues. PPAR β/δ is involved in skin homeostasis, and is involved in HDL metabolism. Use of PPARα and PPARγ agonists may be effective in the management of atherosclerosis (Sagar et al., 2012; Kristina et al., 1996).
4.2. Endoglin receptor
Endoglin (CD 105, TGF-β receptor III) is a homodimeric transmembrane glycoprotein having a regulatory role in transforming growth factor – β (TGF-β) signaling, predominantly expressed on proliferating endothelial cells. This protein is characterized as homodimer of 180 kDA with disulfide attachment. It is primarily observed in endothelial cells, smooth muscle cells, activated macrophages, and fibroblast. Expression of endoglin in vessels increases during different pathological situations including hypoxia or vascular injury. Phosphorylation of endoglin can impede its subcellular localization and cellular migration. In general, endoglin expression is strongly related to cardiovascular system development, vascular development, angiogenesis and vascular homeostasis. Endothelial cells play an important role in the pathogenesis of atherosclerosis. Endothelial dysfunction, characterized by increased expression of inflammatory cell adhesion molecules, altered vasodilation properties and a prothrombogenic state, is very significant in every stage of atherogenesis. SMCs play the essential role in the development of atherosclerotic lesions due to the increment in the extracellular matrix and also for the stabilization of the difficult atherosclerotic plaques because of the fibrous cap which is produced by the collagen formation. Fig. 3 shows involvement of endoglin in the development of atherosclerosis and associated cardiovascular diseases. Endoglin expression is associated with smooth muscle cell migration and proliferation, and the regulation of the function of endothelial cells. Hence, endoglin is necessary for the vascular SMC stability as well as its development by inhibition of SMC migration due to the regulation of TGF-β (Petr et al., 2012, 2008; Francisco and Carmelo, 2012). Reduction of cholesterol level by drugs like statin involves endoglin mediated action.
Figure 3.
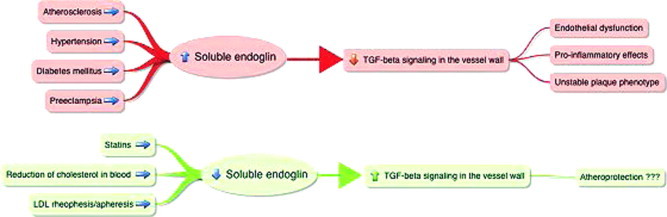
Shows the role of soluble endoglin in atherosclerosis: cardiovascular diseases like atherosclerosis, hypertension, diabetes mellitus and preeclampsia, increase levels of soluble endoglin in blood in humans that leads to increased interactions between endoglin and TGFb1 in blood followed by reduced TGF-b signaling in vessels, thus promoting atherogenesis. Statins, reduction of cholesterol levels and application of extracorporeal procedures decrease soluble endoglin levels in blood, which may result in an increased TGFb1 signaling in the vessel wall and possibly affect atherogenesis (Petr et al., 2012).
4.3. Cholesteryl ester transfer protein inhibitors
Cholesterol ester transfer protein (CETP) is a lipid transfer protein that facilitates the transport of cholesterol ester and TG within the lipoproteins in the blood to mediate the way of transfer of cholesterol ester from the HDL to the LDL and VLDL which should be proatherogenic (Philip and John, 2006). Thus, the movement of cholesteryl esters from HDL to LDL by CETP leads to lowering of HDL. It therefore, follows that inhibition of CETP should lead to elevation of plasma HDL and lowering of plasma LDL, thereby providing a therapeutically beneficial plasma lipid profile. Inhibition of CETP is a choice of treatment in the lipid profile related disorders like atherosclerosis and type II diabetes (Sagar et al., 2012; John et al., 2010).
4.4. Cholesterol absorption inhibitors
Decrease in cholesterol absorption leads to compensatory up-regulation of LDL receptors on the cell surface and increased LDL cholesterol uptake into cells and decreases blood LDL cholesterol content (Sagar et al., 2012; John et al., 2010). Ezetimibe is the only drug of this class having the ability to inhibit absorption of dietary cholesterol without affecting the absorption of fat-soluble vitamins, triglycerides, and bile. Ezetimibe binds to cholesterol transporter Niemann-pick C1-like1 protein in the brush border of the intestine as well as in hepatocytes thereby inhibiting absorption of cholesterol (Sagar et al., 2012; Stuart et al., 1998).
4.5. Cholesterol O-acyltransferase inhibitors
Acyl-CoA: cholesterol O-acyltransferase (ACAT) is a chief enzyme involved in re-esterification of absorbed cholesterol within enterocytes. ACAT has a role in cholesterol metabolism in macrophages, liver, intestine and adrenal cortex and is believed to be involved in secretion of VLDL from liver and development of an atherosclerotic lesion. There are two types of ACAT enzymes, ACAT1 and ACAT2. ACAT1 is present in the ER throughout the body, while ACAT2 found in the ER of liver and intestinal tissues and may be responsible for the formation of cholesteryl esters. Inhibition of ACAT1 may prevent the transformation of macrophages into foam cells in the vessel wall and, thereby, reduce the progression of atherosclerosis and prevent the development of vulnerable plaque whereas inhibition of ACAT-2 leads to a decrease in serum lipid levels by reducing the synthesis of lipoproteins (Sagar et al., 2012).
4.6. Diacylglycerolacyltransferases inhibitors
Diacylglycerolacyltransferase (DGAT) is an integral membrane protein that catalyzes the final enzymatic step in the production of triacylglycerols by transferring an acyl group from acyl-coenzyme-A to the sn-3 position of 1,2-diacylglycerol (Kathryn et al., 2001). It also plays a role in accumulation of lipid adipocyte (Sagar et al., 2012). DGAT is also involved in lipoprotein assembly and the regulation of plasma triacylglycerol concentration, as well as participates in the regulation of DAG levels (Kathryn et al., 2001). DGAT1 belongs to the same family of proteins as the ACATs and is present in skeletal muscle skin, intestine (ileum, colon), and testis, with lower levels of expression in the liver and adipose tissue, while DGAT2 is ubiquitous with high expression levels in hepatocytes and adipocytes (Sagar et al., 2012). DGAT-1 knockout mice were protected from atherosclerosis development (Chandak et al., 2011). Inhibition of DGAT may reduce the risk associated with atherosclerosis.
4.7. Microsomal TG transfer protein inhibitors
Microsomal TG transfer protein (MTTP) is a heterodimeric lipid transfer protein present in the intestine and liver tissues where it is involved in lipid assembly, transport, and secretion of lipoproteins, TG rich chylomicrons (in enterocytes), and VLDL (in hepatocytes). It also plays a role in transportation of TG, cholesteryl ester, and phosphatidylcholine between the membranes. In vitro studies show that MTTP catalyzes the transport of molecules between phospholipid membranes and is also involved in the synthesis of nascent lipoprotein particles within the lumen and ER (Sagar et al., 2012). Defects in MTTP cause a beta-lipoproteinemia, a disorder characterized by production of VLDL and chylomicrons is disrupted (John et al., 1998). The inhibition of MTTP causes reduction in plasma TGs and cholesterol levels which thereby reduces the risk of atherosclerosis.
4.8. Squalene synthase inhibitors
Squalene synthase, a key enzyme in the cholesterol biosynthetic pathway, occupies the first and solely committed step in the biosynthesis of the sterol nucleus of cholesterol; hence it is an attractive target to manage elevated serum cholesterol and thereby for atherosclerosis. Squalene synthase catalyzes one of the subsequent reactions in the cholesterol biosynthetic pathway. It reductively dimerizes two farnesyl pyrophosphate molecules to form squalene which is the first intermediate committed to cholesterol (Sagar et al., 2012). It has been shown that squalene synthase inhibitor suppresses triglyceride biosynthesis in hepatocytes but the mechanism is not precisely known (Tohru et al., 2002).
4.9. Thyroid hormone analogues
Thyroid hormone has potential to reduce total serum cholesterol by accelerating LDL clearance rate in hyperthyroidism like conditions (Sagar et al., 2012).This unique property of lowering cholesterol improves cardiac performance (Eugene et al., 2004). It has been suggested that hypercholesterolemia, hypertension and impaired endothelial function in the hypothyroid state augment atherogenesis (Franc¸oise et al., 2009). In addition, recent studies suggest that hypothyroidism is associated with the emerging risk factors for atherosclerosis such as hyperhomocysteinemia and an increase in C-reactive protein level (Toshihiro, 2010). T3 increases levels of both the hepatic LDL receptor and its mRNA. Thyroid hormone acts on lipid by increasing the activity of lipoprotein lipase. More recent understanding of thyroid hormone receptors has led to the development of thyroid hormone mimetics that have selective functions and are potential therapeutic agents to lower cholesterol (Sagar et al., 2012). Current studies have shown that thyroid hormone also has direct effects on the blood vessel, which may attenuate atherogenesis (Toshihiro, 2010).
4.10. Lanosterol synthase inhibitors
Oxido-squalene-cyclase (lanosterol synthase) is the second enzyme below the farnesyl pyrophosphate branch point that has been identified as a target for novel anticholesterolemic drugs that could complement statins (Sagar et al., 2012). Lanosterol synthase is located in the ER and converts 2,3-oxidosqualenetolanosterol, the initial four-ringed sterol intermediate in the cholesterol synthesis pathway. The 24(S), 25-epoxycholesterol is a ligand of liver X receptor (Andrea et al., 2003). It also sets the template for the design of inhibitors with improved pharmacological properties for cholesterol lowering and treatment of atherosclerosis. Lanosterol synthase inhibitors have a potential to decrease plasma levels LDL and prevents cholesterol deposition within macrophages (Sagar et al., 2012).
4.11. Cholesterol modifying cytochrome P450
In mammals, excess cholesterol (5-cholestene-3β-ol) is removed mainly through conversion to bile acids, and small portion is used for the production of steroid hormone by the enzyme called cytochrome P450 (CYP) (Irina, 2006). CYP7A1, 27A1 and 46A1 from the family of P450s are the most important enzymes involved in the control of cholesterol levels in the periphery and brain (Sagar et al., 2012; Irina, 2006). Cholesterol conversion into bile acids is initiated and controlled by a liver-specific enzyme, CYP7A1 that converts cholesterol into 7α-hydroxycholesterol which is the first and rate-limiting step in the classical or neutral bile acid biosynthetic pathway. Abnormality in the CYP7A1 leads to elevation of total and LDL cholesterol levels, substantial accumulation of cholesterol in the liver, and a markedly decreased rate of bile acid excretion (Irina, 2006). CYP7A1 is an important determinant for reducing plasma cholesterol levels and is considered as a target for cholesterol lowering.
4.12. AMP-activated protein kinase activator
AMP-activated protein kinase (AMPK), is a heterotrimetric energy sensing protein, which restores cellular energy balance by promoting ATP-generating pathways (FA oxidation) and inhibiting ATP-utilizing pathways (FA synthesis) (Sagar et al., 2012). Different physiological processes like conditions that lead to alterations of the intracellular AMP/ATP ratio (e.g., hypoxia, glucose deprivation), calcium concentration and action of various hormones, cytokines, and adipokines stimulate AMPK. AMPK phosphorylates and inactivates acetyl-CoA carboxylase 1 and 3-hydroxy-3-methylglutarylCoA reductase thereby inhibiting synthesis of new FA and cholesterol whereas phosphorylation of acetyl-CoA carboxylase 2 by AMPK causes an increase of FA oxidation (Bei et al., 2009). Also AMPK system plays a major role in regulating glucose and lipid metabolism by effect on energy metabolism and long-term effect on gene expression in the liver. In liver, activation of AMPK results in decreased production of plasma TG and cholesterol and enhanced FA oxidation that could be used for inhibition de novo hepatic lipogenesis (Sagar et al., 2012).
4.13. Omega-3 FAs
Omega-3 belongs to the polyunsaturated FA family, which includes the 20-carbon eicosapentanoic acid and 22-carbon docosahexaenoic acid, having an ability to reduce the TG levels and atherogenic remnant lipoproteins (Vasilios et al., 2011). These FAs mainly present in marine sources, especially salmon, mackerel, sardines, tuna and at 4 g/day were found to be effective in lowering TG concentration, particularly in the postprandial state (Sagar et al., 2012). Combination of statins and Omega-3 FAs significantly decreases TG, VLDL, and non-HDL-c levels compared with simvastatin alone (Vasilios et al., 2011). Synthesis of FAs involves an expression of SREBP-1 which is inhibited by Omega-3 FA. Omega-3 FAs also show hypotriglyceridemia, anti-aggregatory, anti-inflammatory, and anti-arrhythmatic responses (Sagar et al., 2012).
4.14. Heat shock protein-65 and CETP vaccine
Atherosclerosis is inflammation mediated disease characterized by the accumulation of lipids and fibrous elements in the arteries. Anti-inflammatory or lipid-lowering treatments could be useful for alleviating morbidity and mortality of atherosclerotic cardiovascular diseases. However, a vaccine designed to target inflammation and lipid simultaneously is found to be more useful for the management of atherosclerosis. Study by Long Jun et al. showed that, a vaccine designed to target heat shock protein-65 and CETP, reduces TC and LDL and size of atherosclerotic plaques. Long term nasal administration of heat shock protein 65 also helps to reduce atherosclerosis through the reduction of immune tolerance against self-antigen and down-regulation of atherosclerotic inflammation (Long et al., 2012).
4.15. Nitric oxide
Hypercholesterolemia and atherosclerosis disturb the endothelium-dependent nitric oxide (NO) mediated regulation of the vascular tone and endothelial dysfunction is an early event in atherogenesis and coronary heart disease (Claudio and Louis, 2001). NO synthase (NOS) synthesizes NO which is a crucial modulator of vascular disease. The protective role of NO is because of its ability to suppress abnormal proliferation of vascular smooth muscle cells. NO is also involved in various intracellular effects that lead to vasorelaxation, inhibition of leukocyte chemotaxis, endothelial regeneration, and platelet adhesion. Endothelial damage in atherosclerosis reduces bioactivity of endothelial NO synthase that subsequent reduces synthesis of NO and stimulates degradation of NO by generating reactive oxygen species (Claudio et al., 2006). Several mechanisms like lipoprotein-induced alterations in signal transduction, increases in superoxide anion generation followed by degradation of NO, reduced affinity of NOS for l-arginine, and/or elevated levels of circulating antagonists enhances derangement of the NOS pathway (Claudio and Louis, 2001; Russo et al., 2002). Various vasculoprotective agents act through increasing NO synthesis. l-Arginine, the precursor of NO, can be used for the management of atherosclerosis and atherosclerotic coronary diseases (Claudio et al., 2006).
4.16. Phospholipase A2 inhibiter
Several types of phospholipases are associated with atherosclerosis and its complications. The body contains five different types of phospholipase A2(PLA2), secreted phospholipaseA2 (sPLA2), cytosolic phospholipase A2 (cPLA2), calcium-independent PLA2s, lysosomal PLA2s (Lp-PLA2) and platelet-activating factor acetylhydrolases (Keith, 2010). These are involved in the formation of pro-inflammatory products and hence may have significant effects on atherogenesis.
4.16.1. sPLA2 inhibiter
This form of PLA2 is associated with a range of inflammatory, autoimmune and allergic disorders with different isoforms being expressed and released by specific inflammatory cells. In addition to hydrolytic activity, it also interacts with high affinity receptors on inflammatory cells or with cell-surface proteoglycans, for example inducing degranulation in mast cells, neutrophils and eosinophils and activating endocytosis in macrophages. sPLA2s is involved in promotion of chemokine production from monocytes and macrophages and modification of plasma lipoproteins, making them more atherogenic. sPLA2s active on circulating lipoproteins exhibits a different effect from those located in the plaque. sPLA2-V is associated with plaque smooth muscle cells and surrounding foam cell which is active on lipoproteins in human serum. sPLA2-X has the highest catalytic activity toward phosphatidylcholine and is present in human lesion. It is observed that LDL concentrations in patients are reduced when a non-selective sPLA2 inhibitor is administered hence, when the overall sPLA activity is inhibited it may lead to reduced clearance of LDL (Keith, 2010).
4.16.2. Lp-PLA2 inhibiter
Lipoprotein-associated PLA2 is an enzyme produced by inflammatory cells, which binds to apolipoprotein B-containing lipoproteins and degrades oxidatively modified phospholipids in low-density lipoprotein cholesterol particles, leading to formation of pro-inflammatory and cytotoxic products. Approximately 80% of Lp-PLA2 circulates bound to LDL cholesterol, specifically to apolipoprotein B-100, and remaining bound to high-density lipoprotein and remnant lipoprotein particles. It is associated with small, dense and electronegative LDL particles that are believed to be more proatherogenic. Lp-LPA2 acts mainly on oxidized LDL. Oxidative modification of polyunsaturated FAs in the sn-2 position of phospholipids within LDL molecules render them susceptible to hydrolysis by Lp-PLA2, resulting in the formation of lysophosphatidylcholine and oxidized nonesterified FAs, pro-inflammatory and cytotoxic products respectively. Lysophosphatidylcholine and oxidized nonesterified FAs further stimulate expression of adhesion molecules and cytokines by endothelial cells, plaque-based macrophages and other leukocytes. Lp-PLA2 is strongly expressed in the necrotic core of coronary lesions and in the surrounding macrophages of vulnerable and ruptured plaques, which is consistent with the proposed mechanism. Inhibitors of this enzyme can be used for the management of atherosclerosis and high-risk coronary artery disease patient (Eva, 2010).
4.17. Scavenger receptor
The uptake of modified LDL is promoted by scavenger receptors that promote the cellular accumulation of cholesterol. Macrophages in lesions have been shown to express more than six structurally different scavenger receptors. These scavenger receptors involved in initiation of signaling cascades regulate activation of macrophage, lipid metabolism, and inflammatory processes that may control the development and stability of the atherosclerotic plaque. These receptors also regulate induction of apoptosis, apoptotic cell clearance, and pathogen recognition that may differentially impact early and more complex lesions. Inhibition of these scavenger receptor pathways may lead to inhibition of various pro-inflammatory events which contribute in progression of atherosclerosis (Kathryn and Mason, 2006; Agnes et al., 2005).
All the drug targets and their possible mechanisms of action for the management of atherosclerosis are summarized in Table 1.
Table 1.
Drug targets and its mechanism of action for atherosclerosis.
| Drug targets | Mechanism of action | References |
|---|---|---|
| PPARγ selective modulator | Decreases insulin resistance | Kristina et al. (1996) |
| Anti-inflammatory effects in macrophage foam cells | Sagar et al. (2012) | |
| Endoglin receptor modulator | Reduces expression of inflammatory cell adhesion molecules | Francisco and Carmelo (2012) |
| Increases vascular smooth muscle cell | Petr et al. (2012, 2008) | |
| Stabilize and reduce prothrombogenic state | ||
| CETP inhibitor | Increases HDL cholesterol levels | John et al. (2010), Philip and John (2006), Sagar et al. (2012) |
| Reverses cholesterol transport system | ||
| ACAT enzyme inhibitor | Prevents transformation of macrophages into foam cells | Sagar et al. (2012) |
| Decreases synthesis of lipoproteins | ||
| PPARα agonist | Decreases TG-rich lipoproteins | Kathryn and Mason (2006) |
| Increases HDL | Kristina et al. (1996), Philip and John (2006), Sagar et al. (2012) | |
| Anti-inflammatory effects in vessel wall | ||
| DGAT enzyme inhibitor | Inhibit transfer of acyl group from acyl-coenzyme-A to the sn-3 position of 1,2-diacylglycerol | Kathryn et al. (2001), Sagar et al. (2012) |
| Reduces triacylglycerol formation | ||
| MTTP protein inhibitor | Prevents lipid assembly, transport, secretion of lipoproteins, triglyceride rich chylomicrons (in enterocytes) and VLDL | Eugene et al. (2004), Jeffrey et al. (2001),Sagar et al. (2012) |
| Modulates transportation of TG, cholesteryl ester, and phosphatidylcholine | ||
| Squalene synthase inhibitor | Prevent biosynthesis of the sterol nucleus of cholesterol | Sagar et al. (2012), Tohru et al. (2002) |
| Thyroid hormone analogues | Increases activity of lipoprotein lipase | Eugene et al. (2004), Paul and Faith (2002), Sagar et al. (2012), Toshihiro (2010) |
| Accelerates LDL-c clearance | ||
| Lanosterol synthase inhibitor | Restrain four-ringed sterol intermediate formation | Andrea et al. (2003), Sagar et al. (2012) |
| Prevent deposition of cholesterol within macrophages | ||
| Cytochrome P450 modulator | Regulate conversion of cholesterol to 7α-hydroxycholesterol | Irina (2006) |
| Sagar et al. (2012) | ||
| AMPK activator | Regulates glucose metabolism | Bei et al. (2009), Sagar et al. (2012) |
| Decreases hepatic lipogenesis, synthesis of new FA and cholesterol | ||
| Increases FA oxidation | ||
| Omega-3 FAs | Inhibit expression of SREBP-1 | Sagar et al. (2012), Vasilios et al. (2011) |
| Reduces TG levels and atherogenic lipoproteins | ||
| Heat shock protein-65 and CETP vaccine | Reduction in immune tolerance against self antigen | Long et al. (2012) |
| Prevent transfer of CE into triglyceride-rich lipoproteins or LDL | ||
| sPLA2 inhibitor | Prevents inflammatory, autoimmune and allergic reactions | Keith et al. (2010) |
| Lp-PLA2 inhibitor | Restrain formation of LysoPC and oxNEFA | Keith et al. (2012), Pabloal and Juan (2006) |
| Reduces proinflammatory and cytotoxic products | Peter et al. (2008) | |
| Scavenger receptor inhibitor | Inhibits macrophage activation, lipid metabolism, and inflammation | Agnes et al. (2005), Kathryn and Mason (2006) |
| Prevent induction of apoptosis, apoptotic cell clearance, and pathogen recognition |
5. Conclusion
Atherosclerosis is a metabolic disorder characterized by hyperlipidemia and chronic inflammation. Although nowadays many therapies like HMG CoA reductase inhibitors, bile acid sequestrants, fibrates, niacin are successfully implicated to treat altered lipid profile associated with atherosclerosis and atherosclerotic coronary disorders, still it lacks in a significant increase in the benefit-to-risk ratio. Hence, the present review mainly gives the idea about the recent receptor and signaling pathways that take part in the starting stage of atherosclerosis formation that is from the plaques form to the fatty streak formation. PPARs are regulators of several metabolic pathways; hence agonists of these receptors can be used as therapeutics of dyslipidemia. Inhibition of CETP elevates plasma HDL cholesterol and reduces plasma LDL cholesterol. Squalene synthase and lanosterol synthase are the enzymes involved in cholesterol synthesis; their inhibitors will be the possible target for the reduction of hyperlipidemia. The inhibitors of ACAT, DGAT, MTTP and PLA2 are also used as targets for hyperlipidemia associated atherosclerotic diseases. Lipid lowering effect of thyroid hormone analogue will be a possible mechanism to altered lipid profile. Cholesterol modifying CYP P450, AMPK activators, and omega-3 FAs, heat shock proteins-65, will be the new therapeutic drug targets in the management of atherosclerosis.
Footnotes
Peer review under responsibility of King Saud University.

References
- Agnes Boullier, David A.B., Mi-Kyung Chang., Edward A.D., Peter Friedman, Kristin Gillotte-Taylor, Sohvi Hörkkö, Wulf Palinski, Oswald Quehenberger, Peter Shaw, Daniel Steinberg, valeska Terpstra. Annals New York Academy of Sciences; 2005. Scavenger Receptors, Oxidized LDL, and Atherosclerosis. 214–224. [Google Scholar]
- Alexander P.R., Arnold Kahn, Brain H.E., Mark J.P., Natalia Sadetsky, Dale Williams O., Joseph F.P., David R.J., Jr., Marshall L.S. Kidney stones and subclinical atherosclerosis in young adults: the CARDIA study. The Journal of Urology. 2011;185:920–925. doi: 10.1016/j.juro.2010.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amala P.C., Akiko Maehara, Gary S.M., Roxana Mehran, Sunil Kanwal, Ahmed Hassanin, Diaa Hakim, Ning Guo, Usman Baber, Robert Pyo, Jeffrey W.M., Martin Fahy, Jason C.K., George D.D. High platelet reactivity on clopidrogrel therapy correlates with increases coronary atherosclerosis and calcification. JACC: Cardiovascular Imaging. 2012;5:540–549. doi: 10.1016/j.jcmg.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrea H.R., Carmen A.A., Jane Y.E., Cynthia G.S., Olivier H., Morand, Robert A.H., Murray W.H. Enhanced synthesis of the oxysterol 24(s), 25-epoxycholesterol in macrophages by inhibitors of 2,3-oxidosqualene:lanosterol cyclase: a novel mechanism for the attenuation of foam cell formation. Circulation Research. 2003;93:717–725. doi: 10.1161/01.RES.0000097606.43659.F4. [DOI] [PubMed] [Google Scholar]
- Anthony Colpo. LDL cholesterol: “bad” cholesterol, or bad science? Journal of American Physicians and Surgeons. 2005;10(3):83–89. [Google Scholar]
- Anthony J.Y., John D.D., Kimberly A.B., Patrick Y.Lee. Adventitial dysfunction: an evolutionary model for understanding atherosclerosis. Medical Hypotheses. 2005;65:962–965. doi: 10.1016/j.mehy.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Barbara M.M., Marian U.C., Alan J.G., Fiona Mitchell, Hema Navaratnam, Rosemary O.H., Psych D., Peter C.E., Michael R., Le Grande. Lifestyle and physiological risk factor profiles six weeks after an acute cardiac event: are patients achieving recommended targets for secondary prevention? Heart, Lung and Circulation. 2011;20:446–451. doi: 10.1016/j.hlc.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Bei Zhang B., Zhou Gaochao, Li Cai. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metabolism. 2009;9:407–416. doi: 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Bradford B.W., Thomas J.D. The genetics of cerebrovascular atherosclerosis. Seminars in Cerebrovascular Diseases and Stroke. 2002;2:13–14. [Google Scholar]
- Wong Brian.W., Meredith Anna., Lin David., McManus Bruce.M. The biological role of inflammation in atherosclerosis. Canadial Journal of Cardiology. 2012;28(6):631–641. doi: 10.1016/j.cjca.2012.06.023. [DOI] [PubMed] [Google Scholar]
- Chandak P.G., Obrowsky S., Radovic B., Doddapattar P., Aflaki E., Kratzer A., Doshi L.S., Povoden S., Ahammer H., Hoefler G., Levak-Frank S., Kratky D. Lack of acyl CoA: diacylglycerolacyltransferase 1 reduces intestinal cholesterol absorption and attenuates atherosclerosis in apolipoprotein E knockout mice. Biochimica et Biophysica Acta. 2011;1811(12):1011–1020. doi: 10.1016/j.bbalip.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio Napoli, Ignarro Louis J. Nitric oxide and atherosclerosis. Nitric Oxide. 2001;5(2):88–97. doi: 10.1006/niox.2001.0337. [DOI] [PubMed] [Google Scholar]
- Claudio Napoli, Filomena de Nigris, Sharon Williams-lgnarro, Vincenzo Sica, Ignarro Louis J. Nitric oxide and atherosclerosis: an update. Journal of Nitric Oxide. 2006;15(4):265–279. doi: 10.1016/j.niox.2006.03.011. [DOI] [PubMed] [Google Scholar]
- David Spence J. Intensive management of risk factors for accelerated atherosclerosis: the role of multiple interventions. Current Neurology and Neuroscience Reports. 2007;7:42–48. doi: 10.1007/s11910-007-0020-8. [DOI] [PubMed] [Google Scholar]
- Elstad M.R., La Pine T.R., Cowley F.S., Mc Ever R.P. P-selectin regulates platelet-activating factor synthesis and phagocytosis by monocytes. Journal of Immunology. 1995;155:2109–2122. [PubMed] [Google Scholar]
- Eugene Morkin, Paul Ladenson, Steven Goldman, Cynthia Adamson. Thyroid hormone analogs for treatment of hypercholesterolemia and heart failure: past, present and future prospects. Journal of Molecular and Cellular Cardiology. 2004;37:1137–1146. doi: 10.1016/j.yjmcc.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Eva Lonn. Lipoprotein-associated phospholipase A2: a new therapeutic target. Canadian Journal of Cardiology. 2010;26:27A–37A. doi: 10.1016/s0828-282x(10)71058-8. [DOI] [PubMed] [Google Scholar]
- Franc¸oise Chiche, Christel Jublanc, Mathieu Coudert, Valérie Carreau, Jean-Franc¸ois Kahn, Eric Bruckert. Hypothyroidism is not associated with increased carotid atherosclerosis when cardiovascular risk factors are accounted for in hyperlipidemic patients. Atherosclerosis. 2009;203:269–276. doi: 10.1016/j.atherosclerosis.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Francisco J.B., Carmelo Bernabéu. Alternative splicing in endothelial senescence: role of the TGF-ß co-receptor endoglin. Senescence. 2012;203:953–978. [Google Scholar]
- Gerrity R.G., Antonoy A.S. The pathogenesis of atherosclerosis. Diabetologia. 1997;40:S108–S110. doi: 10.1007/s001250051419. [DOI] [PubMed] [Google Scholar]
- Pikuleva Irina A. Cholesterol-metabolizing cytochromes P450. Drug Metabolism and Disposition. 2006;34(4):513–520. doi: 10.1124/dmd.105.008789. [DOI] [PubMed] [Google Scholar]
- James B.M., Brian K.C. Chlamydia pneumoniae and atherosclerosis: does the evidence support a causal or contributory role? FEMS Microbiology Letters. 2001;197:1–9. doi: 10.1111/j.1574-6968.2001.tb10574.x. [DOI] [PubMed] [Google Scholar]
- Jeffrey A.R., Richard Sulsky, Chong-Qing Sun, Ligaya M.S., Tammy Wang, John K.D., Ying Chen, David R.M., Prakash Taunk, William A.S., Scott A.B., Shih-Jung Lan, Fergal Connolly, Lori K.K., Talal Sabrah, Haris Jamil, David Gordon, Thomas W.H., John R.W. A novel series of highly potent benzimidazole-based microsomal triglyceride transfer protein inhibitors. Journal of Medicinal Chemistry. 2001;44(6):851–856. doi: 10.1021/jm000494a. [DOI] [PubMed] [Google Scholar]
- Jerzy Beltowski. Leptin and atherosclerosis. Atherosclerosis. 2006;189:47–60. doi: 10.1016/j.atherosclerosis.2006.03.003. [DOI] [PubMed] [Google Scholar]
- John F.K. Atherosclerosis: from lesion formation to plaque activation and endothelial function. Molecular Aspects of Medicine. 2000;21:99–166. doi: 10.1016/s0098-2997(00)00005-4. [DOI] [PubMed] [Google Scholar]
- John B.S. Hypertension and atherosclerosis: clinical implications from the ALLHAT trial. Current Atherosclerosis Reports. 2005;7:132–139. doi: 10.1007/s11883-005-0036-y. [DOI] [PubMed] [Google Scholar]
- John R.W., Richard E.G., Thomas W.H., Cynthia Arbeeny, Michael Cap, Fergal Connolly, Ching-Hsuen Chu, Rocco J.G., David A.G., Haris Jamil, Kern G.J., Lori K.K., Shih-Jung Lan, Thomas J.M., Beverly Ricci, Mujing Yan, Douglas Young, Ying Chen, Olga M.F., Janette V.H., Christa L.M., Michael A.P., Jeffrey A.R., Ligaya M.S., William A.S., Richard Sulsky, Prakash Taunk, David R.M., Joseph A.T., Michael Lawrence R., John K.D., Scott A.B. An MTP inhibitor that normalizes atherogenic lipoprotein levels in WHHL rabbits. Science. 1998;282:751–754. doi: 10.1126/science.282.5389.751. [DOI] [PubMed] [Google Scholar]
- John M. Chapman, Jan S. Redfern, Mark E. McGovern, Philippe Giral. Niacin and fibrates in atherogenic dyslipidemia: pharmacotherapy to reduce cardiovascular risk. Pharmacology & Therapeutics. 2010;126:314–345. doi: 10.1016/j.pharmthera.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Rubin Jonathan., Nambi Vijay., Lloyd E.C., Michael W.S., Stephen P.J., Josef Coresh A., Sharrett Richey., Selvin Elizabeth. Hyperglycemia and arterial stiffness: the atherosclerosis risk in the communities study. Atherosclerosis. 2012;225(1):246–251. doi: 10.1016/j.atherosclerosis.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathryn J.M., Mason W.Freeman. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arteriosclerosis Thrombosis Vascular Biology. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43. [DOI] [PubMed] [Google Scholar]
- Kathryn D.L., Jennifer T.M., Nicholas W.W., Annette Wyrick, Toni Voelker, Deborah J.H. DGAT2 is a new diacylglycerol acyltransferase gene family: purification, cloning, and expression in insect cells of two polypeptides from mortierella ramanniana with Diacylglycerol acyltransferase activity. The Journal of Biological Chemistry. 2001;276:38862–38869. doi: 10.1074/jbc.M106168200. [DOI] [PubMed] [Google Scholar]
- Keith Suckling. Phospholipase A2s: developing drug targets for atherosclerosis. Atherosclerosis. 2010;212:357–366. doi: 10.1016/j.atherosclerosis.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Kristina Schoonjans, Bart Staels, Johan Auwerx. Role of the peroxisome proliferator-activated Receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. Journalof Lipid Research. 1996;37:907–925. [PubMed] [Google Scholar]
- Li A.C., Glass C.K. The macrophage foam cell as a target for therapeutic intervention. Nature Medicine. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- Long Juna, Lin Jie, Yuan Dongping, Yang Xin, Li Taiming, Cao Rongyue, Wu Jie, Liu Jingjing. Effects of nasal immunization of multi-target preventive vaccines on atherosclerosis. Vaccine. 2012;30:1029–1037. doi: 10.1016/j.vaccine.2011.12.043. [DOI] [PubMed] [Google Scholar]
- Margaret Chan. Burden: mortality, morbidity and risk factors. Global Status Report On Noncommunicable Diseases. 2010 [Google Scholar]
- Markus Juonalab, Mika Kähönen, Risto Huupponen, Jorma S.A. Viikari, Olli T. Raitakari. Associations between serum uric acid and markers of subclinical atherosclerosis in young adults. The cardiovascular risk in young finns study. Atherosclerosis. 2012;223:497–503. doi: 10.1016/j.atherosclerosis.2012.05.036. [DOI] [PubMed] [Google Scholar]
- Mervi Oikonen, Maria Wendelin-Saarenhovi, Leo-Pekka Lyytikäinen, Niina Siitonen, Britt-Marie Loo, Antti Jula, Ilkka Seppälä, Liisa Saarikoski, Terho Lehtimäki, Nina Hutri-Kähönen, Mohammed Al-Omran. Atherosclerotic disease and risk factor modification in Saudi Arabia: a call to action. Journal of Vascular Health and Risk Management. 2012;8:349–355. doi: 10.2147/VHRM.S32783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pablo Piñón, Juan C.K. Inflammation, atherosclerosis, and cardiovascular diseaserisk: PAPP-A, Lp-PLA2, and Cystatin C. new insights or redundant information? Revista Española de Cardiologia. 2006;59(3):247–258. [PubMed] [Google Scholar]
- Pao-Ling Torng a, Ta-Chen Su b, Fung C Sung, Kuo.-Liong Chien, Chien Su-Cheng Huang, Song.-Nan Chow, Yuan.-Teh. Lee. Effects of menopause on intraindividual changes in serum lipids, blood pressure, and body weight- the Chin-Shan community cardiovascular cohort study. Atherosclerosis. 2002;161:409–415. doi: 10.1016/s0021-9150(01)00644-x. [DOI] [PubMed] [Google Scholar]
- Paolo Puddu, Giovanni MPuddu, Luciana Basagli, Giorgio Massarelli, Antonio Muscari. Coronary and cerebrovascular atherosclerosis: two aspects of the same disease or two different pathologies? Archives of Gerontology and Geriatrics. 1995;20:5–22. doi: 10.1016/0167-4943(94)00600-c. [DOI] [PubMed] [Google Scholar]
- Paul Holvoet, Desire Collen. Oxidation of low density lipoproteins in the pathogenesis of Atherosclerosis. Atherosclerosis. 1998;137:S33–S38. doi: 10.1016/s0021-9150(97)00305-5. [DOI] [PubMed] [Google Scholar]
- Paul J.D., Faith B.D. Nongenomic actions of thyroid hormone on the heart. Thyroid. 2002;12(6):459–466. doi: 10.1089/105072502760143827. [DOI] [PubMed] [Google Scholar]
- Peter ten Dijke, Marie-Jose Goumans, Evangelia Pardali. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11:79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- Petr Nachtigal, Lenka Zemankova, Jana Rathous, Zbynek Strasky. The role of endoglin in atherosclerosis. Atherosclerosis. 2012;224:4–11. doi: 10.1016/j.atherosclerosis.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Philip J. Barter, John J.P. Kastelein. Targeting cholesteryl ester transfer protein for the prevention and management of cardiovascular disease. Journal of the American College of Cardiology. 2006;47(3):492–499. doi: 10.1016/j.jacc.2005.09.042. [DOI] [PubMed] [Google Scholar]
- Pieter A.D., Wouter Jukemab, Wilko Spiering, Joep C.D., Kastelein John J.P. Molecular genetics and gene expression in atherosclerosis. International Journal of Cardiology. 2001;80:161–172. doi: 10.1016/s0167-5273(01)00466-1. [DOI] [PubMed] [Google Scholar]
- Qi Wei a, Doris a Peter A., Martin V.P., Boerwinkle Eric, Jacobs David R., Jr., Siscovick David S., Fornage Myriam. Sequence variation in the soluble epoxide hydrolase gene and subclinical coronary atherosclerosis: Interaction with cigarette smoking. Atherosclerosis. 2007;190:26–34. doi: 10.1016/j.atherosclerosis.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Qian Wang, Xuedong Zhou, Dingming Huang. Role for porphyromonasgingivalis in the progression of atherosclerosis. Medical Hypotheses. 2009;72:71–73. doi: 10.1016/j.mehy.2008.04.030. [DOI] [PubMed] [Google Scholar]
- Robert S.R. HDL-C and diabetic patient: target for therapeutic intervention. Diabetes Research and Clinical Practice. 2005;68S2:S36–S42. doi: 10.1016/j.diabres.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Ross R., Glomset J., Harker L. Response to injury and atherogenesis. TheAmerican Journal of Pathophysiology. 1977;86:675–684. [PMC free article] [PubMed] [Google Scholar]
- Russo Guilia, Jane A.Leopold., Joseph Loscalzo. Vasoactive substances: nitric oxide and endothelial dysfunction in atherosclerosis. Vascular Pharmacology. 2002;38(5):259–269. doi: 10.1016/s1537-1891(02)00250-1. [DOI] [PubMed] [Google Scholar]
- Sagar P.M., Rekha D.K., Menaa Farid., Sachin L.B. Therapeutic approaches to drug targets in hyperlipidemia. Biomedicine. 2012;2(4):137–146. [Google Scholar]
- Sainani, Gs., Talwalkar, Pg., Wadia, Rs., Keshvani, A.A., (2008) Hyperhomocysteinemia and its implications in atherosclerosis the Indian Scenario. Medicine, update. 12–20.
- Sandra N.V., Frank L.J. Visseren. Perivascular adipose tissue as a cause of atherosclerosis. Atherosclerosis. 2011;214:3–10. doi: 10.1016/j.atherosclerosis.2010.05.034. [DOI] [PubMed] [Google Scholar]
- Sarah Song, Bruce Ovbiagele. Management of risk factors for accelerated atherosclerosis. Current Treatment Options in Neurology. 2009;11:460–472. doi: 10.1007/s11940-009-0050-4. [DOI] [PubMed] [Google Scholar]
- Steve E.H., Laleh M. Genetic risk factors for stroke and carotidatherosclerosis: insights into pathophysiologyfrom candidate gene approaches. Lancet Neurology. 2004;3:227–236. doi: 10.1016/S1474-4422(04)00708-2. [DOI] [PubMed] [Google Scholar]
- Stuart B.R., Tram Huynh, Adriano Afonso, Harry R.D., Nathan Yumibe, John W.C., Duane A.B. Discovery of 1-(4 Fluorophenyl)-(3R)-[3-(4-fluorophenyl)-(3S)-hydroxypropyl]-(4S)-(4-hydroxyphenyl) 2-azetidinone (SCH 58235): a designed, potent, orally active inhibitor of cholesterol absorption. Journal of Medicinal Chemistry. 1998;41(6):973–980. doi: 10.1021/jm970701f. [DOI] [PubMed] [Google Scholar]
- Tohru Ugawa, Hirotoshi Kakuta, Hiroshi Moritani, Osamu Inagaki. Effect of YM-53601, a novel squalene synthase inhibitor, on the clearance rate of plasma LDL and VLDL in hamsters. British Journal of Pharmacology. 2002;137:561–567. doi: 10.1038/sj.bjp.0704906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshihiro Ichiki. Thyroid hormone and atherosclerosis. Vascular Pharmacology. 2010;52:151–156. doi: 10.1016/j.vph.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Valentin S.Z., Nils H.S. Monitoring of atherosclerosis. International Journal of Cardiology. 2004;95:39–42. doi: 10.1016/j.ijcard.2003.03.018. [DOI] [PubMed] [Google Scholar]
- Vasilios G. Athyros, Konstantinos Tziomalos, Asterios Karagiannis, Dimitri P. Mikhailidis. Dyslipidaemia of obesity, metabolic syndrome and type 2 diabetes mellitus: the case for residual risk reduction after statin treatment. The Open Cardiovascular Medicine Journal. 2011;5:24–34. doi: 10.2174/1874192401105010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen-Harn Pan, Benjam Plasma lipid profiles and epidemiology of atherosclerotic diseases in Taiwan – a unique experience. Atherosclerosis. 1995;118:285–295. doi: 10.1016/0021-9150(95)05616-5. [DOI] [PubMed] [Google Scholar]
- Weyrich A.S., McIntyre T.M., McEver R.P., Prescott S.M., Zimmerman G.A. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and NF-kappa B translocation. Journal of Clinical Investigation. 1995;95:2297–2303. doi: 10.1172/JCI117921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilensky R.L., Hamamdzic D. The molecular basis of vulnerable plaque: potential therapeutic role for immunomodulation. Current Opinion in Cardiology. 2007;22:545–551. doi: 10.1097/HCO.0b013e3282f028fe. [DOI] [PubMed] [Google Scholar]
- Williams K.J., Tabas I. The response-to-retention hypothesis of early atherogenesis. Atherosclerosis, Thrombosis, and Vascular Biology. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]