

Abstract
Background: Mitochondrial permeability transition pore (mPTP) opening is a terminal event leading to mitochondrial dysfunction and cell death under conditions of oxidative stress (OS). However, mPTP blockade with cyclosporine A (CsA) has shown variable efficacy in limiting post-ischemic dysfunction and arrhythmias. We hypothesized that strong feedback between energy dissipating (mPTP) and cardioprotective (mKATP) channels determine vulnerability to OS.
Methods and Results: Guinea pig hearts (N = 61) were challenged with H2O2 (200 μM) to elicit mitochondrial membrane potential (ΔΨm) depolarization. High-resolution optical mapping was used to measure ΔΨm or action potentials (AP) across the intact heart. Hearts were treated with CsA (0.1 μM) under conditions that altered the activity of mKATP channels either directly or indirectly via its regulation by protein kinase C. mPTP blockade with CsA markedly blunted (P < 0.01) OS-induced ΔΨm depolarization and delayed loss of LV pressure (LVP), but did not affect arrhythmia propensity. Surprisingly, prevention of mKATP activation with the chemical phosphatase BDM reversed the protective effect of CsA, paradoxically exacerbating OS-induced ΔΨm depolarization and accelerating arrhythmia onset in CsA treated compared to untreated hearts (P < 0.05). To elucidate the putative molecular mechanisms, mPTP inhibition by CsA was tested during conditions of selective PKC inhibition or direct mKATP channel activation or blockade. Similar to BDM, the specific PKC inhibitor, CHE (10 μM) did not alter OS-induced ΔΨm depolarization directly. However, it completely abrogated CsA-mediated protection against OS. Direct pharmacological blockade of mKATP, a mitochondrial target of PKC signaling, equally abolished the protective effect of CsA on ΔΨm depolarization, whereas channel activation with 30 μM Diazoxide protected against ΔΨm depolarization (P < 0.0001). Conditions that prevented mKATP activation either directly or indirectly via PKC inhibition led to accelerated ΔΨm depolarization and early onset of VF in response to OS. Investigation of the electrophysiological substrate revealed accelerated APD shortening in response to OS in arrhythmia-prone hearts.
Conclusions: Cardioprotection by CsA requires mKATP channel activation through a PKC-dependent pathway. Increasing mKATP activity during CsA administration is required for limiting OS-induced electrical dysfunction.
Keywords: mitochondria, oxidative stress, permeability transition pore, mitochondrial KATP channel, arrhythmia
Introduction
Mitochondria are central mediators of the cardiac response to oxidative stress (OS), as they respond to reactive oxidative species (ROS) through a host of ROS sensitive channels, which can either amplify or limit ROS-induced injury (O'Rourke et al., 2007). Of key importance to OS-induced mitochondrial dysfunction are the inner membrane anion channel (IMAC) and components of the mitochondrial permeability transition pore (mPTP). Both channel complexes activate in response to rising ROS levels. However, as described by Aon and colleagues, they exhibit a hierarchal activation pattern (Aon et al., 2007): IMAC activates first in response to moderate levels of OS followed by the activation of the large conductance mPTP, which leads to irreversible mitochondrial membrane potential (ΔΨm) depolarization (i.e., induction of the mitochondrial permeability transition, MPT) (Aon et al., 2007). Indeed, both channels have been implicated in mitochondrial dysfunction through a regenerative, autocatalytic process known as ROS-induced ROS-release (RIRR) which can culminate in electrical dysfunction or cell death (Zorov et al., 2000, 2006; Yang et al., 2010; Biary et al., 2011; Akar, 2013).
While the role of the mPTP in the activation of necrotic cell death pathways is well established, we and others have demonstrated the importance of IMAC in OS-induced arrhythmias (Akar et al., 2005; Akar and O'Rourke, 2011). In those studies, IMAC (but not mPTP) blockade effectively abrogated pathological OS-induced ΔΨm and action potential (AP) oscillations and prevented post-ischemic arrhythmias (Akar et al., 2005). It is important to note, however, that our previous studies focused on relatively short episodes of ischemia-reperfusion (I/R) injury which did not result in myocardial infarction (MI) (Akar et al., 2005; Lyon et al., 2010). Given the hierarchal nature of mitochondrial channel activation (Aon et al., 2007), we hypothesized that the mPTP may only play a prominent role under conditions of more extreme OS. Indeed, the immunosuppressive agent, Cyclosporin A (CsA), a desensitizer of the mPTP in the heart through its effect on Cyclophilin-D (CyP-D), has been shown to be effective in reducing infarct size in patients (Piot et al., 2008; Hausenloy et al., 2012). Despite these encouraging clinical findings, the efficacy of CsA in preventing arrhythmias is unclear (Arteaga et al., 1992; Ko et al., 1997; Schreiner et al., 2004), and recent experimental, preclinical (Lie et al., 2008), and clinical findings (Ghaffari et al., 2013) have cast new doubts regarding the overall utility and safety profile of CsA.
Mitochondria play a dual role: on the one hand, they initiate cell death and injury pathways through energy dissipating channels, such as the mPTP, but on the other, they act as central mediators of cardioprotection (Penna et al., 2013). Indeed, multiple stimuli (i.e., ischemic pre- and post-conditioning protocols, pharmacological agents and volatile anesthetics) limit cardiac damage by activating powerful cardioprotective signaling cascades which converge on mitochondria, in large part, through mitochondrial ATP-sensitive K (mKATP) channels (Liu et al., 1998, 1999; Sato and Marban, 2000; Garlid et al., 2009). Whether mKATP channels functionally interact with components of the mPTP in a manner that modulates the response of the heart to OS is unclear. In the present study, we set out to address this issue directly in a model of acute OS that was specifically designed to elicit significant ΔΨm depolarization and electrical dysfunction. We found that the efficacy of CsA in limiting OS-induced mitochondrial and electrical dysfunction was dictated by strong functional cross-talk between the mPTP and mKATP channels through a protein kinase C (PKC)-dependent pathway. Our findings highlight the importance of enhancing mKATP channel activity during CsA administration for limiting OS-induced electrical dysfunction, and may explain discrepant reports of the utility and potential toxicity of CsA.
Materials and methods
All procedures involving the handling of animals were approved by the Animal Care and Use Committee of the Mount Sinai School of Medicine and adhered with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Guinea pig hearts (N = 61) were rapidly excised, washed with ice cold cardioplegic solution, transferred to a Langendorff apparatus, and retrogradely perfused through the aorta with oxygenized (95% O2–5% CO2) Tyrodes solution containing (in mM): 130 NaCl, 1.2 MgSO4, 25 NaHCO3, 4.75 KCl, 5 Dextrose, and 1.25 CaCl2 at 36 ± 1°C. Perfusion pressure was maintained at 60–65 mmHg by adjusting perfusion flow rate. Hearts were suspended in the buffer filled, temperature controlled chamber, as we have recently reported (Jin et al., 2010; Lyon et al., 2010). Volume-conducted electrocardiograms were recorded for rhythm analysis using non-contact silver electrodes placed within the chamber. ECG signals were recorded continuously throughout the entire ex vivo perfusion protocol. Left ventricular (LV) cavity pressure (LVP) was measured using a buffer filled latex balloon (Harvard apparatus) that was carefully inserted through the mitral valve into the LV cavity. Signals were amplified (ECG100-MP150 Amplifier, Biopac Systems, CA, USA) and displayed in real-time using the AcqKnowledge 3.9 software package (Biopac Systems). Hearts were positioned such that the mapping field was centered over a 4 × 4-mm2 region of LV epicardium, midway between apex and base. These preparations remain stable for over 4 h of perfusion.
High-resolution optical ΔΨm imaging in ex vivo perfused guinea pig hearts
We used a validated semi-quantitative imaging technique of optical ΔΨm mapping using the ΔΨm-sensitive dye tetramethylrhodamine methylester (TMRM) (Jin et al., 2010; Lyon et al., 2010; Smeele et al., 2011; Nederlof et al., 2013). This method allows the assessment of mitochondrial function at a subcellular resolution within the intact organ (Jin et al., 2010; Lyon et al., 2010). Briefly, following cannulation, hearts were allowed to stabilize for 20 min at physiological temperature. Hearts were then stained with TMRM (250 nM; Molecular Probes Inc.) mixed in a 500 mL volume of Tyrodes solution (dye loading phase) for 20 min. This was followed by a 20–30 min dye washout phase. TMRM background fluorescence intensity was measured periodically (in 1 min intervals) throughout the entire experiment using a 6400 pixel CCD based optical imaging approach that allowed the measurement of normalized ΔΨm with subcellular resolution (50 μm) over a 4 × 4-mm2 window of the epicardial surface. To measure TMRM background fluorescence, hearts were excited with filtered light (525 ± 20 nm) emitted from a quartz tungsten halogen lamp (Newport Corporation, CT, USA). Emitted fluorescence was filtered (585 ± 20 nm for TMRM) and focused onto a high-resolution CCD camera (Scimeasure, GA, USA). During dye washout, the stability of TMRM background fluorescence was evaluated in real-time, as this baseline level served for normalization purposes during OS. In all experiments, the dye washout phase was associated with stable signal intensity.
High-throughput analysis of optical signals was performed using custom designed software. Peak emitted TMRM fluorescence signal from each of 6400 pixels was measured before and after excitation. TMRM background fluorescence was baseline corrected by subtracting fluorescence levels before dye staining (<0.1%) for each pixel. Background corrected TMRM fluorescence (ΔΨm) during the OS protocol was then normalized to the value of steady-state TMRM fluorescence achieved during the dye washout phase for each of the 6400 individual pixels. Normalized ΔΨm measurements during OS across the imaged 4 × 4-mm2 region of the heart were plotted as contour maps using Delta Graph 5.6 (Red Rock Software).
High-resolution optical action potential mapping in ex vivo perfused hearts
For optical AP mapping studies, hearts were stained with di-4-ANEPPS for 10 min as previously described (Akar et al., 2005; Xie et al., 2013). Hearts were paced at a steady-state pacing cycle length (PCL) of 300 ms. Unlike ΔΨm imaging, optical AP mapping requires motion suppression; hence 10 mM BDM was used in this subset of studies.
Ex vivo models of acute OS leading to irreversible ΔΨm depolarization
Perfusion of hearts with H2O2 is a well-established model of acute OS that results in triggered activity (Sato et al., 2009) as well as sustained atrial (Morita et al., 2010) and ventricular tachyarrhythmias (Morita et al., 2009; Biary et al., 2011). Following dye washout and stabilization, hearts were perfused with 200 μM H2O2 (Sigma-Aldrich) in Tyrodes for 30 min to elicit OS. We found that this model consistently gives rise to the regenerative process of RIRR (Biary et al., 2011), which culminates in significant ΔΨm depolarization, contractile and electrical dysfunction (Figure 1). This model served as the platform for investigating the role of mitochondrial ion channel complexes in the modulation of OS-induced ΔΨm and arrhythmias. Specifically, we focused on the role of the mPTP and its cross-talk with the mKATP. The following agents and concentrations were used in the present study: (a) CsA (0.1 μM, mPTP blocker), (b) Chelerythrine Chloride (CHE, 10 μM, PKC inhibitor), (c) 5-Hydroxydecanoate (5-HD, 100 μM, mKATP blocker), and (d) diazoxide (DZX, 30μM, mKATP agonist). Drug delivery was initiated 10 min before OS and was maintained throughout the entire protocol.
Figure 1.
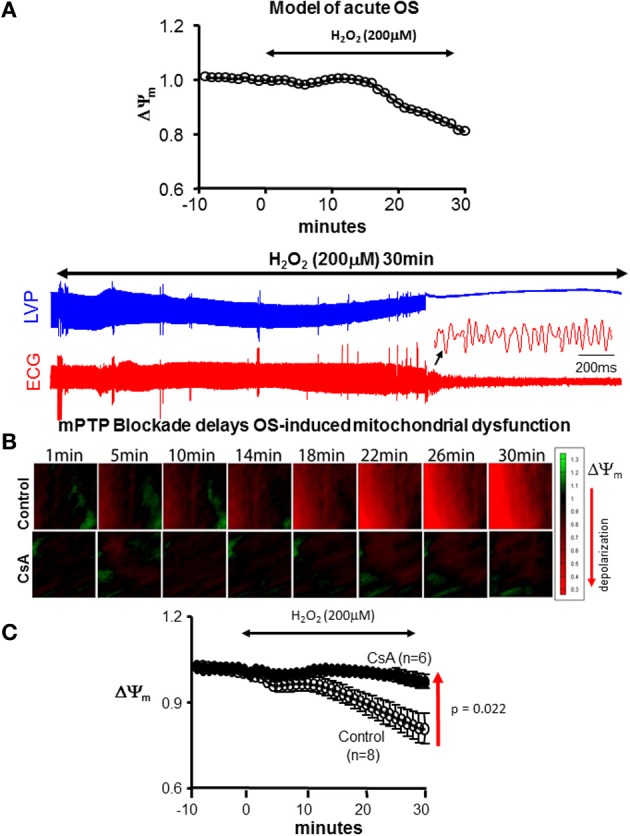
CsA delays OS-induced mitochondrial and contractile dysfunction. (A) Model of acute OS. Continuous 200 μM H2O2 perfusion for 30 min leads to ΔΨm depolarization and initiation of VF. (B) Sequences of isopotential contour maps depicting the spatio-temporal distribution of ΔΨm in representative control (untreated) and CsA-treated hearts following challenge with H2O2 (200 μM) for 30 min. (C) Average ΔΨm response to acute OS in all control and CsA treated hearts.
Statistical analysis
Values were expressed as mean ± SE. Differences between two groups were compared using the Student's t-test and were considered significant for p < 0.05.
Results
Acute model of OS-induced mitochondrial and electrical dysfunction
The main objective of the present work was to test the efficacy of mPTP blockade in protecting against OS-induced mitochondrial and electrical dysfunction. To that end, we used a simple ex vivo model of acute OS by H2O2 challenge (Figure 1A). This consistently resulted in significant ΔΨm depolarization, contractile, and electrical dysfunction. Within 40 min of H2O2, 5/6 hearts underwent spontaneous onset of VF with the remaining heart exhibiting electrical silence. As such, this model served as a reliable platform for investigating the role of mitochondrial ion channel complexes in the functional modulation of OS-induced mitochondrial and electrical dysfunction.
CsA protects against OS-induced mitochondrial and contractile but not electrical dysfunction
We began by investigating the efficacy of CsA in altering the functional response of hearts to acute OS. Shown in Figure 1B are ΔΨm isopotential contour maps from untreated (control) and CsA-treated hearts following challenge with H2O2. Also shown are the average normalized ΔΨm responses from all hearts. As expected, H2O2 challenge resulted in significant ΔΨm depolarization in control hearts. On average, ΔΨm was reduced by 22.1% within 30 min of H2O2 perfusion. Remarkably, CsA treatment completely abolished this response, as ΔΨm remained fully polarized during the same time-course in CsA-treated compared to untreated control hearts (Figure 1). Following 30 min of H2O2 challenge, ΔΨm was 19.3% greater (p = 0.021) in CsA-treated compared to control hearts.
We next tested whether modulation of OS-induced ΔΨm depolarization and its prevention by CsA had a functional impact in terms of contractile (Figures 2A–C) and electrical (Figures 2D,E) properties. As expected, H2O2 perfusion in control hearts resulted in a gradual decrease and eventual loss of contractile function. Interestingly, prevention of ΔΨm depolarization by CsA was associated with relative protection against contractile dysfunction as the loss of LVP was delayed by >8 min, p = 0.01 (Figure 2). We next investigated whether protection against OS-induced mitochondrial dysfunction by CsA translated into an electrical benefit by either preventing or delaying the onset of VF. Surprisingly, we found that CsA treatment failed to protect against the incidence of arrhythmias as the time to onset of VF following H2O2 challenge was comparable (p = NS) in control and CsA-treated hearts (Figures 2D,E).
Figure 2.

CsA does not protect against electrical dysfunction. (A) Representative time-series of LV pressure waveforms during 30 min of H2O2 challenge, indicating reduction and ultimate loss of contractile function. Loss of LVP is delayed in CsA treated hearts. (B,C) Average LVP normalized to the baseline pre-H2O2 value in all control and CsA hearts. Experiments summarized in (A–C) where performed under BDM-free conditions. (D,E) ECG traces in control and CsA treated hearts during H2O2 challenge and time of onset of VF in all hearts irrespective of BDM presence in the purfase. The transition to VF is not delayed by CsA treatment, indicating lack of electrical protection by CsA. BDM, 2,3-Butanedione monoxime; LVP, Left ventricular pressure; ECG, electrocardiogram; VF, ventricular fibrillation. **p < 0.01.
Paradoxical effect of CsA
Previously, we and others have highlighted the importance of maintaining ΔΨm polarization in the protection against OS-induced arrhythmias (Akar et al., 2005; Brown et al., 2010; Lyon et al., 2010). We, therefore, proceeded to investigate the basis for our discrepant findings regarding the role of CsA in protecting against OS-induced ΔΨm depolarization but not electrical dysfunction. We hypothesized that differences in experimental settings, particularly with regards to the use of the electromechanical uncoupling agent BDM in AP but not ΔΨm studies may underlie the discrepant outcomes that we observed. Therefore, we repeated our ΔΨm measurements with and without addition of BDM to the perfusate. Remarkably, we found that BDM completely reversed the protective effect of CsA on OS-induced ΔΨm depolarization which we had initially observed (Figure 3A, gray background), as CsA-treated hearts exhibited a more rapid ΔΨm depolarization compared to untreated controls following H2O2 challenge when BDM was present in the perfusate (Figure 3). As such, the use of BDM revealed a paradoxical effect of CsA which was consistent with exacerbation rather than protection against OS-induced mitochondrial dysfunction. Importantly, BDM alone (i.e., without CsA) did not alter the ΔΨm response of the heart to OS.
Figure 3.
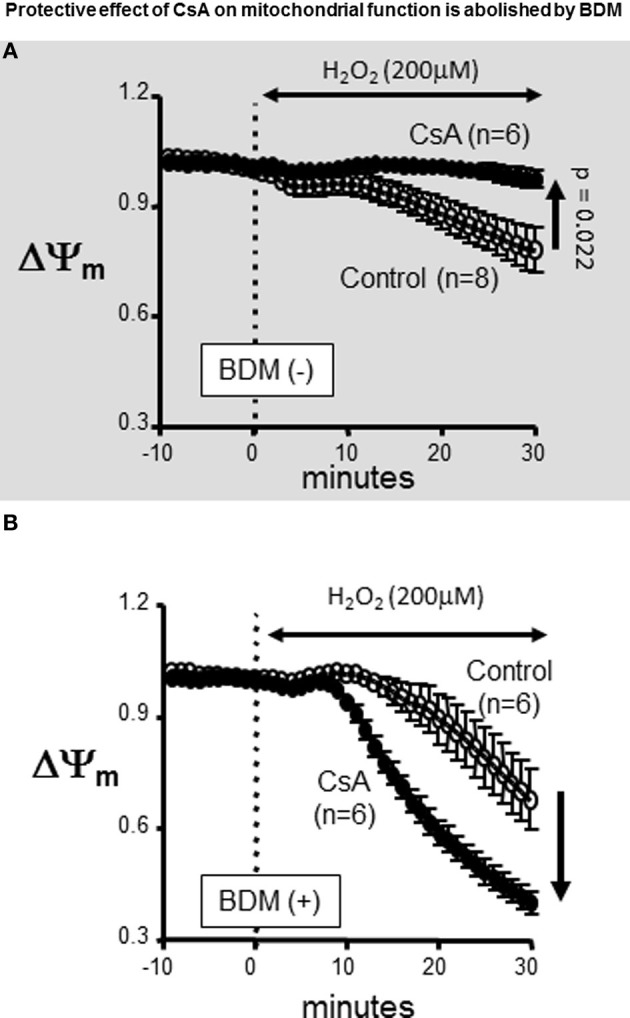
Protective effect of CsA on mitochondrial function is abolished by BDM. Treatment with the chemical phosphatase BDM abolishes the protective effects of CsA. (A) For reference, ΔΨm response presented in Figure 1 indicating protection by CsA against OS-induced ΔΨm depolarization. (B) Addition of the chemical phosphatase BDM (10 mM) reversed the effect of CsA on ΔΨm. BDM, 2,3-Butanedione monoxime.
Protein kinase C inhibition abrogates the protective effect of CsA on OS-induced mitochondrial dysfunction
BDM is a strong chemical phosphatase, which is known to oppose the phosphorylation of serine/threonine target proteins and to increase ATP depletion in metabolically challenged cardiomyocytes (Stapleton et al., 1998). Therefore, we hypothesized that the paradoxical effect that was unmasked by BDM in terms of CsA-mediated dysfunction may be due to its interference with the activity of the cardioprotective mKATP channel which is known to modulate mPTP opening at least in vitro. Since mKATP activity is dependent on PKC-mediated phosphorylation, we replaced BDM with the selective PKC inhibitor, CHE. As shown in Figure 4, addition of CHE completely abolished the protective effect conferred by CsA. Of note, CHE failed to alter the ΔΨm response of the heart to OS when delivered alone (i.e., without CsA) (Figure 4, lower panel).
Figure 4.
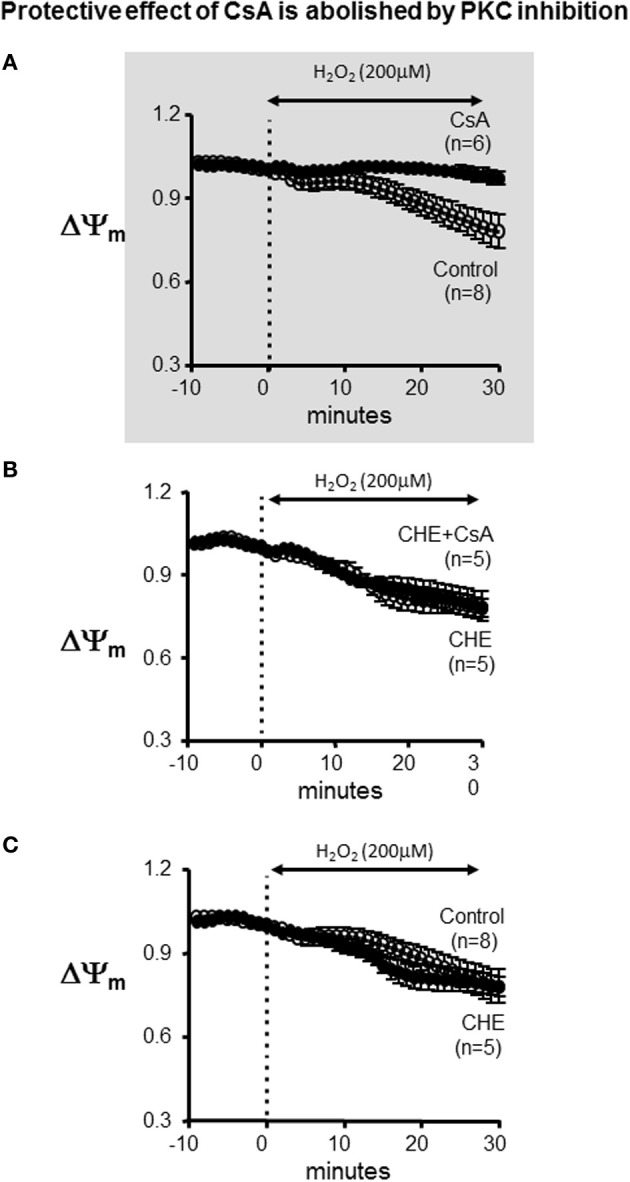
Protective effect of CsA is abolished by PKC inhibition. (A) For reference, ΔΨm response presented in Figure 1 indicating protection by CsA against OS-induced ΔΨm depolarization. (B) Average ΔΨm response to CsA treatment in the presence of the specific PKC inhibitor CHE. (C) Average ΔΨm response in control and CHE treated hearts. CHE alone (i.e., without CsA) did not alter the response. CHE, Chelerythrine; PKC, Protein kinase C.
Direct mKATP blockade abrogates the protective effect of CsA on mitochondrial dysfunction
Since PKC has multiple mitochondrial targets that may alter the response of the heart to OS, we tested whether direct pharmacological blockade of mKATP recapitulated the inhibitory effects of CHE on CSA-mediated cardioprotection. Indeed, addition of 5-HD completely abrogated the protective effect of CsA on ΔΨm depolarization (Figure 5). The ΔΨm response to H2O2 was identical between the control and the combined 5-HD+CsA treated hearts, highlighting the notion that CsA was completely ineffective as a cardioprotective agent under conditions that prevented mKATP channel activation.
Figure 5.

Protective effect of CsA is modulated by mKATP. (A) For reference, ΔΨm response presented in Figure 1 indicating protection by CsA against OS-induced ΔΨm depolarization. (B) Treatment with the mKATP antagonist abrogates the protective effect of CsA on mitochondrial dysfunction. (C) mKATP activation by DZX protects against OS-induced ΔΨm depolarization. (D) BDM abolishes the protective effect of DZX on mitochondrial dysfunction. DZX, Diazoxide.
Pharmacological activation of mKATP protects against mitochondrial dysfunction
To further establish the role of mKATP in the modulation of the ΔΨm response to OS, we used the pharmacological agonist of the channel, DZX (Figure 5). Interestingly, DZX-treated hearts exhibited a markedly blunted ΔΨm response compared to control hearts; thereby, establishing the efficacy of mKATP in modulating the opening of mPTP. Once again, the protective effect of DZX on OS-induced ΔΨm depolarization was prevented by addition of the chemical phosphatase BDM.
Protective effect of CsA on arrhythmias is dependent on mKATP channel activation
Previously, we and others showed that interventions that stabilized ΔΨm were associated with protection against post-ischemic arrhythmias, whereas conditions leading to ΔΨm instability promoted electrical dysfunction (Akar et al., 2005). Therefore, we asked whether modulation of the ΔΨm response in this model of acute OS could also explain differential vulnerability to arrhythmias. Seven groups were examined in terms of their relative sensitivities to OS-induced mitochondrial dysfunction (quantified by the slope of ΔΨm depolarization) and electrical dysfunction (assessed by the time to onset of VF). As shown in Figure 6, conditions that led to accelerated ΔΨm depolarization were indeed associated with enhanced vulnerability to VF as they exhibited significantly (p < 0.05) shorter time to onset of VF in response to OS challenge. While 11/13 BDM (+) hearts exhibited early (within 15 min) onset of VF, only 1/19 BDM (−) hearts were prone to VF within this short time-frame. These findings indicate significantly heightened sensitivity to sustained arrhythmias of hearts treated with the chemical phosphatase (p = 0.000006).
Figure 6.

Cross-talk between mKATPand mPTP modulates mitochondrial and electrical response of hearts to OS. (A) ΔΨm response to OS in CsA-treated hearts without (top) and with (bottom) concomitant perfusion with the chemical phosphatase BDM. The relative sensitivity of hearts to OS-induced mitochondrial dysfunction was quantified by measuring the slope of ΔΨm collapse 10–20 min following H2O2 perfusion (red line). (B) Average slope of OS-induced ΔΨm change in all groups tested. (C) Representative ECG traces from all groups tested, indicating vulnerability to VF in BDM (+) hearts. (D) Time to onset of VF following OS challenge as an index of electrical vulnerability in all hearts from all groups. VF, ventricular fibrillation.
To further address the link between mitochondrial and electrophysiological instability and to elucidate the potential mechanism underlying the CsA mediated pro-arrhythmic effect which we uncovered under conditions that prevented mKATP channel activation and that led to more pronounced ΔΨm depolarization (Figure 7A), we performed detailed optical AP mapping. Analysis of AP properties revealed accelerated shortening of APD in response to OS in CsA-treated hearts compared to controls (Figures 7B–E). These data suggest a heightened electrophysiological sensitivity to OS, particularly with regards to the activation of repolarizing currents as the potential mechanism for CsA-mediated pro-arrhythmia (Figure 7).
Figure 7.

(A) ΔΨm at 15 min of OS normalized to baseline value in control and CsA treated hearts. (B,C) Representative action potentials recorded during early challenge with acute OS in control and CsA treated hearts. AP shortening is more pronounced in CsA compared to control hearts, indicating heightened sensitivity to OS. (D) OS-induced % change in APD relative to baseline (pre-H2O2 value) in control and CsA treated hearts. (E) Representative APD contour maps showing global shortening of APD in response to OS in CsA treated hearts compared to controls.
Discussion
Acute OS manifests in a majority of patients with coronary artery disease, the leading cause of arrhythmic deaths in the United States. Central to the pathology of OS is mitochondrial dysfunction. Although the role of mitochondria as mediators of cell injury is well established, their contribution to arrhythmias is less understood. Indeed, the exact mitochondrial transport pathways that modulate the susceptibility of the heart to electrical dysfunction remain unclear.
In the present work we focused on the mPTP because of its established role in cellular necrosis and MI. Specifically, we investigated the efficacy of CsA in protecting against OS-induced mitochondrial and electrical dysfunction. We chose a simple ex vivo model of H2O2 challenge which reliably and predictably causes ΔΨm depolarization and VF within a relatively short (<30 min) time-frame. Our experiments revealed important discrepancies which initially appeared to discredit our central hypothesis that stabilization of ΔΨm is anti-arrhythmic, as CsA seemed to protect against ΔΨm depolarization but worsen electrical dysfunction. Further analysis revealed the basis of these discrepant observations, as we uncovered a dual role for CsA in either protecting or impairing cardiac function depending on the cellular milieu. As will be discussed below, our initial findings led us to refine our central hypothesis by examining the functional cross-talk between the mPTP and the cardioprotective mKATP channels in ultimately mediating the response of the heart to OS. In the present work, we highlight the importance of mKATP channel availability in determining the efficacy of CsA as a cardioprotective agent.
Mitochondrial ion channels as mediators of cardiac dysfunction: role of mPTP
ΔΨm is a key metric of mitochondrial function as it forms the proton-motive force used for ATP synthesis. In normal hearts, ΔΨm is tightly regulated such that ATP synthesis and ROS generation are maintained within a physiological range. In response to OS, ΔΨm is disrupted, altering over-all energy and redox balance within cardiac myocytes. Specifically, under these conditions, ROS build-up can exceed a threshold level that triggers the sequential opening of mitochondrial channels in a hierarchal manner (IMAC followed by mPTP) (Aon et al., 2007), which in turn leads to ΔΨm instability. ΔΨm instability can lead to inexcitability at the cellular level and conduction block and arrhythmias at the organ level, via a mechanism termed “metabolic sink” (Akar et al., 2005). Furthermore, pharmacological blockade of IMAC which blunted ΔΨm depolarization improved electrical and functional recovery of the heart following IR injury (Akar et al., 2005; Brown et al., 2008; Aon et al., 2009). That work, however, focused on relatively mild levels of OS produced by short episodes of IR injury. Since energy dissipating mitochondrial channels exhibit a hierarchal activation pattern in response to rising ROS levels (Aon et al., 2007), we focused in the present work on the efficacy of mPTP blockade by CsA in a model that was tailored to reliably depolarize ΔΨm and generate VF via MPT formation.
Cyclosporin a as a cardioprotective agent
The initial success of the immunosuppressive agent CsA in reducing infarct size in patients with coronary artery disease through its potent CycP-D inhibitory activity has fueled considerable interest in its potential use as a therapeutic agent for a wide variety of cardiovascular disorders (Piot et al., 2008; Hausenloy et al., 2012). Despite these encouraging clinical findings, the efficacy of CsA in preventing arrhythmias is unclear (Arteaga et al., 1992; Ko et al., 1997; Schreiner et al., 2004), and recent experimental, preclinical (Lie et al., 2008), and clinical findings (Ghaffari et al., 2013) have cast new doubts regarding the overall utility of CsA. Our current work was designed to directly address issues related to CsA efficacy in improving metabolo-electrical function under conditions of OS.
Consistent with the expected therapeutic benefit of preventing irreversible mPTP opening, CsA treatment in our experiments significantly delayed the onset of OS-induced ΔΨm collapse and the loss of contractile function in ex vivo perfused hearts (Figures 1, 2). However, we found that this metabolo-contractile improvement did not translate into an electrical benefit (Figure 2D). Rather, we found evidence of compromised electrical function with no improvement in the onset of VF. As such, our findings regarding lack of arrhythmic protection are fully consistent with those of Artega et al. who reported impairment rather than protection against reperfusion arrhythmias (Arteaga et al., 1992).
Our overarching hypothesis is that stabilization of ΔΨm is anti-arrhythmic. However, our initial findings regarding improved mitochondrial but not electrophysiological function by CsA appeared to disprove this premise. This prompted us to examine this issue in greater detail. As will be discussed next, our subsequent experiments led to the discovery of an intricate cross-talk within a mitochondrial macromolecular complex that ultimately dictated the functional response of the heart to OS, and conferred upon CsA a dual role as a mediator of protection or dysfunction depending on the specific cellular milieu.
Cross-talk between mPTP and mKATP in modulating ΔΨm and arrhythmias
A major finding of the present report is the demonstration that the cardiac response to OS is dictated by complex cross-talk between multiple mitochondrial transport mechanisms. Indeed, we found that OS is mediated through an intra-mitochondrial signaling pathway that can either worsen or protect against arrhythmias depending on the nature of its activation. We first showed that BDM, a classical electromechanical uncoupling agent, not only abrogated the protective effect afforded by CsA but rather led to an acceleration of OS-induced mitochondrial depolarization in response to CsA treatment. This paradoxical effect can explain the worsened electrical outcome in terms of APD shortening that we saw upon CsA treatment. Indeed, the synergy between accelerated mitochondrial depolarization and APD shortening is fully consistent with previous cellular and organ level findings (Akar et al., 2005; Aon et al., 2007). It is important to note that BDM alone (without CsA) did not alter the mitochondrial response to OS. This highlights an interaction of BDM with key proteins that modulate the mPTP (the main target of CsA).
Since BDM inhibits phosphokinases and actively dephosphorylates serine/threonine residues on multiple proteins (Stapleton et al., 1998), we hypothesized that the phosphorylation state of a certain target protein which interacts with elements of the mPTP may be critical for mediating the protective effects of CsA. Given the established role of PKC in mediating cardioprotection by ischemic pre and post-conditioning (Inagaki et al., 2006), we tested whether the detrimental effects of BDM could be explained (at least partially) through its PKC inhibitory activity. We addressed this issue by replacing BDM with the specific PKC inhibitor, CHE. Here too, protection against OS-induced mitochondrial dysfunction by CsA was completely abolished. As such, our findings are consistent with elegant work by the Mochly-Rosen group who highlighted the importance of PKC mediated signaling in the protection against mitochondrial dysfunction in multiple organ systems, including the heart, as well as pioneering molecular and biochemical work from the Garlid laboratory demonstrating the desensitization of mPTP to ROS by PKCε. Unlike BDM, however, CHE did not accelerate (i.e., worsen) the rate of mitochondrial depolarization suggesting involvement of additional off target effects by BDM that adversely impact mitochondrial function. These may include the effects of BDM on a variety of tyrosine kinases as well as its reported role in depleting ATP levels in myocytes under conditions of metabolic challenge (Stapleton et al., 1998). Our present findings not only inform regarding the signaling pathways involved in CsA mediated cardioprotection but also serve to emphasize the need to exercise caution when interpreting findings of studies in which BDM is used, especially those addressing issues relating to metabolic stress.
PKC has numerous target substrates that can impact mitochondrial function either directly or indirectly. One critical target of PKC signaling which has emerged from elegant work by the Marban group and others is the mKATP channel (Sato et al., 1998). We tested whether direct pharmacological modulation of the channel could potentially explain the detrimental effects of PKC inhibition which we observed. Indeed, we found that 5-HD was as effective as CHE in fully abrogating the protective effects of CsA on OS-induced ΔΨm depolarization. Although Baines et al. demonstrated that PKCε interacts with multiple key components of the mPTP, including VDAC, ANT, and HKII independently of its interaction with mKATP(Baines et al., 2003), our present work argues that such mKATP-independent interactions do not impact the functional response of the intact heart to OS. Indeed, we provide functional data that extend previous in vitro studies and give credence to the notion that mKATP is the central mediator of the cardioprotective effects of PKC on the heart. In light of the importance of mKATP we went on to investigate the functional consequences of channel activation and found that DZX treatment was as protective as CsA in preventing ΔΨm collapse. Once again, that protection was prevented by the chemical phosphatase BDM, consistent with the notion that PKC mediated phosphorylation of mKATP is critical to its opening and therefore efficacy in cardioprotection.
Limitations
Our study has several important limitations that require mention. For one, we relied upon a pharmacological strategy using DZX and 5-HD to modulate the activity of mKATP. Although this standard approach which included carefully chosen concentrations was based on numerous published reports, we cannot fully exclude the possibility that minor mKATP-independent effects may have contributed to our findings.
Moreover, we used a non-ratiometric, semi-quantitative method of TMRM imaging to assess relative (not absolute) changes in mitochondrial function in ex vivo perfused hearts. Using this validated method, changes in TMRM fluorescence signal caused by altered cellular membrane potential as opposed to ΔΨm are negligible.
Finally, we used CsA to inhibit the mPTP. While this is a widely accepted and effective strategy in the heart, Li et al. have shown tissue-specific differences in CyP-D expression and therefore sensitivity to CsA (Li et al., 2012). In particular, they reported that mPTP inhibition in tissues exhibiting low expression of CyP-D, is achieved more effectively using Rotenone than CsA (Li et al., 2012).
Conclusion
In summary, our current findings highlight the notion that CsA-mediated cardioprotection against OS requires mKATP channel activation through a PKC-dependent pathway. Increasing mKATP activity during CsA administration is required for limiting OS-induced electrical dysfunction. On the other hand, CsA administration during conditions that may prevent mKATP channel activation may exert unintended pro-arrhythmic consequences through accelerated APD shortening. Our findings may explain existing controversy in the basic and clinical literature surrounding the utility of CsA as a cardioprotective agent.
Conflict of interest statement
The Associate Editor Miguel A. Aon declares that, despite publishing articles in the past with author(s) Fadi G. Akar, Chaoqin Xie, and Justin Kauffman, the review process was handled objectively and no conflict of interest exists. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL114378) and the American Heart Association (13GRNT17000046) to Fadi G. Akar.
References
- Akar F. G. (2013). Mitochondrial targets for arrhythmia suppression: is there a role for pharmacological intervention? J. Interv. Card. Electrophysiol. 37, 249–258 10.1007/s10840-013-9809-3 [DOI] [PubMed] [Google Scholar]
- Akar F. G., Aon M. A., Tomaselli G. F., O'Rourke B. (2005). The mitochondrial origin of postischemic arrhythmias. J. Clin. Invest. 115, 3527–3535 10.1172/JCI25371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akar F. G., O'Rourke B. (2011). Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol. Ther. 131, 287–294 10.1016/j.pharmthera.2011.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon M. A., Cortassa S., Akar F. G., Brown D. A., Zhou L., O'Rourke B. (2009). From mitochondrial dynamics to arrhythmias. Int. J. Biochem. Cell Biol. 41, 1940–1948 10.1016/j.biocel.2009.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon M. A., Cortassa S., Maack C., O'Rourke B. (2007). Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 282, 21889–21900 10.1074/jbc.M702841200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga D., Odor A., Lopez R. M., Contreras G., Pichardo J., Garcia E., et al. (1992). Impairment by cyclosporin a of reperfusion-induced arrhythmias. Life Sci. 51, 1127–1134 10.1016/0024-3205(92)90514-P [DOI] [PubMed] [Google Scholar]
- Baines C. P., Song C. X., Zheng Y. T., Wang G. W., Zhang J., Wang O. L., et al. (2003). Protein kinase cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 92, 873–880 10.1161/01.RES.0000069215.36389.8D [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biary N., Xie C., Kauffman J., Akar F. G. (2011). Biophysical properties and functional consequences of reactive oxygen species (ros)-induced ros release in intact myocardium. J. Physiol. 589, 5167–5179 10.1113/jphysiol.2011.214239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. A., Aon M. A., Akar F. G., Liu T., Sorarrain N., O'Rourke B. (2008). Effects of 4′-chlorodiazepam on cellular excitation-contraction coupling and ischaemia-reperfusion injury in rabbit heart. Cardiovasc. Res. 79, 141–149 10.1093/cvr/cvn053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. A., Aon M. A., Frasier C. R., Sloan R. C., Maloney A. H., Anderson E. J., et al. (2010). Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J. Mol. Cell. Cardiol. 48, 673–679 10.1016/j.yjmcc.2009.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlid K. D., Costa A. D., Quinlan C. L., Pierre S. V., Dos Santos P. (2009). Cardioprotective signaling to mitochondria. J. Mol. Cell. Cardiol. 46, 858–866 10.1016/j.yjmcc.2008.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffari S., Kazemi B., Toluey M., Sepehrvand N. (2013). The effect of prethrombolytic cyclosporine-a injection on clinical outcome of acute anterior st-elevation myocardial infarction. Cardiovasc. Ther. 31, e34–e39 10.1111/1755-5922.12010 [DOI] [PubMed] [Google Scholar]
- Hausenloy D. J., Boston-Griffiths E. A., Yellon D. M. (2012). Cyclosporin a and cardioprotection: From investigative tool to therapeutic agent. Br. J. Pharmacol. 165, 1235–1245 10.1111/j.1476-5381.2011.01700.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K., Churchill E., Mochly-Rosen D. (2006). Epsilon protein kinase c as a potential therapeutic target for the ischemic heart. Cardiovasc. Res. 70, 222–230 10.1016/j.cardiores.2006.02.015 [DOI] [PubMed] [Google Scholar]
- Jin H., Nass R. D., Joudrey P. J., Lyon A. R., Chemaly E. R., Rapti K., et al. (2010). Altered spatiotemporal dynamics of the mitochondrial membrane potential in the hypertrophied heart. Biophys. J. 98, 2063–2071 10.1016/j.bpj.2010.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko W. J., Lin F. L., Wang S. S., Chu S. H. (1997). Hypomagnesia and arrhythmia corrected by replacing cyclosporine with fk506 in a heart transplant recipient. J. Heart Lung Transplant. 16, 980–982 [PubMed] [Google Scholar]
- Li B., Chauvin C., De Paulis D., De Oliveira F., Gharib A., Vial G., et al. (2012). Inhibition of complex i regulates the mitochondrial permeability transition through a phosphate-sensitive inhibitory site masked by cyclophilin d. Biochim. Biophys. Acta 1817, 1628–1634 10.1016/j.bbabio.2012.05.011 [DOI] [PubMed] [Google Scholar]
- Lie R. H., Hasenkam J. M., Nielsen T. T., Poulsen R., Sloth E. (2008). Post-conditioning reduces infarct size in an open-chest porcine acute ischemia-reperfusion model. Acta Anaesthesiol. Scand. 52, 1188–1193 10.1111/j.1399-6576.2008.01756.x [DOI] [PubMed] [Google Scholar]
- Liu Y., Sato T., O'Rourke B., Marban E. (1998). Mitochondrial atp-dependent potassium channels: novel effectors of cardioprotection? Circulation 97, 2463–2469 10.1161/01.CIR.97.24.2463 [DOI] [PubMed] [Google Scholar]
- Liu Y., Sato T., Seharaseyon J., Szewczyk A., O'Rourke B., Marban E. (1999). Mitochondrial atp-dependent potassium channels. Viable candidate effectors of ischemic preconditioning. Ann. N.Y. Acad. Sci. 874, 27–37 10.1111/j.1749-6632.1999.tb09222.x [DOI] [PubMed] [Google Scholar]
- Lyon A. R., Joudrey P. J., Jin D., Nass R. D., Aon M. A., O'Rourke B., et al. (2010). Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. J. Mol. Cell. Cardiol. 49, 565–575 10.1016/j.yjmcc.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita N., Lee J. H., Xie Y., Sovari A., Qu Z., Weiss J. N., et al. (2010). Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J. Am. Coll. Cardiol. 57, 366–375 10.1016/j.jacc.2010.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita N., Sovari A. A., Xie Y., Fishbein M. C., Mandel W. J., Garfinkel A., et al. (2009). Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 297, H1594–H1605 10.1152/ajpheart.00579.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nederlof R., Xie C., Eerbeek O., Koeman A., Milstein D. M., Hollmann M. W., et al. (2013). Pathophysiological consequences of tat-hkii peptide administration are independent of impaired vascular function and ensuing ischemia. Circ. Res. 112, e8–e13 10.1161/CIRCRESAHA.112.274308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke B., Cortassa S., Akar F., Aon M. (2007). Mitochondrial ion channels in cardiac function and dysfunction. Novartis Found. Symp. 287, 140–151 discussion: 152–146. 10.1002/9780470725207.ch10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna C., Perrelli M. G., Pagliaro P. (2013). Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxid. Redox Signal. 18, 556–599 10.1089/ars.2011.4459 [DOI] [PubMed] [Google Scholar]
- Piot C., Croisille P., Staat P., Thibault H., Rioufol G., Mewton N., et al. (2008). Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359, 473–481 10.1056/NEJMoa071142 [DOI] [PubMed] [Google Scholar]
- Sato D., Xie L. H., Sovari A. A., Tran D. X., Morita N., Xie F., et al. (2009). Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc. Natl. Acad. Sci. U.S.A. 106, 2983–2988 10.1073/pnas.0809148106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Marban E. (2000). The role of mitochondrial k(atp) channels in cardioprotection. Basic Res. Cardiol. 95, 285–289 10.1007/s003950070047 [DOI] [PubMed] [Google Scholar]
- Sato T., O'Rourke B., Marban E. (1998). Modulation of mitochondrial atp-dependent k+ channels by protein kinase c. Circ. Res. 83, 110–114 10.1161/01.RES.83.1.110 [DOI] [PubMed] [Google Scholar]
- Schreiner K. D., Kelemen K., Zehelein J., Becker R., Senges J. C., Bauer A., et al. (2004). Biventricular hypertrophy in dogs with chronic av block: effects of cyclosporin a on morphology and electrophysiology. Am. J. Physiol. Heart Circ. Physiol. 287, H2891–H2898 10.1152/ajpheart.01051.2003 [DOI] [PubMed] [Google Scholar]
- Smeele K. M., Southworth R., Wu R., Xie C., Nederlof R., Warley A., et al. (2011). Disruption of hexokinase II-mitochondrial binding blocks ischemic preconditioning and causes rapid cardiac necrosis. Circ. Res. 108, 1165–1169 10.1161/CIRCRESAHA.111.244962 [DOI] [PubMed] [Google Scholar]
- Stapleton M. T., Fuchsbauer C. M., Allshire A. P. (1998). Bdm drives protein dephosphorylation and inhibits adenine nucleotide exchange in cardiomyocytes. Am. J. Physiol. 275, H1260–H1266 [DOI] [PubMed] [Google Scholar]
- Xie C., Biary N., Tocchetti C. G., Aon M. A., Paolocci N., Kauffman J., et al. (2013). Glutathione oxidation unmasks proarrhythmic vulnerability of chronically hyperglycemic guinea pigs. Am. J. Physiol. Heart Circ. Physiol. 304, H916–H926 10.1152/ajpheart.00026.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Korge P., Weiss J. N., Qu Z. (2010). Mitochondrial oscillations and waves in cardiac myocytes: insights from computational models. Biophys. J. 98, 1428–1438 10.1016/j.bpj.2009.12.4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov D. B., Filburn C. R., Klotz L. O., Zweier J. L., Sollott S. J. (2000). Reactive oxygen species (ros)-induced ros release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014 10.1084/jem.192.7.1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov D. B., Juhaszova M., Sollott S. J. (2006). Mitochondrial ros-induced ros release: an update and review. Biochim. Biophys. Acta 1757, 509–517 10.1016/j.bbabio.2006.04.029 [DOI] [PubMed] [Google Scholar]