

Abstract
Chromatin-regulating proteins represent a large class of novel targets for cancer therapy. In the context of radiotherapy, acetylation and deacetylation of histones by histone acetyltransferases (HATs) and histone deacetylases (HDACs) play important roles in the repair of DNA double-strand breaks generated by ionizing irradiation, and are therefore attractive targets for radiosensitization. Small-molecule inhibitors of HATs (garcinol, anacardic acid and curcumin) and HDACs (vorinostat, sodium butyrate and valproic acid) have been shown to sensitize cancer cells to ionizing irradiation in preclinical models, and some of these molecules are being tested in clinical trials, either alone or in combination with radiotherapy. Meanwhile, recent large-scale genome analyses have identified frequent mutations in genes encoding chromatin-regulating proteins, especially in those encoding subunits of the SWI/SNF chromatin-remodeling complex, in various human cancers. These observations have driven researchers toward development of targeted therapies against cancers carrying these mutations. DOT1L inhibition in MLL-rearranged leukemia, EZH2 inhibition in EZH2-mutant or MLL-rearranged hematologic malignancies and SNF5-deficient tumors, BRD4 inhibition in various hematologic malignancies, and BRM inhibition in BRG1-deficient tumors have demonstrated promising anti-tumor effects in preclinical models, and these strategies are currently awaiting clinical application. Overall, the data collected so far suggest that targeting chromatin-regulating proteins is a promising strategy for tomorrow's cancer therapy, including radiotherapy and molecularly targeted chemotherapy.
Keywords: chromatin remodeling, histone modification, histone acetyltransferase, SWI/SNF complex, synthetic lethality, BRM
INTRODUCTION
Nucleosomes, which consist of chromosomal DNA and histone proteins, form a highly condensed structure known as chromatin. Recently, proteins involved in the regulation of nucleosome structure (i.e. chromatin-regulating proteins) have emerged as novel targets for cancer therapy. Recent studies have revealed that several chromatin-regulating proteins play pivotal roles in the repair of DNA double-strand breaks (DSBs) generated by ionizing irradiation (IR), and that inhibition of their activities leads to radiosensitization of cancer cells in preclinical models. Furthermore, recent large-scale genome analyses have identified frequent mutations in genes encoding chromatin-regulating proteins, especially those encoding subunits of the SWI/SNF chromatin-remodeling complex, in various human cancers. These findings have driven development of personalized treatment strategies based on the gene-mutation status of individual tumors. To date, several attractive treatment strategies targeting chromatin-regulating proteins have been proposed, and some of them are being tested in the clinic. In this article, we discuss the emerging strategies for targeting chromatin-regulating proteins in cancer therapy, including radiotherapy and molecularly targeted chemotherapy.
CHROMATIN STRUCTURE AND ITS ALTERATION BY CHROMATIN-REGULATING PROTEINS
Chromosomal DNA and histone proteins form a highly condensed structure known as chromatin (Fig. 1a). The basic unit of chromatin is the nucleosome, which consists of DNA wound around an octamer of histones H2A, H2B, H3 and H4. In human cells, many activities essential for cell survival, such as DNA transcription, synthesis and repair, are mediated by dynamic changes in nucleosome structure that facilitate access of DNA-binding proteins to double-stranded DNA [1]. Proteins that regulate the change in nucleosome structure are called chromatin-regulating proteins. These proteins can be classified into two groups that take part in distinct mechanisms: histone modification (Fig. 1b) and chromatin remodeling (Fig. 1c). Some histone modifiers attach substrates, such as phosphate, poly-ADP-ribosyl, acetyl, methyl, SUMOyl and ubiquityl groups to histone tails by covalent interaction, whereas other histone modifiers detach these groups from previously modified histones. Chromatin remodelers usually function as complexes that change nucleosome structure (e.g. by forming a DNA-loop or sliding a nucleosome) in an ATP-dependent manner. At present, several chromatin-remodeling complexes have been identified, including SWI/SNF, ISWI, INO80, SWR1, NURD/Mi2/CHD and NURF [1].
Fig. 1.

Chromatin structure and its alteration by two distinct mechanisms: histone modification and chromatin remodeling. (a) Structure of chromatin and nucleosome. (b) Examples of histone modification. Ac = acetylation, PAR = poly-ADP-ribosylation, P = phosphorylation, SUMO = SUMOylation, Ub = ubiquitination. (c) Chromatin remodeling: DNA-loop formation (left) and nucleosome sliding (right).
CHROMATIN-REGULATING PROTEINS AS TARGETS FOR RADIOSENSITIZATION
IR kills cancer cells by generating DNA damage. Among the types of DNA damage, DNA DSBs are considered to be most lethal. Repair of IR-generated DSBs requires alteration of chromatin structure at DSB sites by chromatin-regulating proteins [2]. Therefore, chromatin-regulating proteins involved in DSB repair are potential targets for radiosensitization. Recent findings regarding radiosensitization of cancer cells by targeting chromatin-regulating proteins are summarized below.
Histone acetyltransferases
Histone acetyltransferases (HATs) are a class of histone modifiers that attach acetyl units to specific lysine residues of nucleosomal histones. To date, several distinct families of HATs have been identified: the homologs CBP and p300, PCAF, GCN5, and the MYST family (which includes TIP60) [3]. Recent studies have demonstrated that histone acetylation by HATs at DSB sites facilitates alteration of chromatin structure to an ‘open’ state, allowing repair proteins to access the DNA ends [2]. We and others have revealed that CBP, p300 and TIP60 promote non-homologous end joining (NHEJ) and homologous recombination (HR), two major pathways for DSB repair, through histone acetylation at DSB sites in human cells [4–7]. Furthermore, we showed that acetylation of histone H3 and H4 by CBP and p300 at DSB sites is essential for the recruitment of KU70 and KU80, two key proteins involved in NHEJ [4]. Murr et al. demonstrated that acetylation of histone H4 by TIP60 is required for the accumulation of proteins involved in HR at DSB sites [5]. Bird et al. reported that ESA1, a HAT responsible for acetylation of histone H4 in budding yeast, is required for NHEJ and replication-coupled repair [6]. CBP and p300 also function in HR by regulating transcriptional activity of the BRCA1 and RAD51 genes [7]. These findings predict that inhibition of such HATs leads to radiosensitization.
Several known compounds derived from natural ingredients exhibit HAT-inhibitory activity (Table 1). Curcumin, a major curcuminoid found in the spice turmeric, is a specific inhibitor of CBP and p300 [8]. Anacardic acid, derived from the shell of Anacardium occidentale (cashew nut), inhibits p300, PCAF and TIP60 [9–11], whereas garcinol, found in the rind of Garcinia indica (mangosteen), inhibits p300 and PCAF [12]. Recently, curcumin, anacardic acid and garcinol were shown to suppress NHEJ in an assay system in which NHEJ activity against DSBs on chromosomal DNA generated by I-SceI restriction enzyme can be evaluated in living human cancer cells by detecting expression of green fluorescent protein [4, 13]. Curcumin also suppresses HR in two ways: (i) by reducing expression of the BRCA1 gene, which regulates HR, by impairing histone acetylation at the BRCA1 promoter; and (ii) by inhibiting ataxia telangiectasia and Rad3-related protein (ATR) kinase, resulting in impaired activation of ATR-CHK1 signaling, which is necessary for HR and the DNA damage checkpoint pathway [14].
Table 1.
HAT inhibitors that suppress NHEJ in human cancer cells
| Compound | MW | Target HATs | Other target proteins/pathways | References |
|---|---|---|---|---|
| Curcumin | 368.38 | CBP, p300 | NF-kB pathway, AP-1, PI3K/Akt pathway | [22–24] |
| Anacardic acid | 342.47 | p300, PCAF, TIP60 | NF-kB pathway, LOX-1, Xanthine oxidase | [10] |
| Garcinol | 602.80 | p300, PCAF | NF-kB pathway, Src, MAPK/ERK, PI3K/Akt pathways | [25, 26] |
HAT = histone acetyltransferase, NHEJ = non-homologous end joining, MW = molecular weight.
As predicted, curcumin, anacardic acid and garcinol sensitize cancer cells to IR (Table 2) [4, 9, 13, 15–19]. In our study, the radiosensitizing effect of garcinol was strongest, comparable with the effects of well-known radiosensitizers that target DNA repair proteins, e.g. olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, and NU7026, a DNA-dependent protein kinase catalytic subunit (DNA-PKcs) inhibitor (Table 3) [13, 20, 21]. However, because curcumin, anacardic acid and garcinol may also affect many other proteins or pathways associated with cancer cell survival, it is not clear that the observed radiosensitizing effects of these compounds are entirely due to their HAT-inhibitory activity (Table 1) [10, 22–26].
Table 2.
Radiosensitization by HAT inhibitors
| Compound | Cells/mice | Cell lines | References |
|---|---|---|---|
| Curcumin | Cells, mice | SCC1 | [15] |
| Cells | H1299 | [4] | |
| Cells | HCT116 | [16] | |
| Cells | PC-3 | [17] | |
| Cells | PC-3 | [18] | |
| Anacardic acid | Cells | H1299 | [4] |
| Cells | SQ20B, SCC35, HeLa | [9] | |
| Cells | U2OS | [19] | |
| Garcinol | Cells | A549, HeLa | [13] |
HAT = histone acetyltransferase.
Table 3.
Radiosensitizing effect of HAT, PARP and DNA-PKcs inhibitors
| Compound | DER |
|---|---|
| Curcumin | 1.23 |
| Anacardic acid | 1.51 |
| Garcinol | 1.61 |
| Olaparib (PARP inhibitor) | 1.67 |
| NU7026 (DNA-PKcs inhibitor) | 1.77 |
HAT = histone acetyltransferase, PARP = poly (ADP-ribose) polymerase, DNA-PKcs = DNA-dependent protein kinase catalytic subunit, DER = dose enhancement ratio. DER was evaluated using the Celltiter-Glo assay [13].
Histone deacetylases
Histone deacetylases (HDACs) are another class of histone modifiers that remove acetyl units from lysine residues on histones. To date, 18 HDACs have been identified and classified into four groups (HDAC I, II, III and IV) according to their similarity, with reference to their analogues in yeast [27]. Several known compounds exhibit HDAC-inhibitory activity, and these compounds can be classified according to their structures and their inhibitory specificities relative to the HDAC classes. Hydroxamic acids, including suberoylanilide hydroxamic acid (SAHA or vorinostat) and panobinostat (LB589), inhibit class I and II HDACs. Short-chain fatty acids, such as sodium butyrate (NaB) and valproic acid (VPA), are active against class I and IIa HDACs. Cyclic peptides such as romidepsin mainly act against class I HDACs, but can also inhibit class II HDACs at higher concentrations. Benzamides, including entinostat and mocetinostat, inhibit class I HDACs.
Recent studies have implicated direct and indirect involvement of HDACs in both NHEJ and HR. Miller et al. showed that HDAC1 and HDAC2 are recruited to DNA DSB sites, where they reduce H3K56 acetylation and regulate recruitment of KU70 and Artemis, which are involved in NHEJ [28]. Meanwhile, several studies have shown that HDAC inhibitors suppress expression of DNA repair-related proteins such as KU70, KU80, KU86, DNA-PKcs and RAD51 (Table 4) [29–33]. These observations predict that HDAC inhibitors have a radiosensitizing effect. Accordingly, several recent studies support this idea (Table 4). Vorinostat radiosensitizes prostate cancer, glioma, multiple myeloma, osteosarcoma and rhabdomyosarcoma cell lines [29–31]; NaB radiosensitizes melanoma cells [32]; and VPA radiosensitizes colon cancer cells [33].
Table 4.
Radiosensitization by HDAC inhibitors in vitro
| Compound | Cell lines | Target DNA repair proteins | References |
|---|---|---|---|
| Vorinostat | DU145, U373 | DNA-PKcs, RAD51 | [29] |
| RPMI8226, U266B1, KMS-1, MM1.s | RAD51 | [30] | |
| KHOS-242OS, SAOS2, A-204, RD, hFOB1.19 | KU80, RAD51 | [31] | |
| NaB | A375, MeWo | KU70, KU80, DNA-PKcs | [32] |
| VPA | LS174T, HCT116 | [33] |
DNA-PKcs = DNA-dependent protein kinase catalytic subunit.
Even though HATs and HDACs seemingly act in an opposing manner, both HAT and HDAC inhibitors exert radiosensitizing effects, at least in vitro. A clue toward resolving this apparent paradox has emerged from recent studies suggesting that HATs and HDACs act coordinately before DSB repair [4, 5, 28]. Further investigation will elucidate the detailed mechanisms of the coordination of HATs and HDACs in DSB repair following IR, and this knowledge will facilitate development of methods for radiosensitization.
Chromatin remodelers
Several studies have demonstrated the involvement of chromatin remodelers in DSB repair after IR, and the radiosensitizing effects of inhibiting the activities of these proteins. Lan et al. demonstrated in osteosarcoma cells that ACF1 and SNF2H, subunits of the CHRAC chromatin-remodeling complex, engage in an interaction with KU70, which is required for repair of DSBs generated by IR, and that knockdown of ACF1 or SHF2H results in radiosensitization [34]. Meanwhile, we showed that ablation of BRM, a catalytic subunit of the SWI/SNF chromatin-remodeling complex, leads to suppression of recruitment of KU70 to DSB sites after laser micro-irradiation in lung cancer cells [4], suggesting BRM as a target for radiosensitizing agents. Furthermore, our preliminary results indicate that heterozygous knockout of BRG1, another catalytic subunit of the SWI/SNF complex, leads to radiosensitization in lymphoma cells (unpublished data). These data indicate that not only histone modifiers, but also chromatin remodelers, are candidate targets for radiosensitization.
Toward clinical applications
Historically, extensive research has been carried out to develop radiosensitizers. Even though a large number of known compounds exert radiosensitizing effects in preclinical models, most of them have been judged inadequate for clinical application due to their toxicity in normal human tissues [35]. Therefore, it is essential to develop radiosensitizers with satisfactory therapeutic windows, i.e. sufficient anti-tumor effects coupled with limited toxicity to normal tissues. It is considered that cancer cells have defects in the DNA damage response (DDR), including DNA repair and cell-cycle arrest, that makes them more vulnerable to IR than normal cells [36, 37]. In this regard, targeting chromatin-regulating proteins involved in DDR after IR may be a reasonable strategy for radiosensitization of cancer cells, which may result in a large therapeutic window. Several results from basic research support this speculation: neither garcinol nor NaB exert radiosensitizing effects on fibroblasts at concentrations that sensitize cancer cells to IR [13, 32]. However, because the mechanisms underlying their relatively stronger radiosensitizing effect in cancer cells have not been elucidated, further studies are warranted.
Several clinical studies have administered HAT- or HDAC-inhibitory compounds to humans. The HAT inhibitors include curcumin, used either alone or combined with radiotherapy and chemotherapy to treat cancer patients, and garcinol, used for weight-loss therapy [38]. In both of those cases, the side effects of these compounds were tolerable, at least when the drugs were used alone. Regarding HDAC inhibitors, several retrospective studies have shown that administration of VPA, alone or with chemotherapeutic agents, to patients with thyroid or brain tumors caused no increase in adverse effects [39, 40]. Moreover, in the first published prospective clinical trial using an HDAC inhibitor and radiation, combined therapy with vorinostat (100–400 mg daily) and palliative X-ray irradiation (a total of 30 Gy in 10 fractions) was well tolerated in 16 gastrointestinal carcinoma patients [41]. Future clinical trials should explore the optimal way to improve the efficacy of radiation therapy by targeting HATs and HDACs.
MUTATIONS IN CHROMATIN-REGULATING GENES IN HUMAN CANCERS
Recent large-scale genome analyses have identified frequent mutations in genes encoding chromatin-regulating proteins in human cancers. Thus, aberrations in histone modification and chromatin remodeling play important roles in the genesis and development of malignant tumors. In this review, we summarize known mutations in chromatin-regulating genes (Table 5). We focus in particular on mutations in genes encoding the SWI/SNF chromatin-remodeling complex because the mutation rates in these genes are extremely high in human cancers.
Table 5.
Mutation in chromatin-modifying genes in human cancers
| Gene | Function of gene product | Cancer type | Frequency (%) | References |
|---|---|---|---|---|
| Inactivating mutation |
||||
| BRG1/SMARCA4 | SWI/SNF catalytic subunit | Non-small-cell lung carcinoma | 35 | [54] |
| Lung adenocarcinoma | 10 | [55] | ||
| Burkitt lymphoma | 15 | [56] | ||
| Medulloblastoma | 4–13 | [57–59] | ||
| Esophageal cancer | 6 | [60] | ||
| Lung cancer (reduced expression) | 15–50 | [61–63] | ||
| BRM/SMARCA2 | SWI/SNF catalytic subunit | Lung cancer (reduced expression) | 38–75 | [61–63] |
| Gastric cancer (reduced expression) | 42 | [66] | ||
| Prostate cancer (reduced expression) | [67] | |||
| SNF5/SMARCB1/BAF47/INI1 | SWI/SNF core regulatory subunit | Rhabdoid tumor | 98 | [68, 69] |
| Meningioma | [70, 71] | |||
| Shwannoma | [70, 72] | |||
| ARID1A/BAF250A | SWI/SNF variant regulatory subunit | Ovarian clear cell carcinoma | 46–57 | [75, 76] |
| Endometriosis-associated ovarian carcinoma | 30 | [75, 76] | ||
| Renal clear cell carcinoma | 34 | [77] | ||
| Burkitt lymphoma | 15 | [56] | ||
| Hepatocellular carcinoma | 13 | [78] | ||
| Transitional cell carcinoma of the bladder | 13 | [79] | ||
| Gastric cancer | 10 | [80] | ||
| Esophageal cancer | 8 | [60] | ||
| Serous endometrial cancer of uterine | 6 | [81] | ||
| Neuroblastoma | 6 | [82] | ||
| Pancreatic cancer | 4 | [83] | ||
| Malignant melanoma | 3 | [84] | ||
| Medulloblastoma | [85] | |||
| Lung adenocarcinoma | [55] | |||
| ARID1B/BAF250B | SWI/SNF variant regulatory subunit | Hepatocellular carcinoma | 11 | [78] |
| Neuroblastoma | 7 | [82] | ||
| Breast cancer | 4 | [86] | ||
| Malignant melanoma | 2 | [84] | ||
| ARID2/BAF200 | SWI/SNF variant regulatory subunit | Hepatocellular carcinoma | 6–11 | [78, 87] |
| Malignant melanoma | 7 | [84] | ||
| Esophageal cancer | 5 | [60] | ||
| PBRM1/BAF180 | SWI/SNF variant regulatory subunit | Renal clear cell carcinoma | 41 | [77] |
| Esophageal cancer | 3 | [60] | ||
| Breast cancer | [88] | |||
| CPB/CREBBP | H3H4 acetyltransferase | B-cell lymphoma | 40 | [89] |
| Acute lymphoblastic leukemia | 18 | [90] | ||
| Small-cell lung carcinoma | 17 | [91] | ||
| Transitional cell carcinoma of the bladder | 13 | [79] | ||
| Lung cancer | 10 | [92] | ||
| p300 | H3H4 acetyltransferase | Transitional cell carcinoma of the bladder | 13 | [79] |
| Small-cell lung carcinoma | 10 | [91] | ||
| Colorectal, breast and pancreatic cancer | [93] | |||
| MLL | H3K4 methyltransferase | Acute leukemia | 5–10 | [94] |
| Small-cell lung carcinoma | 10 | [91] | ||
| Transitional cell carcinoma of the bladder | 7 | [79] | ||
| SETD2 | H3K36 methyltransferase | Lung adenocarcinoma | 5 | [55] |
| UTX/KDM6A | H3K27 demethylase | Transitional cell carcinoma of the bladder | 21 | [79] |
| Various types of cancer | 3 | [99] | ||
| JARID1C/KDM5C | H3K4 demethylase | Renal carcinoma | 3 | [98] |
| Activating mutation |
||||
| EZH2 | H3K27 methyltransferase | Diffuse large B-cell lymphoma (germinal center type) | 22 | [95] |
| Diffuse large B-cell lymphoma, other lymphomas | 15 | [96] | ||
| Follicular lymphoma | 7 | [95] | ||
| Myelodysplastic syndrome | 12 | [97] | ||
| Esophageal cancer | 3 | [60] |
SWI/SNF chromatin-remodeling complex
The SWI/SNF complex is a chromatin remodeler that utilizes the energy of ATP to disrupt contact between nucleosomal DNA and histones, leading to sliding, ejection and/or insertion of histone proteins [42, 43]. Although the mechanisms by which the SWI/SNF complex elicits biological effects are still not fully understood, previous studies have shown that the SWI/SNF complex regulates the expression of numerous genes. For example, Medina et al. showed that exogenous expression of BRG1, a catalytic subunit of the SWI/SNF complex, resulted in altered gene expression of ∼1% of the entire genome in a BRG1-null cancer cell line [44]. Recent studies identified several transcriptional target genes of the SWI/SNF complex, including RB, MYC and RHOA, all of which are involved in carcinogenesis and tumor progression. The SWI/SNF complex directly interacts with RB, a tumor suppressor, to repress RB target genes including E2F and CCND, leading to cell-cycle arrest at the G1/S transition [45]. The expression of E2F target genes in mouse embryonic fibroblasts is upregulated following inactivation of Snf5 [46]. These data suggest that the SWI/SNF complex negatively regulates cell-cycle progression. The SWI/SNF complex also interacts with MYC, an oncogenic transcription factor that regulates gene expression during cell-cycle progression, apoptosis and differentiation. The SWI/SNF complex mediates the transcription activity of MYC through a direct interaction between SNF5 and MYC [47]. Meanwhile, RHOA regulates cell migration by stimulating stress-fiber formation and contractility. Knockdown of SNF5 increases RHOA enzyme activity and stimulates cell migration, suggesting that the SWI/SNF complex suppresses the migration and metastasis of cancer cells [48].
There are two distinct SWI/SNF complexes, BAF and PBAF, both of which consist of various subunits categorized as catalytic ATPase subunits, core subunits, or variant subunits (Fig. 2) [49–51]. The catalytic ATPase subunits include BRG1 and BRM, whose presence in a given SWI/SNF complex is mutually exclusive. Core subunits include SNF5, BAF155 and BAF170. Variant subunits include ARID1A and ARID1B, mutually exclusive components of the BAF complex, and BAF180, BAF200 and BRD7, which are specific to the PBAF complex. In mammals, different types of the SWI/SNF complex with distinct subunit compositions contribute to the regulation of gene expression in a tissue-specific manner [52].
Fig. 2.

Composition of the SWI/SNF chromatin remodeling complex.
Accumulating evidence suggests that the SWI/SNF genes are frequently mutated and inactivated in various human cancers. Shain et al. analyzed whole-exome sequencing data from 24 published studies encompassing 669 cases with 18 neoplastic diagnoses; their results demonstrated that mutations in the SWI/SNF genes are widespread across various cancers, with an overall frequency approaching that of TP53 mutations (19% in the SWI/SNF complex, 26% in TP53) [53]. Mutations in the SWI/SNF genes were significantly skewed toward deleterious forms (e.g. frameshift, nonsense or splice-site mutations), consistent with the inactivated nature of mutated SWI/SNF subunits. Together, these data suggest that the SWI/SNF complex is a bona fide tumor suppressor and may have a significant impact on the properties of cancer cells, as in the case of TP53. Below, mutations identified in the SWI/SNF genes are summarized with respect to each subunit.
BRG1/SMARCA4
BRG1 encodes a catalytic ATPase subunit of the SWI/SNF complex. Sequencing of the BRG1 gene from various cancer cell lines has demonstrated that BRG1 is mutated in a variety of human cancers, including >30% of non-small-cell lung carcinoma (NSCLC) [54]. Notably, most of the mutations identified are homozygous mutations and deletions, indicating that BRG1 plays a role as a tumor suppressor. A recent large-scale genome analysis confirmed that BRG1 mutations play a prominent role in aberrant chromatin remodeling in lung adenocarcinoma [55]. BRG1 mutations were also identified recently in Burkitt lymphoma [56], medulloblastoma [57–59], and esophageal cancer [60]. Furthermore, loss of BRG1 protein expression is observed in 10–50% of surgical lung cancer specimens [61–63]. A recent report showed that both genetic and epigenetic alterations are involved in the loss of BRG1 expression [55].
Patients with rhabdoid tumor (RT) predisposition syndrome, in which RTs occur on a familial basis, harbor a heterozygous germline BRG1 mutation that truncates the encoded protein, and their RTs are homozygous for this mutation [64]. Mutations in SNF5, which are present in the germline of most cases of RT predisposition syndrome, were not detected in the patients analyzed in that study. These results suggest that deficiencies of the SWI/SNF complex may play a role in the predisposition of RT, and support the idea that BRG1 is a tumor suppressor.
BRM/SMARCA2
BRM encodes another catalytic ATPase subunit of the SWI/SNF complex. In contrast to BRG1, somatic mutations in BRM are rarely identified in human cancers. However, in a mouse model in which lung tumors are induced by exposure to carbamate ethyl, inactivation of one or both Brm alleles led to a significant increase in the number of tumors, indicating that Brm plays a role as a tumor suppressor [65]. In fact, BRM protein expression is absent in human lung, gastric and prostate cancers [61–63, 66, 67]. Furthermore, low BRM expression in NSCLC correlates with a worse prognosis [62, 63]. These findings suggest that epigenetic silencing of BRM may be critical for the development of a subset of cancers.
SNF5/SMARCB1/BAF47/INI1
The SNF5 gene encodes a core regulatory subunit of the SWI/SNF complex and is inactivated via biallelic genetic alterations, including deletions and nonsense, missense and frameshift mutations, in nearly all RTs [68]. Thus, SNF5 is considered a major driver gene of RTs. Consistent with this, a significant proportion of RT predisposition syndrome patients harbor heterozygous SNF5 deleterious germline mutations, and RTs arise via loss of the remaining wild-type SNF5 allele [68, 69]. Germline SNF5 mutations also result in predisposition to meningioma and schwannoma [70–72].
Genetically engineered Snf5-heterozygous mice develop sarcomas that closely resemble human RTs [73]. Furthermore, biallelic inactivation of Snf5 results in the development of sarcoma and lymphoma with a median onset of only 11 weeks, which is a remarkably rapid rate considering that the median onset of tumors induced by biallelic inactivation of p53 and Rb is 16 weeks in the same experimental model. These data strongly suggest that SNF5 acts as a tumor suppressor. However, somatic mutation of SNF5 is rarely detected in common cancers [49, 51, 74], indicating that, in contrast to mutations in other SWI/SNF genes detected in wide range of cancers, the SNF5 mutation is specific to a subset of non-epithelial malignancies.
ARID1A/BAF250A and ARID1B/BAF250B
Recent genome-wide sequencing studies identified ARID1A, which encodes a subunit of the SWI/SNF complex, as one of the most frequently mutated genes in a variety of cancers (Table 5). ARID1A is mutated in 46–57% of ovarian clear cell carcinomas, one of the most lethal subtypes of ovarian cancer, and in 30% of endometriosis-associated ovarian carcinomas [75, 76]. ARID1A mutations have also been detected in a variety of cancers including renal clear cell carcinoma [77], Burkitt lymphoma [56], hepatocellular carcinoma [78], transient cell carcinoma of the bladder [79], gastric adenocarcinoma [80], esophageal cancer [60], uterine serous endometrial cancer [81], neuroblastoma [82], pancreatic cancer [83], malignant melanoma [84], and medulloblastoma [85]. ARID1A is also mutated in a subset of lung adenocarcinomas, although at a lower frequency than BRG1 [55]. Most ARID1A mutations detected in cancer cells to date are inactivating mutations, indicating that ARID1A has a tumor-suppressive function.
ARID1B is mutated in a small subset of patients with hepatocellular carcinoma [78], neuroblastoma [82], breast cancer [86], and malignant melanoma [84].
ARID2/BAF200
Genome sequencing studies conducted by Li et al. and Fujimoto et al. found that ARID2, which encodes a SWI/SNF regulatory subunit, was mutated in ∼10% of surgical specimens of hepatocellular carcinoma [78, 87]. Large-scale exome sequencing in malignant melanoma also revealed that 7.4% of the cases harbored mutations in ARID2, most of which were inactivating mutations. In that study, ARID2 mutation was identified as one of the driver mutations resulting from C-to-T transitions caused by exposure to UV light; this is in contrast to other driver mutations in malignant melanoma, such as those in BRAF and NRAS, that are only weakly associated with UV-induced damage [84]. ARID2 is also mutated in a small subset of esophageal cancers [60].
PBRM1/BAF180
Most recently, mutations in the PBRM1 gene, which encodes a regulatory subunit protein, were identified in 41% of renal clear cell carcinomas [77], making PBRM1 the second most frequently mutated gene in renal cell carcinoma after VHL. PBRM1 mutations have also been detected in esophageal cancer patients [60] and in several breast cancer cell lines [88].
Mutations in histone modifier genes
Recently, mutations in several histone modifier genes have been identified. The gene encoding CBP HAT is mutated in B-cell lymphoma, acute lymphoblastic leukemia, and carcinomas of the lung and bladder [79, 89–92]. Meanwhile, the gene encoding p300 HAT is mutated in bladder carcinoma, small-cell lung carcinoma, colorectal cancer, breast cancer and pancreas cancer [79, 91, 93], although at a lower frequency than CBP.
Histone methytransferases (HMTs) are a class of enzymes that catalyze methylation of histones at lysine and arginine residues. In acute leukemia, MLL, an HMT specific for lysine 4 of histone H3 (H3K4), is rearranged to generate fusion proteins with diverse partners. In most cases, the rearrangement results in the loss of the H3K4 HMT enzymatic activity [94]. MLL is also mutated in small-cell lung carcinoma and bladder carcinoma [79, 91]. SETD2, another HMT specific for H3K36, is mutated in a small fraction of lung adenocarcinomas [55]. On the other hand, activating mutations in the gene encoding EZH2, an HMT directed against H3K27 that is the catalytic subunit of the polycomb repressive complex 2 (PRC2), have been identified in diffuse large B-cell lymphoma (DLBCL), follicular lymphoma, myelodysplastic syndrome, and esophageal cancer [60, 95–97].
In contrast to HMTs, histone demethylases (HDMs) are enzymes that remove methyl units from histones. Some tumors contain alterations in HDM-encoding genes. The gene encoding JARID1C, a HDM specific for H3K4, is mutated in renal carcinoma [98], and the gene encoding UTX, a HDM specific for H3K27, is mutated in multiple cancers [79, 99].
To date, the mechanisms by which mutations in histone modifiers are involved in carcinogenesis and tumor growth have not been fully elucidated. Nevertheless, in several cases, recent research has uncovered new leads for the development of treatment strategies targeting these mutations. These investigations will be discussed in the next chapter.
PERSONALIZED TREATMENT STRATEGIES THAT TARGET MUTATIONS IN CHROMATIN-REGULATING GENES
In parallel with the elucidation of the genetic landscape of various human cancers by genome-wide sequencing analyses, large numbers of chemotherapeutic agents targeting gene mutations (i.e. molecularly targeted drugs) have been developed, driving the advancement and spread of personalized cancer therapy based on gene-mutation profiles. Cancers harboring activating gene mutations can be treated with specific inhibitors of the mutated gene products, thereby suppressing their abnormally high activity. For example, selective cell killing by the tyrosine kinase inhibitors gefitinib, crizotinib, and vandetanib in cancer cells harboring EGFR mutation, ALK fusion, and RET fusion, respectively, has been demonstrated by our group and others, and these treatment strategies have been applied in the clinic [100–104]. Meanwhile, for the treatment of cancers in which certain proteins or pathways are inactivated by genetic and/or epigenetic causes, strategies based on synthetic lethality have attracted a great deal of attention [105]. Furthermore, chromatin-regulating proteins involved in the transcriptional regulation of genes essential for cancer cell survival can be also targeted. The emerging strategies for personalized cancer therapy targeting chromatin-regulating proteins are summarized below.
Drugs targeting activating mutations in chromatin-regulating genes
MLL-rearranged leukemia is a subset of acute myeloid leukemia (AML) with poor prognosis. Recently, inhibition of DOT1L, a HMT specific for H3K79, has emerged as a promising strategy for the treatment of MLL-rearranged leukemia. MLL regulates the transcription of various genes involved in cellular development. The carboxyl-terminal portion of the MLL protein contains a domain with HMT activity specific for H3K4 [94]. Rearrangement of MLL results in the loss of the H3K4 HMT domain and in-frame fusion of the amino-terminal region of MLL to diverse partner proteins. In most cases, these fusions lead to more efficient recruitment of DOT1L to MLL target genes. The resulting hypermethylation at H3K79 by DOT1L leads to aberrant expression of a characteristic set of genes, including HOXA9 and MEIS1, which drive leukemogenesis. In MLL-rearranged leukemia, DOT1L is also required for the development and maintenance of leukemia cells. Daigle et al. developed a compound, EPZ00477, which selectively inhibits DOT1L HMT [106]. They reported that EPZ00477 selectively killed MLL-rearranged leukemia cells in culture and prolonged survival in a mouse xenograft model. Moreover, they further identified another selective inhibitor of DOT1L HMT, EPZ-5676, which possesses superior pharmacokinetic properties [107]. Continuous intravenous infusion of EPZ-5676 in a rat xenograft model of MLL-rearranged leukemia caused complete tumor regressions. The state of complete tumor response was sustained beyond the period of compound infusion with no significant toxicity. EPZ-5676 is now the subject of a clinical trial for MLL-rearranged leukemia patients, as the first inhibitor of an HMT to be tested in humans.
Activating mutations of EZH2 in non-Hodgkin lymphoma have provoked interest in this protein as a therapeutic target. Y641 and A677 are hot spots for lymphoma-associated EZH2 mutations [95, 96]. These mutations increase the activity of the encoded proteins, leading to elevated levels of trimethylated H3K27, thereby promoting proliferation of lymphoma cells [96, 108, 109]. Several groups have developed selective small-molecule inhibitors of EZH2 HMT and demonstrated promising preclinical results. McCabe et al. showed that the compound GSK126 decreased global methylation at H3K27 and reactivated silenced PRC2 target genes in EZH2-mutant DLBCL cell lines [110]. Furthermore, this compound effectively inhibited the proliferation of the EZH2-mutant DLBCL cells, and suppressed tumor growth in a mouse xenograft model. Qi et al. developed another compound, El1 [111], which when administered to DLBCL cells carrying the Y641 mutation also decreased the H3K27 methylation level genome-wide, activated PRC2 target genes, and decreased cell proliferation. Knutson et al. reported that the compound EPZ005687 induced apoptotic cell death in lymphoma cells with heterozygous mutations in Y641 or A677, with minimal effect on the proliferation of wild-type cells [112]. On the other hand, several studies have demonstrated that AML cells harboring MLL-AF9 fusion require EZH2 for survival, suggesting that EZH2 inhibition is also a candidate strategy to treat this disease [113, 114]. Most recently, Kim et al. developed a peptide called stabilized α-helix of EZH2 (SAH-EZH2), which inhibits EZH2 inhibition by a different mechanism from GSK126, El1, and EPZ005687 [115]. SAH-EZH2 selectively disrupts the contact between EZH2 and EED, another subunit in the PRC2 complex, whereas the other EZH2 inhibitors target the HMT catalytic domain. As in the case of GSK126, SAH-EZH2 decreases the H3K27 trimethylation level, resulting in growth arrest of PRC2-dependent MLL-AF9 leukemia cells. Together, these studies provide hope for the establishment of novel treatment strategies against EZH2-mutant hematologic malignancies. Moreover, recent studies have revealed the overexpression of various HMTs in human cancers [116]. It is speculated that the resulting histone hypermethylation patterns are associated with the malignant features of cancer cells in a cancer-type specific manner. Further studies of these cancer-specific patterns and the roles of histone hypermethylation should lead to the development of novel targeted therapies.
Synthetic lethality therapy targeting inactivation of chromatin-regulating proteins
As discussed, cancers with gain-of-function gene mutations can be treated by inhibition of the mutated gene products. However, in many cases, tumor growth is also driven by loss-of-function mutations in tumor-suppressor genes. Previous studies have demonstrated that the restoration of the lost function of tumor-suppressor proteins is not a simple process [117–119]. Therefore, it is necessary to develop alternative strategies to treat cancers harboring inactivating mutations in tumor-suppressor genes. Synthetic lethality holds great promise as a strategy for specific killing of cancer cells possessing inactivating mutations that are not present in normal cells [105]. Synthetic lethality is based on genetic interactions between two mutations in which the presence of either mutation alone has little or no effect on cell viability, but the combination of mutations in both genes becomes lethal. The presence of one of these mutations in cancer cells, but not in normal cells, presents opportunities to selectively kill cancer cells by mimicking the effect of the second genetic mutation with targeted therapy. The synthetic-lethal relationship between PARP1 and BRCA1 or BRCA2, genes involved in DNA repair, was demonstrated in preclinical models in 2005 [120, 121] and translated to the clinic in 2009 through the application of PARP inhibitors to BRCA1/BRCA2-deficient tumors [122]. These advances have driven researchers to explore novel combinations of genes to identify additional synthetic-lethal relationships [123–125].
Most recently, synthetic-lethal relationships in chromatin-regulating genes have been identified, e.g. between BRG1 and BRM [61] and between SNF5 and EZH2 [126]. BRG1 or BRM are present in a mutually exclusive manner as catalytic subunits in the SWI/SNF chromatin-remodeling complex. Previous studies of the embryos of genetically engineered mice have indicated that BRG1 and BRM play complementary roles in development [127]. Together, these data suggest that BRG1 and BRM have a synthetic-lethal relationship. Accordingly, silencing of BRM suppresses the growth of BRG1-deficient cancer cells relative to BRG1-proficient cancer cells (Fig. 3), by inducing senescence via activation of p21/CDKN1A. Moreover, in a conditional RNAi experiment using a mouse xenograft model, BRM depletion suppressed the growth of BRG1-deficient tumors [61]. These results offer a rationale for treating BRG1-deficient cancers, including subsets of NSCLCs, medulloblastoma, and Burkitt lymphoma, by inhibition of the BRM ATPase. To this end, screening for small-molecule compounds with selective BRM ATPase inhibitory activity is currently underway.
Fig. 3.
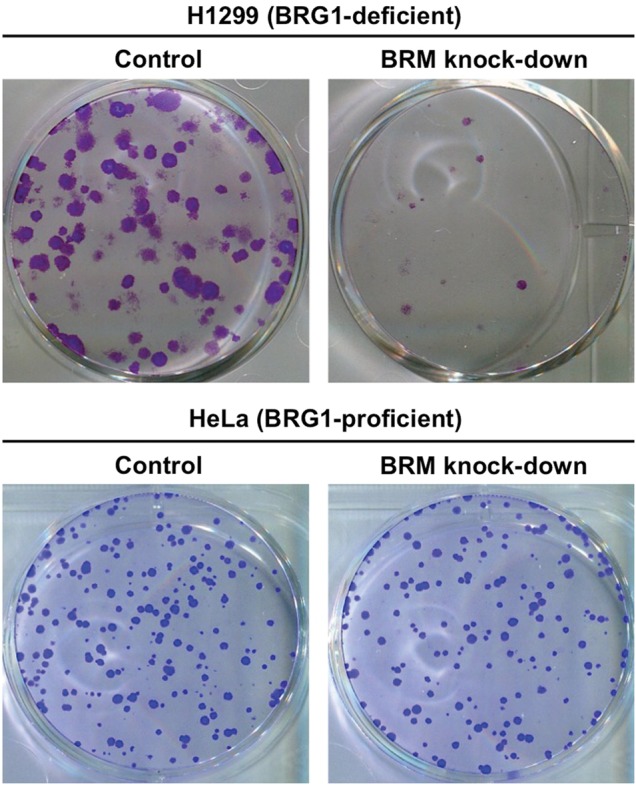
Specific cell killing of BRG1-deficient H1299 cells, but not BRG-proficient HeLa cells, by BRM knockdown based on synthetic lethality. Cells were treated with BRM-siRNA (BRM knockdown) or non-targeting siRNA (control) (Dharmacon). After 48 h, the cells were subjected to clonogenic survival assays. Colonies fixed and stained after incubation for an additional 10 d are shown.
SNF5 is a core subunit in the SWI/SNF complex. SNF5-heterozygous mice develop sarcomas that closely resemble human malignant RTs in which the second allele of SNF5 is spontaneously lost. The genesis of the sarcoma in SNF5-heterozygous mice can be completely suppressed by deletion of EZH2. These data suggest a synthetic-lethal relationship between SNF5 and EZH2. In line with this concept, Alimova et al. showed that disruption of EZH2 by RNAi and/or 3-deazaneplanocin A, an indirect and general inhibitor of methyltransferases, impaired growth of SNF5-mutant atypical teratoid RT cells [128]. Furthermore, Knutson et al. developed EPZ-6438, a selective small-molecule inhibitor of EZH2, and demonstrated that EPZ-6438 specifically killed SNF5-mutant malignant RT cells both in vitro and in vivo [126]. EPZ-6438 decreased cellular H3K27 methylation levels and activated the PRC2 target gene CDKN2A in SNF5-mutant malignant RT cells, but not in wild-type cells. Because the PRC2 complex regulates gene expression in several cancer-associated pathways such as RB, Cyclin D1, MYC [129] and Hedgehog [130] in a manner that antagonizes the function of the SWI/SNF complex, EZH2 inhibition by EPZ-6438 might contribute to restoration of the balance between the PRC2 and SWI/SNF complexes, leading to an anti-tumor effect. SNF5 is inactivated through biallelic mutation in nearly all cases of malignant RTs and atypical teratoid RTs [131]. Synthetic lethality therapy using EZH2 inhibitors in such tumors is awaiting clinical application.
Targeting BRD4, which is involved in transcriptional regulation of cancer-related genes
BRD4 is a member of the bromodomain and extraterminal (BET) subfamily. These proteins recognize acetylated lysine residues on chromatin and bind to them via bromodomains, facilitating activation of transcription along surrounding DNA. JQ1, a small-molecule inhibitor of BRD4, competitively binds to the bromodomain in BRD4, leading to displacement of BRD4 from chromatin [132]. Recent studies have revealed the promising anti-tumor effects of JQ1 in various hematologic malignancies [133–135]. In an AML mouse model, Zuber et al. performed a screen for epigenetic vulnerability using short-hairpin RNAs targeting known chromatin-regulating proteins and identified BRD4 as a key hit [133]. Consistent with this, they showed that JQ1 inhibits proliferation of diverse subtypes of AML cells. Merz et al. demonstrated that cell lines derived from various hematologic malignancies were highly susceptible to JQ1 [134]. Delmore et al. showed that JQ1 prolongs survival of mice bearing multiple myeloma [135]. Furthermore, Dawson et al. developed another BRD4 inhibitor, I-BET151, and showed that this compound prolonged the lifespan of mice with mixed-lineage fusion leukemia treated with I-BET151 [136]. On the other hand, JQ1 also exerts an anti-tumor effect in nuclear protein in testis (NUT) midline carcinoma, a rare subtype of squamous cell carcinoma with an aggressive nature, in which t(15;19) chromosomal translocation results in a fusion between BRD4 and NUT [137].
Previous studies have suggested that the anti-tumor effect of BRD4 inhibition depends on suppression of MYC transcription, leading to genome-wide downregulation of MYC-dependent target genes [134, 135]. In addition to MYC, two other well-known cancer-related genes, BCL2 and CDK6, have recently been identified as targets of transcriptional regulation by BRD4 [136]. The preclinical results are encouraging with regard to potential clinical applications of BRD4 inhibitors. However, because BRD4 is broadly expressed in almost all normal human cells and regulates gene expression throughout the genome, the off-target effects of BRD4 inhibition in humans should be carefully investigated. Most recently, the mechanism underlying selective activation of MYC transcription by BRD4, which has been termed a ‘super-enhancer’, was revealed in multiple myeloma cells [138]. Further studies are needed to establish the optimal way to treat cancer by targeting BRD4.
PERSPECTIVES
The growing incidence of cancer worldwide indicates that radiation therapy will become increasingly significant in cancer treatment [139]. Enhancing the efficacy of IR against cancer cells is paramount if we are to improve local control of tumors. Recent studies have elucidated the involvement of various chromatin-regulating proteins in the DDR after irradiation, and these proteins represent promising targets for radiosensitization. As we discussed, several inhibitors of HATs or HDACs exert radiosensitizing effects in preclinical models. However, the detailed mechanisms by which these compounds sensitize cancer cells to IR are still largely unknown. Moreover, it remains unknown whether these compounds can achieve clinically significant radiosensitization in humans at a dose that causes no (or at least low) toxicity; this issue warrants further investigation.
Large-scale genome sequencing techniques have elucidated the genetic landscape of various cancers, accelerating the development of molecularly targeted drugs. High frequencies of mutation in chromatin-regulating genes, especially in the SWI/SNF genes, highlight the pivotal roles of chromatin-regulating proteins in cancer cells, and thus offer rationales for targeting these mutations in cancer therapy. DOT1L inhibition in MLL-rearranged leukemia, EZH2 inhibition in EZH2-mutant or MLL-rearranged hematologic malignancies and SNF5-deficient tumors, BRD4 inhibition in hematologic malignancies, and BRM inhibition in BRG1-deficient tumors all hold great promise and are awaiting clinical applications. Moreover, considering the role of chromatin-regulating proteins in the DDR [4], elucidation of the association between mutation status in chromatin-regulating genes and the clinical outcome of radiation therapy and/or chemotherapy will facilitate the selection of patients who will (or will not) respond to such therapies. Accordingly, we recently reported that single-nucleotide polymorphisms in the BPTF gene, which encodes a bromodomain PHD finger transcription factor contained in the NURF chromatin-remodeling complex, affect the risk of lung adenocarcinoma in the Japanese population [140]. Future studies will elucidate the molecular mechanisms underlying the effect of aberrant chromatin-regulating proteins on carcinogenesis and cancer progression, and will contribute to the establishment of personalized treatment based on the genetic profiles of tumors.
FUNDING
This work was supported by Grants-in-Aid from the Japan Society for the Promotion of Science for Young Scientists (B) KAKENHI (10643471) and (23701110), and from the Ministry of Education, Culture, Sports, Science and Technology of Japan for Scientific Research on Innovative Areas (22131006).
ACKNOWLEDGEMENTS
We thank Dr Yoshihisa Matsumoto of Tokyo Institute of Technology for technical assistance and sincere encouragement. The content of this paper was partially presented at ASTRO's 54th Annual Meeting (Boston, MA, USA, October 2012) and the AACR Annual Meeting 2012 (Chicago, IL, USA, April 2012).
REFERENCES
- 1.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 2.Rossetto D, Truman AW, Kron SJ, et al. Epigenetic modifications in double-strand break DNA damage signaling and repair. Clin Cancer Res. 2010;15:4543–52. doi: 10.1158/1078-0432.CCR-10-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrozza MJ, Utley RT, Workman JL, et al. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003;19:321–9. doi: 10.1016/S0168-9525(03)00115-X. [DOI] [PubMed] [Google Scholar]
- 4.Ogiwara H, Ui A, Otsuka A, et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;5:2135–46. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]
- 5.Murr R, Loizou JI, Yang YG, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–9. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 6.Bird AW, Yu DY, Pray-Grant MG, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–5. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 7.Ogiwara H, Kohno T. CBP and p300 histone acetyltransferases contribute to homologous recombination by transcriptionally activating the BRCA1 and RAD51 genes. PLOS ONE. 2012;7:e52810. doi: 10.1371/journal.pone.0052810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balasubramanyam K, Varier RA, Altaf M, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–71. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 9.Sun Y, Jiang X, Chen S, et al. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 2006;580:4353–6. doi: 10.1016/j.febslet.2006.06.092. [DOI] [PubMed] [Google Scholar]
- 10.Hemshekhar M, Sebastin SM, Kemparaju K, et al. Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin Pharmacol Toxicol. 21 November 2011 doi: 10.1111/j.1742-7843.2011.00833.x. 10.1111/j.1742-7843.2011.00833.x. [DOI] [PubMed] [Google Scholar]
- 11.Balasubramanyam K, Swaminathan V, Ranganathan A, et al. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134–40. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- 12.Balasubramanyam K, Altaf M, Varier RA, et al. Polyisoprenylated benzphenone, garcinol, anatural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem. 2004;279:33716–26. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- 13.Oike T, Ogiwara H, Torikai K, et al. Garcinol, a histone acetyltransferase inhibitor, radiosensitizes cancer cells by inhibiting non-homologous end joining. Int J Radiat Oncol Biol Phys. 2012;84:815–21. doi: 10.1016/j.ijrobp.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 14.Ogiwara H, Ui A, Shiotani B, et al. Curcumin suppresses multiple DNA damage response pathways and has potency as a sensitizer to PARP inhibitor. Carcinogenesis. 2013;34:2486–97. doi: 10.1093/carcin/bgt240. [DOI] [PubMed] [Google Scholar]
- 15.Khafif A, Lev-Ari S, Vexler A, et al. Curcumin: a potential radio-enhancer in head and neck cancer. Laryngoscope. 2009;119:2019–26. doi: 10.1002/lary.20582. [DOI] [PubMed] [Google Scholar]
- 16.Sandur SK, Deorukhkar A, Pandey MK, et al. Curcumin modulates the radiosensitivity of colorectal cancer cells by suppressing constitutive and inducible NF-kB activity. Int J Radiat Oncol Biol Phys. 2009;75:534–42. doi: 10.1016/j.ijrobp.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Zhang Z, Hill DL, et al. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007;67:1988–96. doi: 10.1158/0008-5472.CAN-06-3066. [DOI] [PubMed] [Google Scholar]
- 18.Chendil D, Ranga RS, Meigooni D, et al. Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene. 2004;23:1599–607. doi: 10.1038/sj.onc.1207284. [DOI] [PubMed] [Google Scholar]
- 19.Cate RT, Krawczwk P, Stap J, et al. Radiosensitizing effect of the histone acetyltransferase inhibitor anacardic acid on various mammalian cell lines. Oncol Lett. 2010;1:756–9. doi: 10.3892/ol_00000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albert JM, Cao C, Kim KW, et al. Inhibition of Poly (ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007;13:3033–42. doi: 10.1158/1078-0432.CCR-06-2872. [DOI] [PubMed] [Google Scholar]
- 21.Veuger SJ, Curtin NJ, Richardson CJ, et al. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly (ADP-ribose) polymerase-1. Cancer Res. 2003;63:6008–15. [PubMed] [Google Scholar]
- 22.Dhandapani KM, Mahesh VB, Brann DW, et al. Curcumin suppresses growth and chemoresistance of human glioblastoma cells via AP-1 and NFkappaB transcription factors. J Neurochem. 2007;102:522–38. doi: 10.1111/j.1471-4159.2007.04633.x. [DOI] [PubMed] [Google Scholar]
- 23.Plummer SM, Holloway KA, Manson MM, et al. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signalling complex. Oncogene. 1999;18:6013–20. doi: 10.1038/sj.onc.1202980. [DOI] [PubMed] [Google Scholar]
- 24.Qiao Q, Jiang Y, Li G, et al. Inhibition of the PI3K/AKT-NF-κB pathway with curcumin enhanced radiation-induced apoptosis in human Burkitt's lymphoma. J Pharmacol Sci. 2013;121:247–56. doi: 10.1254/jphs.12149fp. [DOI] [PubMed] [Google Scholar]
- 25.Padhye S, Ahmad A, Oswal N, et al. Emerging role of Garcinol, the antioxidant chalcone from Garcinia indica Choisy and its synthetic analogs. J Hematol Oncol. 2009;2:38. doi: 10.1186/1756-8722-2-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmad A, Wang Z, Ali R, et al. Apoptosis-inducing effect of garcinol is mediated by NF-kappaB signaling in breast cancer cells. Cell Biochem. 2010;109:1134–41. doi: 10.1002/jcb.22492. [DOI] [PubMed] [Google Scholar]
- 27.Groselj B, Sharma NL, Hamdy FC, et al. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer. 2013;108:748–54. doi: 10.1038/bjc.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–51. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chinnaiyan P, Vallabhaneni G, Armstrong E, et al. Modulation of radiation response by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys. 2005;62:223–9. doi: 10.1016/j.ijrobp.2004.12.088. [DOI] [PubMed] [Google Scholar]
- 30.Chen X, Wong P, Radany EH, et al. Suberoylanilide hydroxamic acid as a radiosensitizer through modulation of RAD51 protein and inhibition of homology-directed repair in multiple myeloma. Mol Cancer Res. 2012;10:1052–64. doi: 10.1158/1541-7786.MCR-11-0587. [DOI] [PubMed] [Google Scholar]
- 31.Blattmann C, Oertel S, Ehemann V, et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys. 2010;78:237–45. doi: 10.1016/j.ijrobp.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Munshi A, Kurland J, Nishikawa T, et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res. 2005;11:4912–22. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Wong P, Radany E, et al. HDAC inhibitor, valproic acid, induces p53–dependent radiosensitization of colon cancer cells. Cancer Biother Radiopharm. 2009;24:689–99. doi: 10.1089/cbr.2009.0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lan L, Ui A, Nakajima S, et al. The ACF1 complex is required for DNA double-strand break repair in human cells. Mol Cell. 2010;40:976–87. doi: 10.1016/j.molcel.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Hall EJ, Giaccia AJ. Radiosensitizers and bioreductive drugs. In: McAllister L, Bierig L, Barret K, editors. Radiobiology for the Radiologist. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 419–31. [Google Scholar]
- 36.Bolderson E, Richard DJ, Zhou BB, et al. Recent advances in cancer therapy targeting proteins involved in DNA double-strand break repair. Clin Cancer Res. 2009;15:6314–20. doi: 10.1158/1078-0432.CCR-09-0096. [DOI] [PubMed] [Google Scholar]
- 37.Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer. 2008;98:523–8. doi: 10.1038/sj.bjc.6604208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oike T, Ogiwara H, Nakano T, et al. Histone acetyltransferases (HATs) involved in non-homologous end joining as a target for radiosensitization. In: Kataria T, editor. Frontiers in Radiation Oncology. Rijeka: Intech; 2013. pp. 3–12. [Google Scholar]
- 39.Noguchi H, Yamashita H, Murakami T, et al. Successful treatment of anaplastic thyroid carcinoma with a combination of oral valproic acid, chemotherapy, radiation and surgery. Endocr J. 2009;56:245–9. doi: 10.1507/endocrj.k08e-016. [DOI] [PubMed] [Google Scholar]
- 40.Masoudi A, Elopre M, Amini E, et al. Influence of valproic acid on outcome of high-grade gliomas in children. Anticancer Res. 2008;28:2437–42. [PubMed] [Google Scholar]
- 41.Ree AH, Dueland S, Folkvord S, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the pelvic radiation and vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010;11:459–64. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- 42.Lorch Y, Maier-Davis B, Kornberg RD. Mechanism of chromatin remodeling. Proc Natl Acad Sci U S A. 2010;107:3458–62. doi: 10.1073/pnas.1000398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol. 2006;7:437–47. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 44.Medina PP, Carretero J, Ballestar E, et al. Transcriptional targets of the chromatin-remodelling factor SMARCA4/BRG1 in lung cancer cells. Hum Mol Genet. 2005;14:973–82. doi: 10.1093/hmg/ddi091. [DOI] [PubMed] [Google Scholar]
- 45.Trouche D, Le Chalony C, Muchardt C, et al. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci U S A. 1997;94:11268–73. doi: 10.1073/pnas.94.21.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isakoff MS, Sansam CG, Tamayo P, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci U S A. 2005;102:17745–50. doi: 10.1073/pnas.0509014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng SW, Davies KP, Yung E, et al. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nature Genet. 1999;22:102–5. doi: 10.1038/8811. [DOI] [PubMed] [Google Scholar]
- 48.Caramel J, Quignon F, Delattre O. RhoA-dependent regulation of cell migration by the tumor suppressor hSNF5/INI1. Cancer Res. 2008;68:6154–61. doi: 10.1158/0008-5472.CAN-08-0115. [DOI] [PubMed] [Google Scholar]
- 49.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–92. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 50.Euskirchen G, Auerbach RK, Snyder M. SWI/SNF chromatin-remodeling factors: multiscale analyses and diverse functions. J Biol Chem. 2012;287:30897–905. doi: 10.1074/jbc.R111.309302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–68. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 52.Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136:200–6. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLOS ONE. 2013;8:e55119. doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medina PP, Romero OA, Kohno T, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008;29:617–22. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 55.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Love C, Sun Z, Jima D, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321–5. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–8. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pugh TJ, Weeraratne SD, Archer TC, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–10. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Northcott PA, Korshunov A, Pfister SM, et al. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol. 2012;8:340–51. doi: 10.1038/nrneurol.2012.78. [DOI] [PubMed] [Google Scholar]
- 60.Dulak AM, Stojanov P, Peng S, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–86. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oike T, Ogiwara H, Tominaga Y, et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73:5508–18. doi: 10.1158/0008-5472.CAN-12-4593. [DOI] [PubMed] [Google Scholar]
- 62.Fukuoka J, Fujii T, Shih JH, et al. Chromatin remodeling factors and BRM/BRG1 expression as prognostic indicators in non-small cell lung cancer. Clin Cancer Res. 2004;10:4314–24. doi: 10.1158/1078-0432.CCR-03-0489. [DOI] [PubMed] [Google Scholar]
- 63.Reisman DN, Sciarrotta J, Wang W, et al. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 2003;63:560–6. [PubMed] [Google Scholar]
- 64.Schneppenheim R, Fruhwald MC, Gesk S, et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86:279–84. doi: 10.1016/j.ajhg.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glaros S, Cirrincione GM, Muchardt C, et al. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene. 2007;26:7058–66. doi: 10.1038/sj.onc.1210514. [DOI] [PubMed] [Google Scholar]
- 66.Yamamichi N, Inada K, Ichinose M, et al. Frequent loss of Brm expression in gastric cancer correlates with histologic features and differentiation state. Cancer Res. 2007;67:10727–35. doi: 10.1158/0008-5472.CAN-07-2601. [DOI] [PubMed] [Google Scholar]
- 67.Shen H, Powers N, Saini N, et al. The SWI/SNF ATPase Brm is a gatekeeper of proliferative control in prostate cancer. Cancer Res. 2008;68:10154–62. doi: 10.1158/0008-5472.CAN-08-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–6. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 69.Biegel JA, Zhou JY, Rorke LB, et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–9. [PubMed] [Google Scholar]
- 70.van den Munckhof P, Christiaans I, Kenter SB, et al. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics. 2012;13:1–7. doi: 10.1007/s10048-011-0300-y. [DOI] [PubMed] [Google Scholar]
- 71.Christiaans I, Kenter SB, Brink HC, et al. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet. 2011;48:93–7. doi: 10.1136/jmg.2010.082420. [DOI] [PubMed] [Google Scholar]
- 72.Hulsebos TJ, Plomp AS, Wolterman RA, et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007;80:805–10. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Williams BO, Remington L, Albert DM, et al. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet. 1994;7:480–4. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- 74.Manda R, Kohno T, Hamada K, et al. Absence of hSNF5/INI1 mutation in human lung cancer. Cancer Lett. 2000;153:57–61. doi: 10.1016/s0304-3835(00)00342-6. [DOI] [PubMed] [Google Scholar]
- 75.Jones S, Wang TL, Shih Ie M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–31. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–43. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Varela I, Tarpey P, Raine K, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–42. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fujimoto A, Totoki Y, Abe T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–4. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 79.Gui Y, Guo G, Huang Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–8. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zang ZJ, Cutcutache I, Poon SL, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–4. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 81.Le Gallo M, O′Hara AJ, Rudd ML, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–5. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sausen M, Leary RJ, Jones S, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45:12–7. doi: 10.1038/ng.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Parsons DW, Li M, Zhang X, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–9. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li M, Zhao H, Zhang X, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–9. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xia W, Nagase S, Montia AG, et al. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res. 2008;68:1667–74. doi: 10.1158/0008-5472.CAN-07-5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pasqualucci L, Dominguez-Sola D, Chiarenza A, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peifer M, Fernandez-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kishimoto M, Kohno T, Okudela K, et al. Mutations and deletions of the CBP gene in human lung cancer. Clin Cancer Res. 2005;11:512–9. [PubMed] [Google Scholar]
- 93.Gayther SA, Batley SJ, Linger L, et al. Mutations truncating the Ep300 acetylase in human cancers. Nat Genet. 2000;24:300–3. doi: 10.1038/73536. [DOI] [PubMed] [Google Scholar]
- 94.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morin RD, Johnson NA, Severson TM, et al. Somatic mutation of EZH2 (Y641) in follicular and diffuse large B-cell lymphomas of germinal center origin. Nat Genet. 2010;42:181–5. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McCabe MT, Graves AP, Ganji G, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27) Proc Natl Acad Sci U S A. 2012;109:2989–94. doi: 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 98.Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Haaften G, Dalgliesh GL, Davies H, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41:521–3. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lovly CM, Carbone DP. Lung cancer in 2010: one size does not fit all. Nat Rev Clin Oncol 2011. 8:68–70. doi: 10.1038/nrclinonc.2010.224. [DOI] [PubMed] [Google Scholar]
- 101.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 102.Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–4. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–7. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kohno T, Tsuta K, Tsuchihara K, et al. RET fusion gene: translation to personalized lung cancer therapy. Cancer Sci. 2013;104:1396–400. doi: 10.1111/cas.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011;10:351–64. doi: 10.1038/nrd3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Daigle SR, Olhava EJ, Therkelsen CA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–25. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sneeringer CJ, Scott MP, Kuntz KW, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107:20980–5. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yap DB, Chu J, Berg T, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–9. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 111.Qi W, Chan H, Teng L, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–5. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Knutson SK, Wigle TJ, Warholic NM, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 113.Neff T, Sinha AU, Kluk MJ, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A. 2012;109:5028–33. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shi J, Wang E, Zuber J, et al. The polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9;Nras(G12D) acute myeloid leukemia. Oncogene. 2013;32:930–8. doi: 10.1038/onc.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim W, Bird GH, Neff T, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9:643–50. doi: 10.1038/nchembio.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–32. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 117.Harris CC, Hollstein M. Clinical implications of the p53 tumor-suppressor gene. N Engl J Med. 1993;329:1318–27. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- 118.Olivier M, Petitjean A, Marcel V, et al. Recent advances in p53 research: an interdisciplinary perspective. Cancer Gene Ther. 2009;16:1–12. doi: 10.1038/cgt.2008.69. [DOI] [PubMed] [Google Scholar]
- 119.Liu TC, Hwang TH, Bell JC, et al. Translation of targeted oncolytic virotherapeutics from the lab into the clinic, and back again: a high-value iterative loop. Mol Ther. 2008;16:1006–8. doi: 10.1038/mt.2008.70. [DOI] [PubMed] [Google Scholar]
- 120.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 121.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 122.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 123.Muller FL, Colla S, Aquilanti E, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337–42. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chan DA, Sutphin PD, Nguyen P, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3002394. 94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Martin SA, McCabe N, Mullarkey M, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17:235–48. doi: 10.1016/j.ccr.2009.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reyes JC, Barra J, Muchardt C, et al. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2α) EMBO J. 1998;17:6979–91. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Alimova I, Birks DK, Harris PS, et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol. 2013;15:149–60. doi: 10.1093/neuonc/nos285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roberts CW, Biegel JA. The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther. 2009;8:412–6. doi: 10.4161/cbt.8.5.8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jagani Z, Mora-Blanco EL, Sansam CG, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. 2010;16:1429–33. doi: 10.1038/nm.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol. 2012;2:114. doi: 10.3389/fonc.2012.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moyer MW. First drugs found to inhibit elusive cancer target. Nat Med. 2011;17:1325. doi: 10.1038/nm1111-1325. [DOI] [PubMed] [Google Scholar]
- 133.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–74. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–33. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Boyle P, Leon B. Lyon: International Agency for Research on Cancer, World Health Organization; 2008. World Cancer Report 2008. [Google Scholar]
- 140.Shiraishi K, Kunitoh H, Daigo Y, et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat Genet. 2012;44:900–3. doi: 10.1038/ng.2353. [DOI] [PubMed] [Google Scholar]