

Abstract
Catalytic reactions have played an indispensable role in organic chemistry for the last several decades. In particular, catalytic multicomponent reactions have attracted a lot of attention due to their efficiency and expediency towards complex molecule synthesis. The presence of bismetallic reagents (e.g. B–B, Si–Si, B–Si, Si–Sn, etc.) in this process renders the products enriched with various functional groups and multiple stereocenters. For this reason, catalytic bismetallative coupling is considered an effective method to generate the functional and stereochemical complexity of simple hydrocarbon substrates. This review highlights key developments of transition-metal catalyzed bismetallative reactions involving multiple π components. Specifically, it will highlight the scope, synthetic applications, and proposed mechanistic pathways of this process.
1. Introduction
Catalytic multicomponent reactions are regarded as one of the most attractive strategies for organic synthesis, since they can generate molecular diversity and complexity from simple substrates.1 Recent investigations in this area have revealed that the incorporation of bismetallic reagents (e.g. R2B–BR2, R3Si–SiR3, R2B–SiR3, R3Si–SnR3, etc.) into this process allows access to functionalized products in a stereo- and regioselective fashion.2, 3, 4 The resulting organometallic compounds are synthetically valuable intermediates due to their versatility and reactivity in organic synthesis. 2 Compared to bismetallation of one π-component (Scheme 1, eq. 1), bismetallation of two (or more) π-components (Scheme 1, eq. 2) are particularly noteworthy due to their applications towards complex molecule syntheses.
Scheme 1.
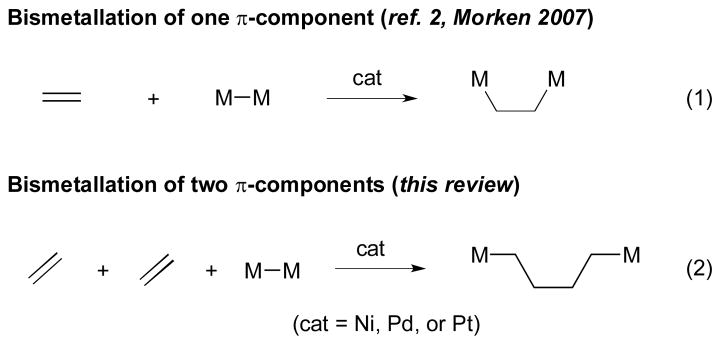
Bismetallative coupling with one vs. two π-components.
The coupling reactions of organometallic reagents with diene components were pioneered by Mori5 and Tamaru6 (Scheme 2, eq. 1). These powerful reductive coupling reactions, involving organometallic reagents (M–R) or metal hydrides (M–H), have attracted much attention and have expanded on earlier reports by Sato and Montgomery.7 However, the coupling of bismetallic reagents (M–M) with two π components (Scheme 2, eq. 2) has gained relatively less attention in spite of its potential for providing an efficient method in multiple bond-forming reactions. This review focuses on the major developments in the bismetallative multicomponent coupling reactions that are catalyzed by group 10 transition metals.
Scheme 2.
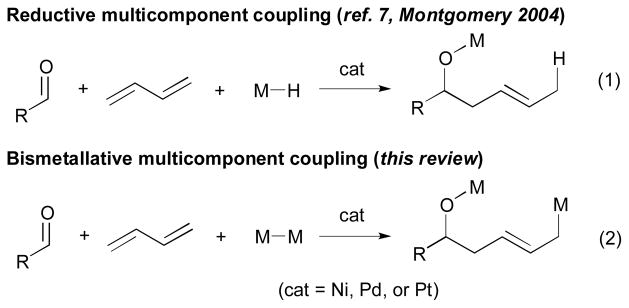
Reductive vs. bismetallative multicomponent coupling.
2. Mechanistic Considerations
Several different mechanistic pathways have been suggested for the bismetallative multicomponent coupling reactions that are catalyzed by Ni, Pd, or Pt. One possible mechanism (oxidative cyclization mechanism)8, 9 commences with the coordination of the π-components with the catalyst 1 and the subsequent formation of a metallocyclic intermediate 2 (Scheme 3).10 In the presence of bismetallic reagents, this cyclic intermediate (2) may undergo σ-bond metathesis forming a bismetallic complex 3. Finally, reductive elimination would afford the product 4 and regenerate the catalyst (1).
Scheme 3.

Oxidative cyclization mechanism.
In some cases, however, the initial oxidation of the catalyst (1) occurs by insertion with bismetallic reagents (oxidative addition mechanism, Scheme 4).11 Next, subsequent insertion of one π-component of substrate 5 would give an intermediary complex 6. The intermediate 6 then can undergo additional insertion reaction with the other π-component of the substrate to generate cyclic intermediate 7. Lastly, reductive elimination of 7 would afford the product 8 and close the catalytic cycle.
Scheme 4.

Oxidative addition mechanism.
The above-described catalytic cycles represent some of the most typical reaction pathways suggested for this process, but they are not meant to be comprehensive. Mechanisms for these reactions can vary depending on catalysts, substrates, and other reaction conditions. More detailed mechanistic considerations for each category of the process will be discussed accordingly in the corresponding sections.
3. Pd-Catalyzed Bismetallative Multicomponent Coupling
3.1. Coupling of Alkyne–Alkyne and Alkyne–Alkene
The Pd-catalyzed bismetallative alkyne–alkyne and alkyne–alkene coupling reactions have been investigated in a variety of contexts by several research groups. One of the earliest observations of this type of coupling was reported by Sakurai (Scheme 5).12 The main goal of this study was to investigate the properties of silicon–silicon bond of 1,2-disilacycloalkanes, focusing on the donor ability of the Si–Si bond. During their study of cycloaddition reactions of organodisilane reagents to various acetylenes, it was observed that the disilane reagent (10) can participate in a coupling reaction between two acetylenes (9) to give 11 (15% yield), along with product (12). Although the yield of the two-π-component-coupling product was not great, its presence demonstrated the potential of bismetallic reagents towards multicomponent reactions.
Scheme 5.
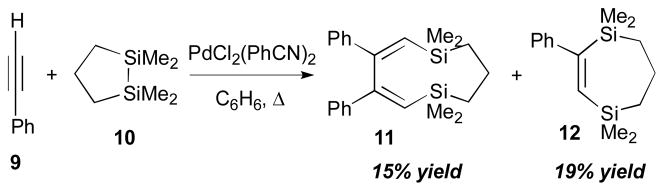
Sakurai’s intermolecular alkyne–alkyne coupling (1975).
More synthetically useful methods in this type of reaction were developed later by Tanaka,13 Lautens,14 Mori,11 and RajanBabu.15 The Tanaka group (Scheme 6) observed intramolecular alkyne–alkene coupling reactions that are promoted by a B–Si reagent in the presence of a palladium catalyst.13a An intramolecular alkyne-alkyne coupling reaction with 4,4-bis(ethoxycarbonyl)hep-6-en-1-yne (13) and 14 was catalyzed by Pd2(dba)3 and ETPO (4-ethyl-2,6,7-trioxa-1-phosphabicyclo[2.2.2]octane) to afford 15 in 84% yield (detected by 1H NMR). Along with the desired cyclization product, the formation of a byproduct (9% yield, not shown), which was speculated to have come from silaboration of the triple bond, was also observed by 1H NMR.
Scheme 6.
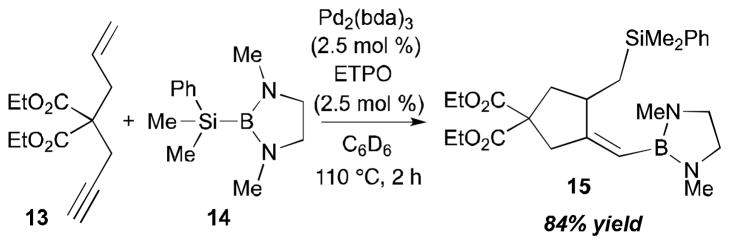
Tanaka’s intramolecular alkyne–alkene coupling (1997).
In Mori’s work (Scheme 7),11 enyne 16 was reacted with 1.5 equivalents of Me3Si–SnBu3 (17) in the presence of 10 mol % of Pd(OH)2 on charcoal at room temperature to furnish the bismetallation product 18 in 82% yield. With the use of Pd2(dba)3 as a ligand (3 mol %) under the same reaction conditions, the product was obtained in a slightly lower yield (57%). Also, a saturated indole moiety 20 was constructed (66% yield, with Pd(OH)2/C) from 3° amine-substituted enyne 19 in a stereospecific fashion. In this study, it was observed that using a palladium catalyst in the absence of phosphine ligands tends to suppress the formation of the bismetallation product of alkyne, which is an undesired product of the reaction.
Scheme 7.
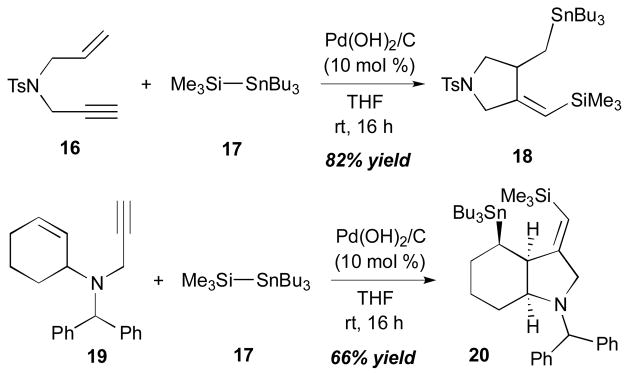
Mori’s intramolecular alkyne–alkene coupling (2001).
Two possible pathways for this process were considered as illustrated in Scheme 8. The reaction commences with oxidative addition of Me3Si–SnBu3 (17) to a Pd catalyst to give 21, which is followed by insertion of the alkyne moiety of 5 into the Pd–Si bond to form intermediate 22. Then, an insertion of the alkene portion of 22 into the Pd–C bond occurs intramolecularly to afford a Pd complex 23. Finally, reductive elimination would furnish cyclized product 24, and the palladium catalyst is regenerated. Mori and co-workers also consider an alternate pathway; it involves the formation of 25, which is the product of insertion of the alkene into the Pd–Sn bond of 22. From the intermediate complex 25 the identical cyclized product 24 would be afforded via reductive elimination.
Scheme 8.

Proposed mechanism for enyne cyclization (Mori, 2001).
One of RajanBabu’s many contributions in this field is the preparation of functionalized bisalkylidenes from 1,6-diynes, as described in Scheme 9.15 The construction of a (Z,Z)-1,3-diene 27 (79% yield) was accomplished by reaction of di-O-methyl dipropargylmalonate (26) with Me3Si–SnBu3 (17) with a catalytic amount of Pd2(dba)3 and P(o-tolyl)3.
Scheme 9.
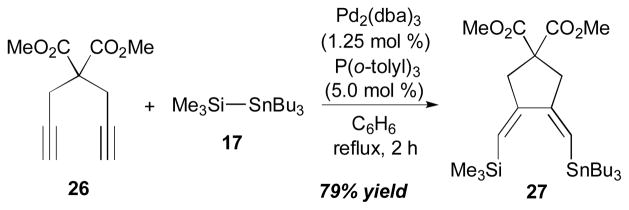
RajanBabu’s intramolecular alkyne–alkyne coupling (2000).
One of the interesting features of molecule 27 is that sterically-encumbered silicon and tin groups enforce a nonplanar/helically chiral structure for a usually planar diene (Scheme 10). The fluxional nature and stereochemistry of the (Z,Z)-1,3-diene 27 were analysed by NMR spectroscopy. The 1H NMR shows two distinctive sets of signals (two quartets) for HA and HB at −40 °C, while broad signals for the ring methylene protons appeared at 20 °C. Except for a highly unlikely conformational equilibrium involving the cyclopentane, such chirality must have originated from the helical arrangement of substituents in the (Z,Z)-diene 27.
Scheme 10.
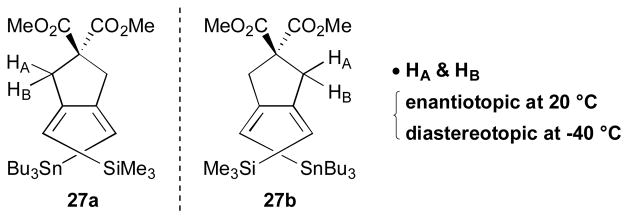
Enantiomers of 27 at –40 °C (RajanBabu, 2000).
In a recent study, the RajanBabu group explored more details of the stereochemistry in these cyclization reactions.16 For this study, cyclization reactions of diynes with a B–Sn reagent 29 were investigated. The regio- and stereoselectivities of this process were rationalized by the assumption that the addition of 29 to the diyne substrate 28 occurs at the less substituted, electron-rich alkyne forming a C–B bond in the product 30 (Scheme 11). The cyclization via carbometalation of the palladium intermediate 31, followed by reductive elimination, will result in the formation of C–Sn bond in the product 30.
Scheme 11.

RajanBabu’s stereoselective diyne cyclization (2012).
In this report, the group speculated that the cyclization step with the carbometalation process is the stereoselectivity-determining step and proposed a possible origin of stereoselectivity (Scheme 12). When the 7,8-bisalkylidenecyclooctadiene moiety of the product 30 is formed via carbometalation, the configuration of the newly-formed axial chiral element (Ra) is determined. The (Sa, Ra) configuration of the product (30-conf A) and, thereby, its transition state have relatively strain-free pseudo-chair conformation, compared to more strained pseudo-boat conformation (30-conf B).
Scheme 12.
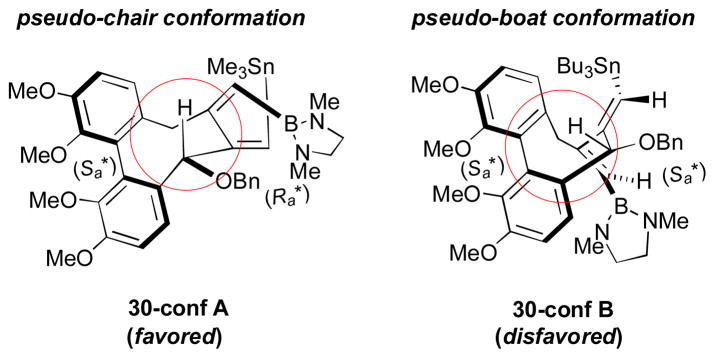
Proposed origin of stereoselectivity (RajanBabu, 2012).
3.2. Coupling of Allene–Allene and Allene–Alkyne
Pd-catalyzed bismetallative allene-allene coupling reactions with bismetallic reagents were first demonstrated by the Kang group.17 In this study, it was demonstrated that silylstannanes (Si–Sn) or distannanes (Sn–Sn) can promote the palladium-catalyzed addition–cyclization reaction of tethered bis(allenes). As described in Scheme 13, the cyclization reaction proceeds with bis(allene) 32 and (trimethylsilyl)tributylstannane (17) in the presence of Pd(Ph3P)4 (5 mol %) to afford a trans-fused cyclized product 33 in 78% yield. On the other hand, when distannane Bu3Sn–SnBu3 (34) was used for this process with the same bis(allene) 32 and Pd(PPh3)4 (5 mol %), a cis-fused distannane 35 was obtained in 73% yield.
Scheme 13.

Kang’s intramolecular allene–allene coupling (2000).
The different stereochemical outcomes between two bismetallic reagents (i.e. cis vs. trans fusion at ring junction) were rationalized in this report (Scheme 14). For the Si–Sn reagent, allylpalladium complex 37 is formed by the addition of the Bu3Sn–Pd–SiMe3 species to the allene moiety. Intermediate 37 should be favoured over 36 due to the steric hindrance of the nearby TMS (trimethylsilyl) group. The cyclization reaction of this intermediate (37) and subsequent reductive elimination would result in the trans bicyclic product 33. In the case of the Si–Sn reagent, however, the chelated σ-allylpalladium intermediate 39 is preferably formed from the coordinated compound 38. Then, the rapid carbocyclization of 39 and reductive elimination would give the cis-fused bicycle 35. It is conjectured that the differentiation between the two types of the reagents comes from the different bond lengths (C–Sn bond vs. C–Si bond). In other words, the steric encumbrance of the TMS group in those allylpalladium complexes is more severe than that of the Bu3Sn group due to the shorter bond length of the C–Si bond.
Scheme 14.
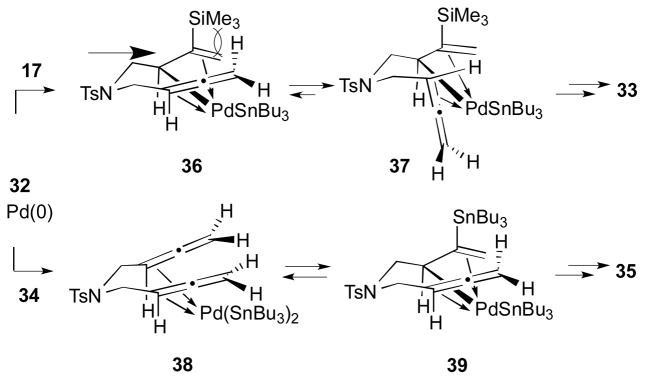
Rationale for stereochemical outcome (Kang, 2000).
With the same types of organometallic reagents (i.e. Si–Sn and Sn–Sn), RajanBabu and co-workers showed that an allene and alkyne can be coupled intramolecularly with a palladium catalyst (Scheme 15).18 In the presence of Ph3Sn–SiMe2tBu (41), Pd2(dba)3·CHCl3 and P(C6F5)3, alleneyne 40 was cleanly transformed into the cyclic product 42 in 80% yield at room temperature. It was found that the source of Pd affects the efficiency of the reaction in the order of the following: (C6F5)3P: (PhCN)2PdCl2 ≈ [Pd(allyl)Cl]2/AgOTf > Pd2(dba)3·CHCl3 ≫ PdCl2. It was also observed that the silyltin reagents are generally superior to other bismetallating reagents (Sn–Sn or Sn–B) for the cyclization of allenynes.
Scheme 15.
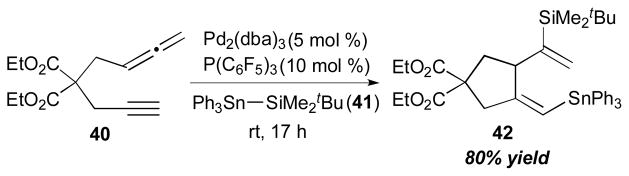
RajanBabu’s intramolecular allene–alkyne coupling (2001).
For allene–allene coupling, Ge–Sn reagents can also participate as a coupling component; this process was investigated by Yu et al (Scheme 16).19 They pointed out that the stereochemistry of the outcomes is dependent on the Ge–Sn reagents and the catalysts that are used in the reactions. Bis(allene) 32 can react with Ph3Ge–SnBu3 (43) with a catalytic amount of (π-allyl)2Pd2Cl2 (5 mol %) to afford the trans-cyclized product 44 (77% yield). On the other hand, the same bis(allene) 32 furnished the cis-cyclized product 46 (51% yield) when it was reacted with Bu3Ge–SnBu3 (45) in the presence of Pd(PPh3)4.
Scheme 16.

Yu’s intramolecular allene–allene coupling (2004).
3.3. Coupling of Diene–Diene
Among bismetallative multicomponent coupling reactions, the Pd-catalyzed diene–diene bismetallative coupling is one of the most frequently-observed reaction categories. Earliest studies were reported by Sakurai et al., in which dienes are intermolecularly coupled in the presence of cyclic disilane reagents to generate allyl silanes (Scheme 17).20 Since the initial Sakurai report, preliminary observations of this type of coupling were reported by a number of research groups including Kumada,21 West,22 Seyferth,23 Manners,24 and Ando25 groups. Then, more synthetically applicable versions of this method were examined by the Tsuji group.26
Scheme 17.
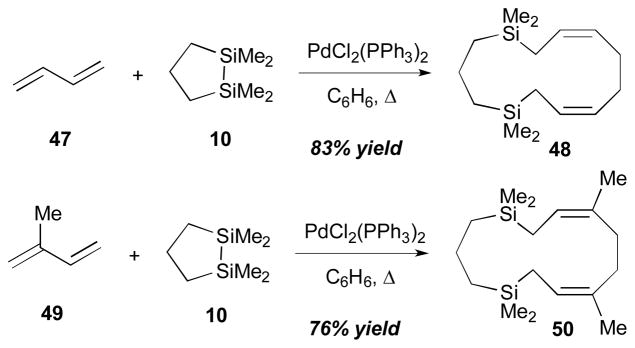
Sakurai’s intermolecular diene–diene coupling (1975).
According to the Sakurai’s account,20 when 1,3-butadiene (47) was treated with 1,1,2,2-tetramethyl-1,2-disilacyclopentane (10) in the presence of a Pd(II) catalyst, 1,1,5,5-tetramethyl-1,5-disilacyclotrideca-7,11-diene (48) was obtained in 83% yield (Scheme 17). Isoprene (49) also gave the corresponding allyl silane 50 in 76% yield under the same reaction conditions.
Besides the cyclic organodisilane 10, acyclic organodisilanes have been utilized for diene–diene coupling reactions by the Sakurai group (Scheme 18).27 The reaction of hexamethyldisilane (51) with isoprene (49) in the presence of a catalytic amount of Pd(OAc)2 afforded 1,8-disilyloctadiene 52 (87% yield) in a regio- and stereoselective fashion. Also, 1,3-butadiene (47) can participate in this coupling reaction with hexamethyldisilane (51) and a palladium catalyst, PdCl2(p-MeOC6H4CN)2, to afford 53 in 85% yield. The coupling product 53 was used as a starting material for the synthesis of dl-muscone (54),28 as described in Scheme 19.
Scheme 18.
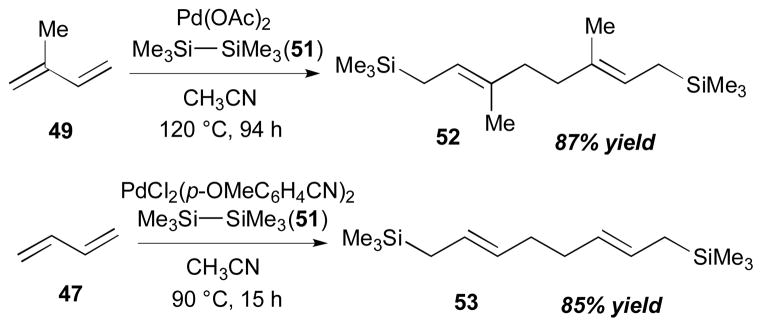
Sakurai’s disilylative dimerization of dienes (1984).
Scheme 19.

Synthesis of dl-muscone 54 (Sakurai, 1984).
The Tsuji group investigated coupling reactions of 1,3-dienes with use of distannane and disilane reagents in the presence of a palladium catalyst (Scheme 20).26 Employing hexamethyldistannane (55), 1,3-butadiene (47), and a catalytic amount of Pd(dba)2, Tsuji and co-workers were able to prepare a single isomer of double-stannated dimer 56 in 89% yield. Under the same conditions, a reaction with isoprene (49) furnished the coupling product 57 (75% yield) in a highly regio- and stereoselective manner.
Scheme 20.
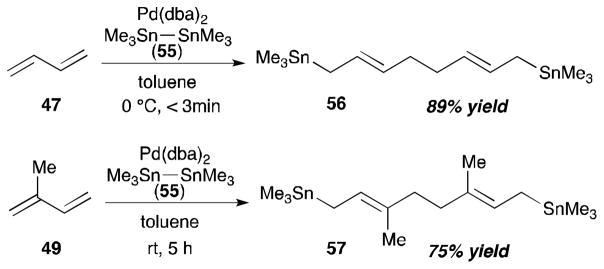
Tsuji’s intermolecular diene–diene coupling (1992).
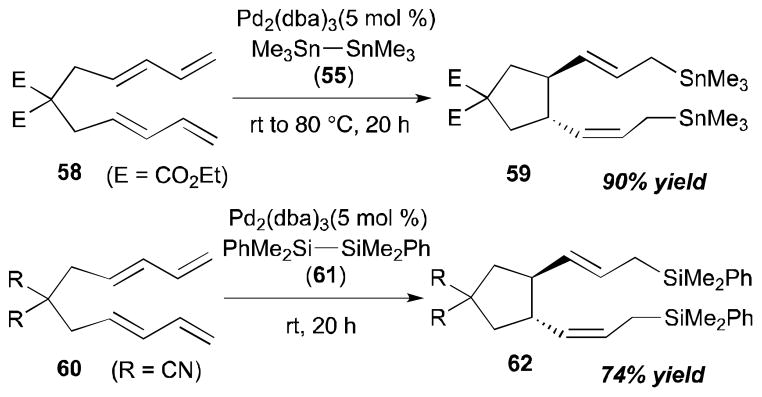
Intramolecular coupling reactions of dienes were also surveyed by the Tsuji group (Scheme 21).29 The Pd-catalyzed reaction of Me3Sn–SnMe3 (55) with ethoxycarbonyl-substituted bisdiene 58 afforded the cyclized/distannylated product 59 in 90% yield in a highly regio- and stereoselective manner. A disilane reagent, Bu3Si–SiBu3 (61), also promoted intramolecular coupling of cyanosubstituted bisdiene 60 to provide 62 in 74% yield with a catalytic amount of Pd(dba)2. The stereochemistry of the alkenes of the product was confirmed by 2D heteronuclear multiple bond coherence (HMBC) spectra; the stereochemistry at the ring was determined by X-ray crystallography of urethane 63, which was transformed from the product 59. The X-ray structure (ORTEP drawing with 30% probability ellipsoids) and the derivatization details of diester 59 to diurethane 63 are described in Scheme 22.
Scheme 21.
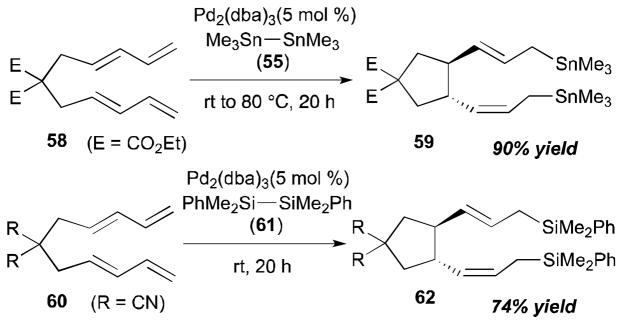
Tsuji’s intramolecular diene–diene coupling (1995).
Scheme 22.
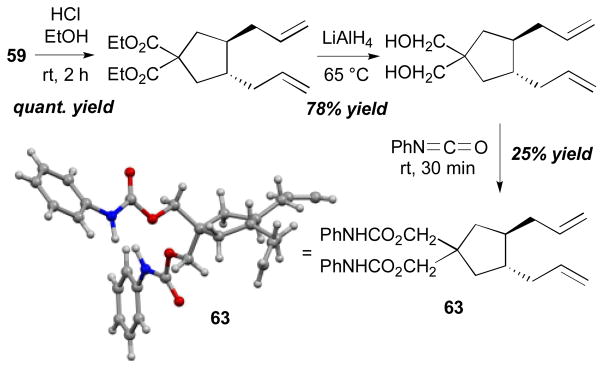
Synthesis and crystal structure of 63 (Tsuji, 1995).
3.4. Coupling of Aldehyde–Allene and Ketone–Allene
The Pd-catalyzed carbonyl–allene coupling reactions with a Si–Sn reagent were demonstrated by Kang et al. (Table 1).30 Allene-aldehyde 64 efficiently underwent silastannylative coupling of multiple π components to produce cis-cyclopentanol 65 (71% yield), when it was treated with Me3Si–SnBu3 in the presence of (π-allyl)2Pd2Cl2 at ambient temperature (entry 1). This method was also effective for the preparation of cyclohexanol derivatives; allene aldehyde 66 was transformed into cis-cyclohexanol 67 under the same reaction conditions in 62% yield (entry 2). In addition to these aldehyde–allene couplings, they tackled more challenging tasks, ketones–allene couplings (entries 3 & 4). More sterically-hindered and less reactive allene ketones, 68 and 70, smoothly cyclized to give the corresponding coupling products 69 and 71, respectively, when they were reacted with 1.1 equivalents of Me3Si–SnBu3 (17) and 5 mol % of (π-allyl)2Pd2Cl2.
Table 1.
Kang’s intramolecular carbonyl–allene coupling (2002).a
| Entry | Allene carbonyl | Product | Yield (%) |
|---|---|---|---|
| 1 |
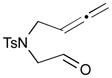 64 |
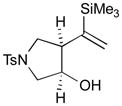 65 |
71 |
| 2 |
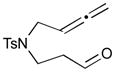 66 |
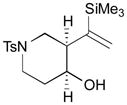 67 |
62 |
| 3 |
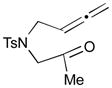 68 |
 69 |
67 |
| 4 |
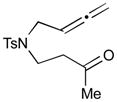 70 |
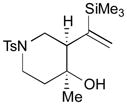 71 |
67 |
Reaction conditions: 1.0 equiv. of allene carbonyls, 1.1 equiv. of Me3Si–SnBu3 (17), 5 mol % of (π-allyl)2Pd2Cl2, THF, room temperature, 10 min.
The presumed mechanism of this process is illustrated in Scheme 23. The Si–Sn reagent 17 will oxidatively add to the palladium catalyst to form Me3Si–Pd–SnBu3 (21); then, it will add to the allene moiety of 68 to give a palladium complex 73, which is in a more stable conformation than 72. Subsequently, the σ- or π-allyl Pd complex 73 would undergo intramolecular allylation to the carbonyl and reductive elimination to afford cis-cyclopentanol 69. It is speculated that the stereochemistry of the reaction originated from the energy difference between intermediates 72 and 73. In other words, the steric interference between the TMS and the methyl group may render intermediate 73 energetically more stable than 72.
Scheme 23.

Rationale for stereochemical outcome (Kang, 2002).
4. Pt-Catalyzed Bismetallative Multicomponent Coupling
Platinum (Pt) catalyzed bismetallative multicomponent reactions are relatively sparse, compared to those catalyzed by other group 10 transition metal complexes (Pd or Ni). The main contributors in this area are the Miyaura group who studied diene–diene coupling (section 4.1), and the Ito group for aldehyde–diene coupling reactions (section 4.2).
4.1. Coupling of Diene–Diene
Pt-catalyzed diborylative diene–diene coupling reactions were examined by Miyaura et al. (Scheme 24).31 The reaction of isoprene (49) with bis(pinacolato)diboron (74) that is catalyzed by Pt(dba)2 afforded a borylated dimer product 75 in 94% yield. The process shows high stereoselectivity as well as great regioselectivity; only the (E,E) isomer was observed in this reaction (eq. 1). This three-component coupling product was obtained as a major product only when Pt(dba)2 was used as a catalyst. When Pt(PPh3)4 is employed as a catalyst in the same reaction, on the other hand, the 1:1 adduct of B2(pin)2 and diene (76) is formed exclusively (eq. 2).
Scheme 24.
Miyaura’s intermolecular diene–diene coupling (1996).
4.2. Coupling of Aldehyde–Diene
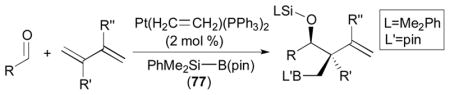
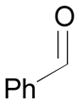
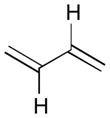
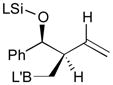
Pt-catalyzed silaborative intermolecular aldehyde–diene coupling reactions were accomplished by the Ito group (Table 2).32 It was observed that benzaldehyde (78), 2,3-dimethyl-1,3-butadiene (79), and silylborane 77 can be coupled in the presence of Pt(CH2=CH2)(PPh3)2 at 120 °C to give 80 in 85% yield (entry 1). An unsymmetrical diene, 2-phenyl-1,3-butadiene (82), can also be employed in this process to be coupled with both aromatic and aliphatic aldehydes (81 and 84) under similar reaction conditions to afford the coupling products 83 and 85 (entries 2 & 3). Also, this coupling reaction occurs under butadiene (47) atmosphere (1 atm) to give the correspondent product 86 in 63% yield with excellent stereoselectivity (entry 4). Notably, a cyclic diene 87 that is fixed in an s-cis conformation turned out to be an efficient coupling partner in this process to furnish 88 as a single diastereomer (entry 5).
Table 2.
Ito’s intermolecular aldehyde–diene coupling (1998).a
| |||||
|---|---|---|---|---|---|
| Entry | Aldehyde | Diene | Product | Yield | d.r. |
| 1 |
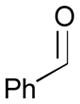 78 |
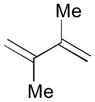 79 |
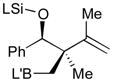 80 |
85 | 96:4 |
| 2 |
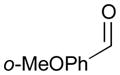 81 |
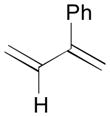 82 |
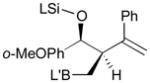 83 |
83 | 99:1 |
| 3 |
 84 |
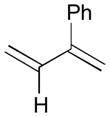 82 |
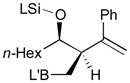 85 |
71 | 93:7 |
| 4 |
 78 |
 47 |
 86 |
63 | 95:5 |
| 5 |
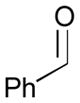 78 |
 87 |
 88 |
60 | 99:1 |
Reaction conditions: 2 mol % Pt(0), 1.5–3.0 equiv. aldehyde, hexane or octane, 80–120 °C.
The proposed mechanism for the Pt-catalyzed silaborative aldehyde–diene coupling reaction is illustrated in Scheme 25. The Si–B bond of 77 will be oxidatively added to the platinum(0) catalyst, and subsequent coordination of a diene will form a platinum(II) complex 89. Then, insertion of the diene to the Pt–B bond can occur to give cis-crotylplatinum complex 90 forming a C–B bond at the terminal carbon of the less substituted alkene. Reaction of the platinum complex 90 with an aldehyde will form a C–C bond at the γ position to the Pt atom, which will generate an (alkoxy)(silyl)platinum(II) complex 91. Finally, reductive elimination will afford the coupling product, in which a Si–O bond is present.
Scheme 25.

Proposed mechanism for Pt-catalyzed coupling (Ito, 1998).
5. Ni-Catalyzed Bismetallative Multicomponent Coupling
5.1. Coupling of Alkyne–Alkyne
The Ni-catalyzed bismetallative alkyne–alkyne coupling reactions was introduced by the Ito group.33 It was demonstrated that a nickel(0) catalyst can couple two 1-hexyne (92) molecules with a B–Si reagent (77) in a regio- and stereoselective manner to provide 93 as a major product. This silaborative dimerization was applied to intramolecular cyclization of diyne (94) to afford 95 in 55% yield. The diyne dimerization with a germylborane (Ge–B) reagent 96 was also reported; a head-to-head dimer 97 was obtained as a major product.
Based on the observed experimental results, they suggest the following catalytic cycle for a plausible mechanism for this process (Scheme 27). According to their speculation, an oxidative addition of the Si–B bond onto the Ni(0) complex generates the (silyl)(boryl)Ni(II) intermediate (98). Then, an alkyne undergoes cis-insertion into the B–Ni bond of the Ni(II) complex. The resulting vinyl-substituted nickel(II) intermediate (99) gets involved in another cis-insertion event to furnish 100. Finally, reductive elimination would give the product.
Scheme 27.

Proposed mechanism for alkyne dimerization (Ito, 1998).
5.2. Coupling of Aldehyde–Diene
5.2.1. Intramolecular Aldehyde–Diene Coupling
The intramolecular coupling of a 1,3-diene and a tethered aldehyde was studied by the Mori group (Scheme 28).34 With the use of Ni(cod)2 and PMe2Ph, a cyclic alcohol 102 (23% yield) was formed from a reaction of 101 and Bu3Sn–SiMe3 (17) in toluene. The product form of this process turned out to be sensitive to the ligands and solvents that are used in the reactions. The coupling reaction from the same starting material (101) with DMF as a solvent in the absence of any phosphine ligands afforded 103 as a sole product in 55% yield.
Scheme 28.
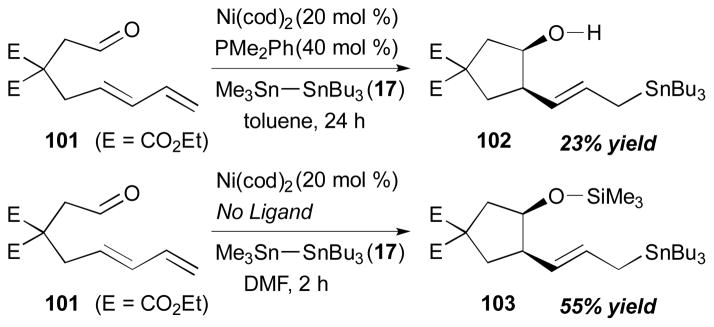
Mori’s intramolecular aldehyde–diene coupling (2002).
The Yu group examined sequential four-component coupling reactions (i.e. coupling of an aldehyde, a diene, and a diboron reagent followed by allylboration with another aldehyde) as shown in Scheme 29.35, 36 The reaction of a diene-aldehyde 104, benzaldehyde (78), and a diboron reagent 105 at 20 °C in the presence of Ni(cod)2 and P(2-furyl)3 afforded 106 in 87% yield. The unexpected stereochemical inversion of the product was observed at different reaction temperatures; when the subsequent allylation with benzaldehyde (78) was carried out at −78 °C (instead of at 20 °C), compound 107 was obtained as a single isomer. Additionally, a six-membered ring moiety (109) can be also prepared by this process from a diene-aldehyde 108, benzaldehyde (78), and 105 in the presence of a nickel catalyst (74% yield). This method is particularly noteworthy given its efficiency for installing four contiguous stereogenic centers in a single operation.
Scheme 29.

Yu’s sequential four-component coupling (2005).
The origin of the contrasting stereochemical outcomes, which vary depending on the temperatures, is speculated by the proposed pathways that are depicted in Scheme 30. The reaction begins with oxidative addition of the nickel catalyst to the diboron reagent. Then, diene insertion giving a π-allyl complex 110 and carbonyl insertion forming 111 would occur sequentially. After reductive elimination to give 112, a subsequent intermolecular allylation with benzaldehyde (78) will furnish either 106 or 107 depending on the temperature for this step. The source of the stereochemical control is not revealed in this report, but subtle geometrical preferences of two intermediates 113 and 114 at two different temperatures are described in Scheme 30.
Scheme 30.

Rationale for stereochemical outcome (Yu, 2005).
The Mori and Sato’s laboratories investigated intramolecular disilylative and silastannylative coupling reactions of 1,3-diene and a tethered aldehyde (Scheme 31).37 As for disilane reagents, it is known that halogenated disilane reagents tend to enhance the reactivity toward nickel catalysts.38 Disilylative coupling of 115 and PhF2Si–SiMe3 (116) in the presence of 20 mol % of Ni(cod)2 and 40 mol % of PPh3 furnished 117 (45% yield) in a completely regio- and stereoselective manner. It is worth mentioning that catalytic enantioselective cyclizations were attempted in this study. With the use of Ni(cod)2 and a chiral ligand 118,39 the reaction of 115 and Me3Si–SnBu3 (17) in DMF afforded 119 as a mixture of diastereomers.
Scheme 31.

Mori and Sato’s aldehyde–diene coupling (2007)
5.2.2. Intermolecular Aldehyde–Diene Coupling
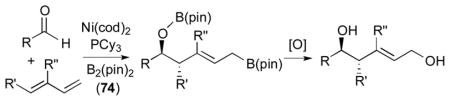
The intermolecular Ni-catalyzed diastereoselective bismetallative aldehyde–diene coupling reactions were demonstrated by the authors’ laboratory.8, 40 The coupling reactions of aldehydes, dienes, and B2(pin)2 (74) led to stereoselective formation of homoallylic boronic esters, which are synthetically valuable motifs (Table 3).8 Various aromatic aldehydes (78, 122, and 124) are coupled a 1,3-diene 120 and B2(pin)2 (74) to give 121, 123, and 125, respectively (entries 1–3); a reaction with an aliphatic aldehyde 126 affords 127 in a moderate yield (entry 4). Different diene substrates (128 and 49) also undergo the diboron-promoted three-component coupling reactions giving 129 and 130 under the same reaction conditions (entries 5–6). These reactions feature efficient preparatory methods for functionally and stereochemically enriched allylboronates.
Table 3.
Intermolecular aldehyde–diene coupling.a
| |||||
|---|---|---|---|---|---|
| Entry | Aldehyde | Diene | Product | Yield | d.r. |
| 1 |
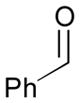 78 |
120 |
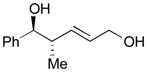 121 |
66 | >20:1 |
| 2 |
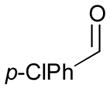 122 |
120 |
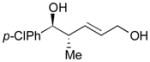 123 |
63 | >20:1 |
| 3 |
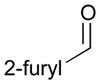 124 |
120 |
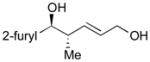 125 |
63 | >20:1 |
| 4 |
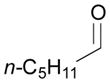 126 |
120 |
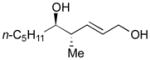 127 |
47 | >20:1 |
| 5 |
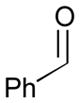 78 |
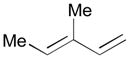 128 |
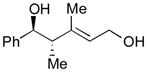 129 |
70 | >20:1 |
| 6b |
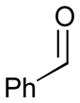 78 |
 49 |
130 |
68 | N/A |
Reaction conditions: 5 mol % Ni(cod)2, 10 mol % PCy3, 1.1 equiv. diene, 1.2 equiv. B2(pin)2, 0.2 M THF, rt, 6 h. Then, oxidation with H2O2 and NaOH. (b) Modified reaction conditions: 10 mol % Ni(cod)2, 10 mol % P(OEt)3. Acetylation improved the isolated yield.
Based on the mechanistic studies on these coupling reactions as well as relevant reductive coupling reactions, it was proposed the following catalytic cycle operates (Scheme 32). Initial oxidative cyclization will lead to the formation of nickelacyclic intermediate 132 from a nickel complex 131. In the presence of diboron reagent, subsequent σ-bond metathesis will yield intermediate 133. Then, reductive elimination will generate an allylic boronic ester 134 and restart the catalytic cycle.
Scheme 32.

Proposed mechanism for borylative aldehyde-diene coupling.
Surprisingly, the regioselectivity of the products in this process is reversed with the replacement of the ligand. In the presence of P(SiMe3)3 as a ligand, the nickel-catalyzed coupling of benzaldehyde (78), 1,3-pentadiene (120), and B2(pin)2 (74) afforded terminal boronate 137, which is regioisomeric to the product 136 that comes from the reaction with PCy3 (Scheme 33).40 The ligand effect that is shown in this study is rationalized by unique characteristics of P(SiMe3)3. The electron accepting ability of P(SiMe3)3 was observed by both Bartik41 and Helm.42 It is plausible that the large cone angle of the ligand, together with its electron accepting property, would accelerate reductive elimination of 135 to form 137, ahead of allyl isomerization that will lead to the formation of 136.
Scheme 33.
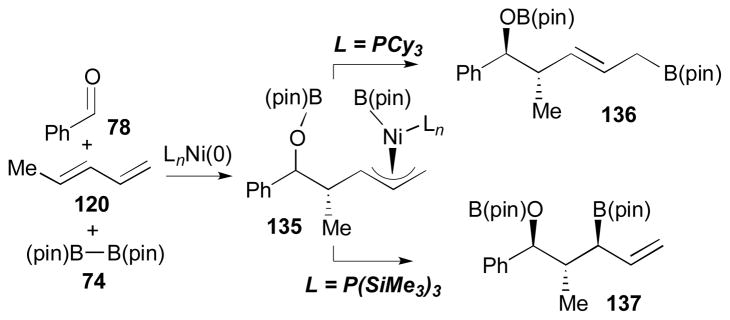
Ligand effect on regioselectivity.
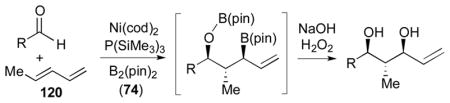
Using P(SiMe3)3 as a ligand, various aldehydes can be coupled with 1,3-pentadiene and B2(pin)2 (74) with excellent stereoselectivity by this process (Table 4). Generally, aromatic and heteroaromatic aldehydes (78 and 139) efficiently undergo the Ni-catalyzed coupling reactions with trans-piperylene (120) and B2(pin)2 (74) to provide 138 and 140 with great diastereoselectivities (entries 1–2). The coupling with aliphatic aldehydes (126 and 142) was also observed to be effective and afforded 141 and 143 under the same reaction conditions (entries 3–4). In addition, an α-chiral aldehyde (144) reacted with the Felkin selectivity to give 145 thereby revealing the potential for asymmetric synthesis (entry 5).
Table 4.
Aldehyde–diene coupling for 1,3-diols.a
| ||||
|---|---|---|---|---|
| Entry | Aldehyde | Product | Yield | d.r. |
| 1 |
 78 |
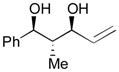 138 |
67 | >20:1 |
| 2 |
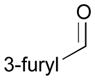 139 |
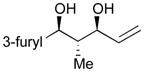 140 |
55 | >20:1 |
| 3 |
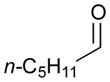 126 |
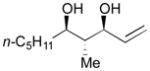 141 |
58 | >20:1 |
| 4 |
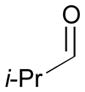 142 |
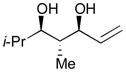 143 |
50 | >20:1 |
| 5 |
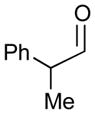 144 |
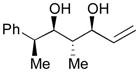 145 |
49 | 6:1 |
Reaction conditions: 10 mol % Ni(cod)2, 15 mol % P(t-Bu)3, 3.0 equiv. diene, 3.0 equiv. B2(pin)2 (74), 0.2 M THF, rt, 12 h. Then, oxidative workup with H2O2 and NaOH.
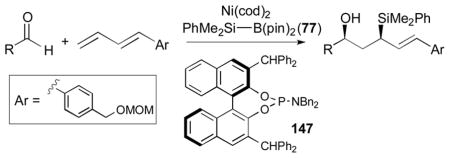
An outstanding accomplishment in this area was made by Saito and Sato in 2012; Ni-catalyzed enantio- and diastereoselective bismetallative aldehyde–diene couplings were demonstrated (Table 5).43 A silylborane reagent, PhMe2Si–B(pin) (77), was employed in this process along with 1,3-dienes and aldehydes in the presence of a nickel catalyst and a chiral phosphine ligand (147). Reactions with aldehydes that possess electron-donating groups (148 and 150) furnished the corresponding products (149 and 151) in good yields and with high enantioselectivities (entries 1–2). However, a reaction of an aldehyde with an electron-withdrawing group (152) exhibited a decreased yield (29% yield) giving 153, even though the enantiopurity was still good (85% ee, entry 3). Aliphatic aldehydes (142 and 155) also participate in this process to give the corresponding products (154 and 156) with great enantioselectivities (entries 4–5). These reactions provide a new tool to prepare optically active α-chiral allylsilanes.
Table 5.
Saito and Sato’s enantioselective coupling reactions (2012).a
| ||||
|---|---|---|---|---|
| Entry | Aldehyde | Product | Yield | ee |
| 1 |
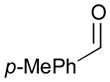 148 |
149 |
89 | 96 |
| 2 |
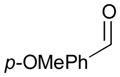 150 |
151 |
92 | 92 |
| 3 |
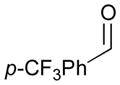 152 |
153 |
29 | 85 |
| 4 |
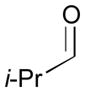 142 |
154 |
68 | 96 |
| 5 |
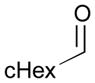 155 |
156 |
74 | 94 |
Reaction conditions: 10 mol % Ni(cod)2, 10 mol % 147, 1 equiv. diene, 2.5 equiv. aldehyde, 2.5 equiv. PhMe2Si–B(pin) (77), DMF, room temperature.
5.3. Coupling of Ketone–Diene
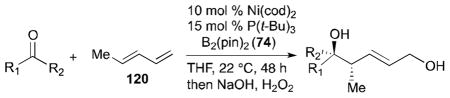
Ni-catalyzed ketone–diene diborylative coupling reactions have been reported by the authors’ laboratory (Scheme 34).44 The study has shown that ketones can participate in this process with high regio- and stereoselectivities to afford tertiary alcohols. Initial inquiry involved acetophenone, (E)-1,3-pentadiene (120), B2(pin)2 (74) with a nickel catalyst, which provided a tertiary alcohol 157 (76% yield) in a highly stereoselective fashion. As described in Scheme 34, halogenated aromatic methyl ketones are accommodated in this reaction (compounds 158, 159) as are the ketones with both electron-donating and electron-withdrawing substituents (compounds 160, 161). In all cases, excellent diastereoselectivity (> 20:1) was observed in the reaction products.
Scheme 34.

Intermolecular ketone–diene coupling.
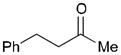
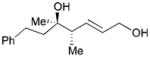
While reactions with aromatic methyl ketones furnish the 1,3-diols regioselectively, reactions of aliphatic ketones afford regioisomeric 1,5-diols (Table 6). For instance, the reaction with 4-phenyl-2-butanone (162) and 1,3-pentadiene gave the derived 1,5-diol 163 in 69% yield (entry 1). Also, constrained cyclic ketones (164 and 166) are found to undergo smooth coupling with the diene to afford 1,5-diols (165 and 167, entries 2–3). Notably, the coupling product 169 from 168 is acquired by preferred equatorial attack of the diene to the ketone electrophile (entry 4).
Table 6.
Ketone–diene coupling for 1,5-diol synthesis.a
| ||||
|---|---|---|---|---|
| Entry | Ketone | Product | Yield | d.r. |
| 1 |
 162 |
 163 |
69 | 7:1 |
| 2 |
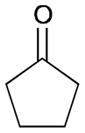 164 |
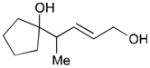 165 |
56 | N/A |
| 3 |
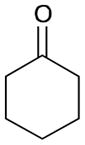 166 |
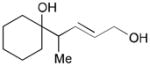 167 |
52 | N/A |
| 4 |
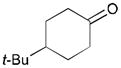 168 |
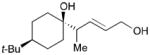 169 |
53 | >20:1 |
Reaction conditions: 10 mol % Ni(cod)2, 15 mol % P(t-Bu)3, 2.0 equiv. diene, 2.0 equiv. B2(pin)2 (74), 0.2 M THF, rt, 48 h. Then, oxidative workup with H2O2 and NaOH.
Based on the observations from these ketone–diene reactions, along with the aldehyde–diene couplings (section 5.2.2.), it was suggested that following mechanistic operates (Scheme 35). With non-hindered carbonyls (i.e. substrates in Tables 3, 4, and 6), reaction will proceed to give nickelacycle 170. Then, subsequent σ-bond metathesis would form π-allyl complex 171. Prior to reductive elimination, this intermediate may undergo π-σ-π isomerization to give 172, which would afford product 173. However, in the case of more hindered carbonyls (i.e. substrates in Scheme 34), the less substituted end of the diene would add to the carbonyl giving 174 due to the steric effects. The 1,3-diol 176 is furnished by subsequent σ-bond metathesis with 174 and reductive elimination of 175. In this case, the π-σ-π isomerization is likely impeded with the substituted π-allyl complex.
Scheme 35.

Mechanistic rationale for borylative ketone-diene coupling.
6. Conclusions
Catalytic bismetallative coupling reactions involving multiple πcomponents are considered an effective method to build complex molecules. This one-step process features C–C bond formation, functionalization with bismetallic reagents, and the control of regio- and stereoselectivities.
The scope of this process in terms of both bismetallic reagents and the π-components are broad enough to be generally applied to more elaborate synthetic sequences. In particular, contemporary applications of the bismetallative multicomponent coupling reactions, in which high enantio- and/or diastereoselectivities are displayed, have enabled the study of this area to make a significant step forward.
Despite of these considerable improvements, there is still much room for further progress in this field. More detailed investigation of the reaction mechanisms and applications to the construction of biologically-active molecules will be of great interest. Additionally, development of catalytic enantioselective versions of many of these process is warranted as is extension of bis-metallative coupling to simple 2π-2π (i.e. alkene-carbonyl) systems.
Scheme 26.

Ito’s Ni-catalyzed alkyne dimerization (1998).
Acknowledgments
H.Y.C. is grateful for a Rodin Fellowship (Boston College) and an ACS Organic Division Graduate Fellowship (Roche). Our research in this area has been supported by the US National Institutes of Health (NIGMS-59417)
Notes and references
- 1.For a recent review on multicomponent reactions, see: de Graaff C, Ruijter E, Orru RVA. Chem Soc Rev. 2012;41:3969. doi: 10.1039/c2cs15361k.
- 2.Burks HE, Morken JP. Chem Commun. 2007:4717. doi: 10.1039/b707779c. [DOI] [PubMed] [Google Scholar]
- 3.Ohmura T, Suginome M. Bull Chem Soc Jpn. 2009;82:29. [Google Scholar]
- 4.Chenard BL, Laganis ED, Davidson F, RajanBabu TV. J Org Chem. 1985;50:3666. [Google Scholar]
- 5.For one of the earlies reports from the Mori group, see: Sato Y, Takimoto M, Hayashi K, Katsuhara T, Takagi K, Mori M. J Am Chem Soc. 1994;116:9771.
- 6.For one of the earlies reports from the Tamaru group, see; Kimura M, Ezoe A, Shibata K, Tamaru Y. J Am Chem Soc. 1998;120:4033.
- 7.Ikeda S, Sato Y. J Am Chem Soc. 1994;116:5975.Montgomery J, Savchenko AV. J Am Chem Soc. 1996;118:2099.For a representative review on catalytic reductive coupling reactions, see: Montgomery J. Angew Chem, Int Ed. 2004;43:3890. doi: 10.1002/anie.200300634.
- 8.Cho HY, Morken JP. J Am Chem Soc. 2008;130:16140. doi: 10.1021/ja806113v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For a DFT study on relevant reductive coupling reactions, see: McCarren PR, Liu P, Cheong PH-Y, Jamison TF, Houk KN. J Am Chem Soc. 2009;131:6654. doi: 10.1021/ja900701g.
- 10.A nickelacyclic complex comprising an aldehyde, a diene, a nickel, and a phosphine ligand was characterized by X-ray crystallography: Ogoshi S, Tonomori K, Oka M, Kurosawa H. J Am Chem Soc. 2006;128:7077. doi: 10.1021/ja060580l.
- 11.One of the earlier reports from the Mori group: Mori M, Hirose T, Wakamatsu H, Imakuni N, Sato Y. Organometallics. 2001;20:1907.
- 12.Sakurai H, Kamiyama Y, Nakadaira Y. J Am Chem Soc. 1975;97:931. [Google Scholar]
- 13.(a) Onozawa S-Y, Hatanaka Y, Tanaka M. Chem Commun. 1997:1229. [Google Scholar]; (b) Onozawa SY, Hatanaka Y, Choi N, Tanaka M. Organometallics. 1997;16:5389. [Google Scholar]
- 14.Lautens M, Mancuso J. Synlett. 2002:394. [Google Scholar]
- 15.One of the earlier reports from the RajanBabu group: Gréau S, Radetich B, RajanBabu TV. J Am Chem Soc. 2000;122:8579.
- 16.Gong W, Singidi RR, Gallucci JC, RajanBabu TV. Chem Sci. 2012;3:1221. and references therein. [Google Scholar]
- 17.Kang SK, Baik TG, Kulak AN, Ha YH, Lim Y, Park J. J Am Chem Soc. 2000;122:11529. [Google Scholar]
- 18.Shin S, RajanBabu TV. J Am Chem Soc. 2001;123:8416. doi: 10.1021/ja011281t. [DOI] [PubMed] [Google Scholar]
- 19.Hong YT, Yoon SK, Kang SK, Yu CM. Eur J Org Chem. 2004:4628. [Google Scholar]
- 20.Sakurai H, Kamiyama Y, Nakadaira Y. Chem Lett. 1975:887. [Google Scholar]
- 21.Tamao K, Okazaki S, Kumada M. J Organomet Chem. 1978;146:87. [Google Scholar]
- 22.Carlson CW, West R. Organometallics. 1983;2:1801. [Google Scholar]
- 23.Seyferth D, Goldman EW, Escudié J. J Organomet Chem. 1984;271:337. [Google Scholar]
- 24.Finckh W, Tang BZ, Lough A, Manners I. Organometallics. 1992;11:2904. [Google Scholar]
- 25.Kusukawa T, Kabe Y, Nestler B, Ando W. Organometallics. 1995;14:2556. [Google Scholar]
- 26.Distannylative coupling: Tsuji Y, Kakehi T. J Chem Soc, Chem Commun. 1992:1000.Disilylative coupling: Obora Y, Tsuji Y, Kawamura T. Organometallics. 1993;12:2853.
- 27.Sakurai H, Eriyama Y, Kamiyama Y, Nakadaira Y. J Organomet Chem. 1984;264:229. [Google Scholar]
- 28.For an enantioselective synthesis of (R)-muscone, see: Kamat VP, Hagiwara H, Katsumi T, Hoshi T, Suzuki T, Ando M. Tetrahedron. 2000;56:4397. doi: 10.1021/jo000785r.
- 29.Obora Y, Tsuji Y, Kakehi T, Kobayashi M, Shinkai Y, Ebihara M, Kawamura T. J Chem Soc, Perkin Trans. 1995;1:599. [Google Scholar]
- 30.Kang SK, Ha YH, Ko BS, Lim Y, Jung J. Angew Chem, Int Ed. 2002;41:343. doi: 10.1002/1521-3773(20020118)41:2<343::aid-anie343>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 31.Ishiyama T, Yamamoto M, Miyaura N. Chem Commun. 1996:2073. [Google Scholar]
- 32.Suginome M, Nakamura H, Matsuda T, Ito Y. J Am Chem Soc. 1998;120:4248. [Google Scholar]
- 33.Suginome M, Matsuda T, Ito Y. Organometallics. 1998;17:5233. [Google Scholar]
- 34.Sato Y, Saito N, Mori M. Chem Lett. 2002;18 [Google Scholar]
- 35.Yu CM, Youn J, Yoon SK, Hong YT. Org Lett. 2005;7:4507. doi: 10.1021/ol051806c. [DOI] [PubMed] [Google Scholar]
- 36.Similar sequential three-component coupling reactions (i.e. coupling of a diene and a diboron reagent followed by allylboration with an aldehyde) are also reported by authors’ laboratory: Morgan JB, Morken JP. Org Lett. 2003;5:2573. doi: 10.1021/ol034936z.
- 37.Saito N, Mori M, Sato Y. J Organomet Chem. 2007;692:460. [Google Scholar]
- 38.(a) Hayashi T, Matsumoto Y, Ito Y. Teterahedron Lett. 1988;29:4147. [Google Scholar]; (b) Ozawa F, Sugawara M, Hayashi T. Organometallics. 1994;13:3237. [Google Scholar]
- 39.Burk MJ, Feaster JE, Harlow RL. Organometallics. 1990;9:2653. [Google Scholar]
- 40.Cho HY, Morken JP. J Am Chem Soc. 2010;132:7576. doi: 10.1021/ja101513d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barkik T, Himmler T, Schulte HG, Seevogel K. J Organomet Chem. 1984;272:29. [Google Scholar]
- 42.McCampbell TA, Kinkel BA, Miller SM, Helm ML. J Chem Crystallogr. 2006;36:271. [Google Scholar]
- 43.Saito N, Kobayashi A, Sato Y. Angew Chem, Int Ed. 2012;51:1228. doi: 10.1002/anie.201107360. [DOI] [PubMed] [Google Scholar]
- 44.Cho HY, Yu Z, Morken JP. Org Lett. 2011;13:5267. doi: 10.1021/ol202138v. [DOI] [PMC free article] [PubMed] [Google Scholar]