

Abstract
Innate lymphoid cells (ILCs) are a group of hematopoietic cells devoid of antigen receptors that have important functions in lymphoid organogenesis, in the defense against extracellular pathogens, and in the maintenance of the epithelial barrier. Three distinct groups of ILCs have been identified on the basis of phenotypic and functional criteria and termed ILCs1, ILCs2, and ILCs3. Specifically, ILCs1 express the transcription factor T-bet and secrete T helper type-1- (Th1-) related cytokines, ILCs2 are dependent on the transcription factor RORα and express Gata-3 and the chemokine receptor homologous molecule (CRTH2) and produce Th2-related cytokines, and ILCs3 express the transcription factor RORγt and synthesize interleukin- (IL-) 17, IL-22, and, under specific stimuli, interferon-γ. ILCs represent a relatively small population in the gut, but accumulating evidence suggests that these cells could play a decisive role in orchestrating both protective and detrimental immune responses. In this review, we will summarize the present knowledge on the distribution of ILCs in the intestinal mucosa, with particular focus on their role in the control of both infections and effector cytokine response in immune-mediated pathologies.
1. Introduction
Cells of the innate immune system (i.e., dendritic cells, macrophages, NK cells, and neutrophils) have the ability to recognize and rapidly respond to pathogens with production of various cytokines, which in turn regulate the antigen-driven differentiation of cells of the adaptive immune system (i.e., T and B lymphocytes). This process triggers an effective and specific response, which eliminates pathogens and resolves inflammation and tissue damage [1]. Effector cytokines are also involved in the orchestration of tissue-damaging immune response in states of chronic inflammation [2]. In recent years, the advent of sophisticated techniques of cellular biology has led to the identification of a new class of innate cells, termed innate lymphoid cells (ILCs), which have the ability to produce a vast array of cytokines mainly depending on their state of differentiation [3, 4]. ILCs play broad roles in lymphoid organogenesis, in the defense against extracellular pathogens, and in the maintenance of the epithelial barrier and are supposed to contribute to the amplification of immune-inflammatory responses in various organs [3, 4]. ILCs lack some cell lineage markers associated with T and B lymphocytes, dendritic cells, macrophages, and granulocytes but express CD90, CD25, and interleukin- (IL-) 7 receptor (R)α (CD127) (Table 1) [3, 4]. ILCs develop from hematopoietic precursors and their development is partially or wholly dependent on the common γ-chain, Notch, the transcription factor inhibitor of DNA binding-2 (Id2), IL-7, a cytokine involved in hematopoietic cell development and proliferation [3–6], and other transcription factors such as T-bet for ILC1, RORα and Gata-3 for ILC2, and TCF1 and Gata-3 for ILC3 (Figure 1). Mice deficient for Id2 show largely normal development of T and B cells but lack all ILC subsets, suggesting the existence of a common Id2-expressing progenitor to ILC subsets [3–6].
Table 1.
Markers expressed by various subsets of innate lymphoid cells (ILCs).
| ILC group | ILC lineage | Marker |
|---|---|---|
| 1 | ILC1 | LIN−, CD25 low, CD56−, IL-7Rα+, CD161+/−, NKp44−, NKp46−, IL-1R+, ICOS+, IL-12Rβ2+, and CRTH2− |
| NK cells | LIN−, CD56+, CD25+/−, IL-7Rα+/−, CD161+/−, NKp44+/−, NKp46+, IL-12Rβ2+, and CRTH2− | |
|
| ||
| 2 | ILC2 | LIN−, CD25 low, CD117+/−, IL-7Rα+, ICOS+, IL-1R+, ST2+, IL-17RB+, CRTH2+, and CD161+ |
|
| ||
| 3 | ILC3 | LIN−, CD25 low, CD117+, IL-7Rα+, CD161+, NKp44+, NKp46+, CRTH−, IL-1R+, IL-23+, and ICOS+ |
| LTi cells | LIN−, CD117+, IL-7Rα+, CD161+/−, IL-1R+, and IL-23+ | |
LIN, lineage; NK, natural killer; LTi, lymphoid tissue-inducer; IL, interleukin; CRTH2, chemoattractant receptor homologous molecule; ICOS, inducible T cell costimulator; R, receptor; ST2, subunit of IL-33R.
Figure 1.
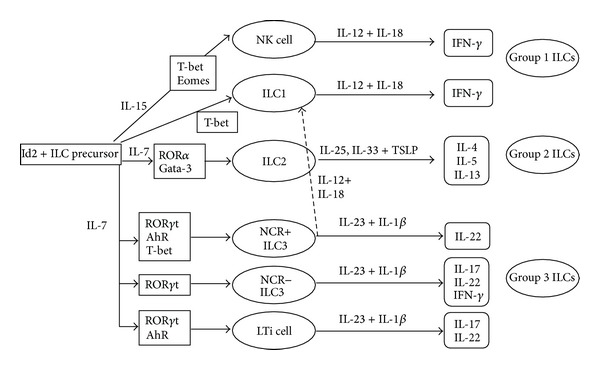
Schematic view of the differentiation of various innate lymphoid cells (ILCs), of the factors involved in such a differentiation, and of the cytokines produced by distinct subsets of ILCs. Id2, inhibitor of DNA binding-2; Gata-3, Gata-binding protein 3; RORα, retinoic acid receptor-related orphan receptor-α; Eomes, eomesodermin; NK, natural killer; LTi, lymphoid tissue-inducer; IL, interleukin; NCR, natural cytotoxicity triggering receptor; IFN-γ, interferon-γ; AhR, aryl hydrocarbon receptor; TSLP, thymic stromal lymphopoietin.
ILCs are currently classified into three distinct populations on the basis of the expression of specific transcription factors and/or cell surface markers and their ability to secrete some profiles of effector cytokines. Thus, they closely resemble the heterogeneity of CD4+ T helper (Th) cell subsets. ILCs1 include classical NK cells and ILCs expressing T-bet, a Th-type-1-associated transcription factor, and producing interferon- (IFN-) γ, a potent stimulator of phagocyte activity against intracellular bacteria [3, 4, 7]. Conventional NK cells differ from ILCs1, because their development is dependent on the transcription factor eomesodermin (Eomes) and independent of Id2 [3, 4, 7, 8]. ILCs2 express the Th2-related transcription factor Gata-3 and, in humans, the chemokine receptor homologous molecule CRTH2 and produce IL-5, IL-9, IL-13, IL-4, and/or amphiregulin [3, 4] and play important roles in immunity to helminth infections [9–11] and in the pathogenesis of asthma and allergies [12–14]. ILCs3 express the transcription factor RORγt, synthesize IL-17A and IL-22, and, under specific stimuli, IFN-γ, and are involved in the recruitment of neutrophils, release of antimicrobial peptides, and epithelial cell proliferation (Figure 1) [3, 4]. Therefore, ILCs3 are required for the defense against bacterial infections [3, 4] and provide “help” to marginal zone B cells [15].
Here we review the available data on the role of ILCs in the control of both intestinal infections and effector cytokine response in immune-mediated pathologies of the gut.
2. ILCs1
At the present time, it is debated if typical ILCs1, which are predicted to be RORγt-independent, really exist in the gut, as IFN-γ-producing ILCs1-like cells described so far seem to originate from ILCs3 that upregulate T-bet and downregulate RORγt. NKp44-negative, c-kit-low ILCs expressing T-bet and IFN-γ and responding to IL-12, but not IL-23, with enhanced IFN-γ production are present in the intestinal lamina propria of patients with Crohn's disease (CD) but not in the fetal gut and in the noninflamed intestine of adults [16]. These cells are not seen in the intestinal lamina propria of alymphoid mice reconstituted with a human immune system under homeostatic conditions, but they appear following induction of colitis by dextran sodium sulphate (DSS) [16]. IFN-γ-producing, T-bet-positive, NKp44-negative, c-kit-low ILCs maintain, however, low levels of RORγt raising the possibility that they differentiate from RORγt-expressing ILCs3 in inflamed tissues. Fuchs and colleagues described an ILCs1-like subset, characterized by the expression of NKp44, NKp46, CD56, CD103, granzyme, and perforin and located in the intestinal epithelial compartment (intraepithelial ILC1-like cells). These cells respond to IL-12 and IL-15, but not IL-18, with enhanced secretion of IFN-γ [17]. Despite sharing the NKp44 marker with human ILCs3, intraepithelial ILC1 subset clearly differs from ILCs3 in terms of phenotype, function, and transcription factors involved in development. In particular, studies in mice demonstrated the requirement of NFIL3 and Tbx21 (the gene encoding T-bet) for their development, while RORc (the gene encoding RORγt) and aryl hydrocarbon receptor (AhR) were dispensable [17]. However, these cells differ also in various aspects from ILCs1, as they did not express IL-7Rα and are independent of IL-15 for development and/or maintenance. Intraepithelial ILCs1 are increased in inflamed gut of CD patients and produce high amounts of IFN-γ in Rag1−/− mice treated with anti-CD40, a model of colitis characterized by wasting syndrome and severe intestinal inflammation [17]. Depletion of such cells ameliorates colon inflammation, supporting their role in orchestrating detrimental responses in the gut [17].
Recently, a new common progenitor to all IL-7R-expressing ILC lineages, expressing Id2, has been identified and named CHILP. CHILP gives rise to peculiar RORγt-independent, T-bet-dependent, Eomes−, NKp46+, NK1.1+, IL-7Rα + ILCs1. This cell type is present in the lamina propria of the small intestine and produces IFN-γ and TNF in response to IL-12 and to intestinal infection with the intracellular parasite Toxoplasma gondii [18]. Moreover, these cells express high levels of CXCR3, CXCR6, and CCR9, all of which are involved in lymphocyte migration to tissues. Studies in mice lacking specific genes revealed also that maintenance or differentiation of these cells requires T-bet, NFIL3, and Gata-3 as well as IL-15, but not IL-7 [18]. Another lymphoid precursor has been described in mouse fetal liver and adult bone marrow. It expresses high amounts of PLZF, a transcription factor previously associated with NK-T cell development, and has the potential to differentiate in ILC1, ILC2, and ILC3 [19].
3. Role of ILCs2 in the Control of Helminth Infections
Different types of Th2 cytokine-producing innate cells (e.g., natural helper cells, nuocytes, and type 2 innate helper cells), which express markers commonly found on ILC subsets (IL-7Rα, c-kit, CD25, and CD90), have been described [5, 9, 10]. These cells, now collectively referred to as ILCs2, act downstream to IL-25 and IL-33 and make a substantial contribution to antihelminth immunity through their ability to produce IL-13, a cytokine that drives many of the physiological responses required for worm expulsion, such as mucus production and smooth muscle contractility [3, 4, 9, 10]. In this context it was shown that adoptive transfer of nuocytes into Nippostrongylus brasiliensis-infected IL17βR/IL1R1 mice (which are severely impaired in their ability to expel worms) enables these animals to efficiently eliminate the parasite and IL-13 secretion from nuocytes is essential for worm expulsion [9]. The ability of ILCs2 to combat parasites is dependent on T cells, because nuocytes fail to undergo sustained expansion in helminth-infected Rag2-deficient mice [9]. Similarly, adoptive transfer of natural helper cells into γ-chain-Rag2-deficient mice infected with Nippostrongylus brasiliensis resulted in the restoration of goblet-cell hyperplasia in the recipient mice [5]. Along the same line are the results obtained with adoptive transfer of type 2 innate helper cells into Nippostrongylus brasiliensis-infected γ-chain-Rag2-deficient mice, in which worm expulsion requires IL-25 administration [20].
ILCs2 also produce amphiregulin, an epithelial growth factor family member that is crucial for tissue repair following virus-induced inflammation [21] as well as enhancing the immunosuppressive properties of regulatory T cells during colitis [22]. ILCs2 express elevated levels of Gata-3, which is crucial for their development and maintenance [23]. Development of ILCs2 is also dependent on RORα as mutant mice lacking this transcription factor do not have ILCs2 but display normal development of ILCs3 [24].
4. Regulation of ILCs3 Development and Activation
The factors/mechanisms that regulate differentiation/activation of ILCs3 are not fully understood, even though there is evidence that these cells can be activated by cytokines released by the intestinal epithelium and antigen presenting cells. While IL-7, stem cell factor, and TSLP are necessary for the development of ILCs3 and, together with IL-1, IL-2, and IL-15, regulate cell proliferation, IL-23 and IL-1β play an important role in inducing ILCs3 effector functions [25–30]. IL-23, a heterodimeric cytokine produced mainly by dendritic cells and macrophages, stimulates production of IL-22 by ILCs3 during intestinal infections [31, 32], but it is not essential for ILCs3 functions at steady state. Indeed, IL-23p19-deficient mice exhibit normal production of IL-22 by ILCs3 [33]. IL-23 induces also IL-17 production in CD56-negative ILCs3 isolated from the gut of patients with CD [34]. Another cytokine involved in the ILCs3 functions is IL-1β. IL-1β induces the accumulation and activation of ILCs3 during the course of Helicobacter (H.) hepaticus infection and synergizes with IL-23 or IL-7 in enhancing ILCs3-derived IL-22 production [35, 36]. Like CD4+Th17 cells, ILCs3 display a certain degree of context-dependent plasticity, as they are capable of acquiring functional characteristics of ILCs1. A subset of intestinal ILCs3, which is negative for CCR6 and either positive or negative for NKp46, coexpresses T-bet and RORγt [37, 38]. T-bet in ILCs3 controls expression of various target genes such as those encoding IFN-γ, Fas ligand, IL-12Rβ1, and CXCR3 [37, 38]. Interestingly, IL-22 production is evident in both T-bet+ and T-bet− ILCs3, albeit at different levels, and T-bet is highly expressed in NKp46+ IL-22-producing ILCs3 in the intestine. Accordingly, mice deficient in Tbx21 have a reduced number of NKp46+ IL-22-producing ILCs3 in the intestinal lamina propria and failed to produce IFN-γ [37].
Collectively the available data indicate that CCR6-negative ILCs3 progress from a T-bet-negative to a T-bet-positive state and then acquire NKp46 expression. This later phenomenon can be followed by downregulation of RORγt [38, 39]. IL-7 is crucial not only for the development of ILCs but also for stabilizing in vivo the expression of RORγt in ILCs3, thus preventing their full conversion into IFN-γ-producing NKp46, T-bet-expressing ILCs [39].
The signals that induce T-bet expression in CCR6-negative ILCs3 are not completely understood. IL-12, the major Th1 inducing factor, seems to be uninvolved as mice deficient for IL-12 signaling have normal numbers of T-bet-expressing ILCs3 [38]. In contrast, IL-23-deficient mice have reduced numbers of T-bet-expressing ILCs3 [38], suggesting a role for IL-23 in inducing or maintaining T-bet expression in ILCs3. ILCs3 can also recognize and directly respond to environmental cues. For example, both mouse and human ILCs3 express AhR, a ligand inducible transcription factor that mediates a wide range of cellular events in response to halogenated aromatic hydrocarbons and nonhalogenated polycyclic aromatic hydrocarbons, small synthetic compounds, and metabolites of tryptophan and arachidonic acid. In its inactive state, AhR resides in the cytosol bound to several cochaperones. Following ligand binding, AhR dissociates from the chaperones and translocates to the nucleus, where it binds to its dimerization partner aryl hydrocarbon receptor nuclear translocator, and this complex activates the expression of a battery of genes with promoters containing a dioxin responsive element consensus sequence or a xenobiotic responsive element consensus sequence [40]. The number of postnatal ILCs3 as well as IL-22 but not IL-17 expression by ILCs3 in small intestine and colon is reduced in AhR-deficient mice [41]. The decrease in ILCs3 observed in AhR-deficient mice is not evident until the third week, suggesting that environmental stimuli may contribute to the differentiation, survival, and postnatal expansion of these cell subsets [41]. The pathways involved in the AhR-dependent ILCs3 development and function in the gut remain to be clarified, even though AhR ligands derived from food components could be involved. Indeed, it was shown that mice fed with phytochemical-free diets have a phenotype similar to AhR-deficient mice [42]. Another possibility is that AhR-dependent modulation of ILCs3 function is mediated by bacterial metabolites, as, under conditions of unrestricted tryptophan availability, Lactobacilli species produce indole-3-aldehyde, an AhR ligand, which enhances IL-22 expression in ILCs3 [43]. Interestingly, human ILCs3 express also RNA transcripts of Toll-like receptors (TLR) 1, 2, 5, 6, 7, and 9, though it seems that only TLR2 agonists induce cytokine production by human ILC3 in the presence of IL-2, IL-15, and IL-23 [44], supporting the hypothesis that bacteria can directly stimulate ILCs3 to synthesize effector cytokines.
Both human and mouse ILCs3 express NK cell activating receptors (e.g., NKG2D, DNAX accessory molecule-1, 2B4, CD94/NKG2C, NKp46, NKp44, and NKp30) that are known to mediate NK cell cytotoxicity and production of cytokines upon recognition of cognate cellular and viral ligands. In particular, NKp44 is detectable on ILCs3 and selectively marks the IL-22-producing subset in human tonsil and gut lamina propria [32, 36]. Engagement of NKp44 in ex vivo isolated ILCs3 selectively induces the expression of TNF and IL-2 while stimulation with IL-23, IL-1, and IL-7 preferentially induces IL-22 and GM-CSF expression [36]. Therefore, ILCs3, whose development is Notch dependent [45], can switch between IL-22 and TNF production, depending on the triggering stimulus.
5. The Role of ILCs3 in the Control of Intestinal Epithelial Barrier, Infections, and Inflammation
ILCs3 are involved in the development of intestinal lymphoid organs such as cryptopatches, which are located in the lamina propria between the gut crypts, and isolated lymphoid follicles, which represent important sites of T-cell-independent IgA production [3, 4]. For a detailed description of the regulatory functions of ILCs3 in the development of intestinal lymphoid organs, the reader is directed towards recent reviews [3, 4].
In healthy mammals, commensal bacteria are anatomically restricted either to the intestinal lumen, to the epithelial surface, or within the underlying gut-associated lymphoid tissues, a process that is essential to limit inflammation and maintain normal systemic immune homeostasis. ILCs3 have a critical role in this phenomenon, as their depletion in mice results in peripheral dissemination of commensal bacteria such as Alcaligenes species, residing within Peyer's patches and mesenteric lymph nodes of healthy humans and mice, and systemic inflammation [46]. RORγt-expressing ILCs3 express major histocompatibility complex class II (MHCII) and can process and present antigen to CD4+ T cells. Among ILCs3, MHCII is highly expressed on cells that lack T-bet and NKp46, while minimal expression occurs in cells positive for those markers. Moreover, MHCII is seen on ILCs2 but not ILCs1 [46]. Interestingly, RORγt-expressing, MHCII-positive ILCs3 lack expression of classical costimulatory molecules, such as CD40, CD80, and CD86, and therefore antigen presentation by these cells limits, rather than promoting, CD4+ T cell responses, through a mechanism that is independent of the ability of ILCs to produce IL-22 or IL-17A [33]. Mice lacking MHCII selectively on ILCs3 exhibit increased frequencies of proliferating CD4+ T cells in the blood, significant increase in commensal bacteria-specific serum IgG, and development of colitis characterized by enhanced production of IFN-γ, IL-17A, and TNF by mucosal CD4+ T cells [46]. All these phenomena can be prevented by administration of antibiotics, demonstrating a crucial role for commensal bacteria in the development of the disease [33, 46]. The ILC-mediated containment of commensal bacteria and regulation of mucosal homeostasis could also rely on the ability of ILCs3 to produce IL-22 in response to AhR-activating stimuli [41]. Indeed, binding of IL-22 to its heterodimeric receptor, comprising IL-10R2 and IL-22R1, on epithelial cells triggers the transcription factor STAT3, thereby promoting synthesis of antimicrobial peptides and proteins (i.e., β-defensins, RegIIIβ and RegIIIγ, calgranulins S100A8 and S100A9, and lipocalin-2) and elevated levels of mucus-associated molecules (i.e., Muc1, Muc3, Muc10, and Muc13), with the downstream effect of limiting the translocation of commensal bacteria across the epithelial barrier during inflammation [47]. Although microbiota can modulate production of IL-22 by ILCs3 [33], the development of such cells seems to be independent of commensal bacteria, as the frequencies of IL-22-producing ILCs3 are similar in conventional versus germ-free mice [46].
ILCs3 also play an important role in the defense against pathogen infections, such as Citrobacter rodentium, a murine pathogen that models human enterohemorrhagic and enteropathogenic Escherichia coli infections [31, 32]. In particular, it was shown that mice lacking T and NK cells but retaining NKp46-expressing ILCs3 developed an IL-23-driven IL-22-mediated response and were resistant to infection [48]. ILCs3 also provide an early source of IL-22 during Candida albicans fungal infection [49]. IFN-γ produced by T-bet-dependent CCR6-ILCs3 has been shown to contribute to the response against Salmonella typhimurium infection in mouse [38].
Due to their ability to modulate epithelial cell functions as well as respond against commensal bacteria and pathogens, it is tempting to speculate that ILCs3 can participate in the complex regulation of IBD-related mucosal response, given that there is evidence that IL-22 is protective in murine models of IBD [27, 50]. On the other hand, as above specified, IL-17A and IFN-γ from NKp46-negative ILCs3 contribute to sustain inflammation in innate IBD models, such as anti-CD40 or H. hepaticus-induced colitis [25, 51]. These later findings are supported by the demonstration that IL-17A and IFN-γ-producing CD127+CD56−ILCs3 accumulate in inflamed gut of patients with CD [52]. However, another study documented a reduced frequency of NKp44+NKp46−ILCs type-3-like cells in CD [34]. Additional support to the hypothesis that ILCs3 can be proinflammatory in the gut comes from research into Tbx21/Rag2 double-knockout mice that develop spontaneous intestinal inflammation resembling UC [53]. In these mutants, ILCs3 produce high levels of IL-17A, a finding not observed in control mice, and depletion of all ILCs or neutralization of IL-17A improves colitis [54]. In addition, alymphoid mice on a Tbx21-deficient background do not develop colitis, indicating that lymphoid cells are required for inflammation [54].
A more complex scenario emerged however from studies in chronic CD45RB (high) CD4+ T cell transfer and anti-CD40 antibody-induced acute innate colitis models in Rag1-deficient mice showing that IL-23R signaling in ILCs3 is protective in the former and pathogenic in the latter [51]. Furthermore, it was shown that IL-23R signaling promotes innate colitis via IL-22 as neutralization of IL-22 protects mice from colitis and the adding back of IL-22 to IL-23R-deficient animals restores the disease [51].
6. Conclusions
In recent years, it became evident that ILCs play a fundamental role in immune responses, not only as first barrier against pathogens but also for their ability to influence downstream adaptive immune steps. These advances have been facilitated by the better characterization of the factors involved in the differentiation and maintenance of such cells as well as identification of the ILC subsets involved in specific immune responses. Environmental cues can promote the activation of ILCs as well as shifting from a subset to another one. In this context, it has been demonstrated that commensal microbiota-driven induction of IL-7 is determinant in the maintenance of ILCs3 and activation of AhR, an intracellular receptor for various environmental molecules, regulates the function of these cells in the gut. The intestine also contains additional stimuli, such as TLR ligands and cytokines, that could contribute to ILC development and activation. Much work remains however to be performed in order to ascertain the exact contribution that each ILC subset plays in the maintenance of immune homeostasis in the gut and how this task is accomplished. Further experimentation will be also needed to investigate whether and which ILCs are involved in the pathogenesis of chronic inflammatory diseases of the gut, such as CD, UC, and celiac disease, since cytokines produced by these cells are known to regulate the tissue-damaging immune responses in these disorders. At the same time, it would be relevant to investigate whether ILCs play a role in the process of colitis-associated colon cancer by either providing an immunosuppressive environment that eventually promotes tumor growth or amplifying cytotoxic pathways that kill tumor cells. In this context, it is noteworthy that IL-22 produced by ILCs facilitates the growth of cancer cells through a STAT3-dependent mechanism in a bacteria-driven mouse model of colorectal cancer [55].
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Silvia Sedda, Irene Marafini, Michele M. Figliuzzi, Francesco Pallone, and Giovanni Monteleone solely contributed to this paper. All authors read and approved the final paper.
References
- 1.Boltjes A, van Wijk F. Human dendritic cell functional specialization in steady-state and inflammation. Frontiers in Immunology. 2014;5, article 131 doi: 10.3389/fimmu.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magombedze G. Cellular and population plasticity of helper CD4+ T cell responses. Frontiers in Physiology. 2013;4(206):1–9. doi: 10.3389/fphys.2013.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annual Review of Immunology. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 4.Spits H, Artis D, Colonna M, et al. Innate lymphoid cells: a proposal for uniform nomenclature. Nature Reviews Immunology. 2013;13(2):145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 5.Moro K, Yamada T, Tanabe M, et al. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+ Sca-1+ lymphoid cells. Nature. 2010;463(7280):540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- 6.Yokota Y, Mansouri A, Mori S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature. 1999;397(6721):702–706. doi: 10.1038/17812. [DOI] [PubMed] [Google Scholar]
- 7.Gordon SM, Chaix J, Rupp LJ, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36(1):55–67. doi: 10.1016/j.immuni.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boos MD, Yokota Y, Eberl G, Kee BL. Mature natural killer cell and lymphoid tissue—inducing cell development requires Id2-mediated suppression of E protein activity. Journal of Experimental Medicine. 2007;204(5):1119–1130. doi: 10.1084/jem.20061959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price AE, Liang H, Sullivan BM, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(25):11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang H, Reinhardt RL, Bando JK, Sullivan BM, Ho I, Locksley RM. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nature Immunology. 2012;13(1):58–66. doi: 10.1038/ni.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barlow JL, Bellosi A, Hardman CS, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. Journal of Allergy and Clinical Immunology. 2012;129(1):191–198. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 13.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage -CD25 +CD44 hi lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. Journal of Immunology. 2012;188(3):1503–1513. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HY, Chang Y, Subramanian S, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. Journal of Allergy and Clinical Immunology. 2012;129(1):216–227. doi: 10.1016/j.jaci.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magri G, Miyajima M, Bascones S, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nature Immunology. 2014;15(4):354–364. doi: 10.1038/ni.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernink JH, Peters CP, Munneke M, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nature Immunology. 2013;14(3):221–229. doi: 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- 17.Fuchs A, Vermi W, Lee JS, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity. 2013;38(4):769–781. doi: 10.1016/j.immuni.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klose CS, Flach M, Möhle L, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157(2):340–356. doi: 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 19.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508(7496):397–401. doi: 10.1038/nature13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saenz SA, Siracusa MC, Perrigoue JG, et al. IL25 elicits a multipotent progenitor cell population that promotes TH2 cytokine responses. Nature. 2010;464(7293):1362–1366. doi: 10.1038/nature08901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monticelli LA, Sonnenberg GF, Abt MC, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nature Immunology. 2011;12(11):1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaiss DMW, van Loosdregt J, Gorlani A, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. 2013;38(2):275–284. doi: 10.1016/j.immuni.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong SH, Walker JA, Jolin HE, et al. Transcription factor RORα is critical for nuocyte development. Nature Immunology. 2012;13(3):229–236. doi: 10.1038/ni.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoyler T, Klose CSN, Souabni A, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37(4):634–648. doi: 10.1016/j.immuni.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buonocore S, Ahern PP, Uhlig HH, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464(7293):1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang YJ, Kim HY, Albacker LA, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nature Immunology. 2011;12(7):631–638. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geremia A, Arancibia-Cárcamo CV, Fleming MPP, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. The Journal of Experimental Medicine. 2011;208(6):1127–1133. doi: 10.1084/jem.20101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. Journal of Experimental Medicine. 2010;207(6):1293–1305. doi: 10.1084/jem.20092054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taube C, Tertilt C, Gyülveszi G, et al. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0021799.e21799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature Medicine. 2008;14(3):282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 32.Cella M, Fuchs A, Vermi W, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457(7230):722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sawa S, Lochner M, Satoh-Takayama N, et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nature Immunology. 2011;12(4):320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 34.Takayama T, Kamada N, Chinen H, et al. Imbalance of NKp44+NKp46− and NKp44−NKp46+ natural killer cells in the intestinal mucosa of patients with Crohn's disease. Gastroenterology. 2010;139(3):882.e3–892.e3. doi: 10.1053/j.gastro.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 35.Coccia M, Harrison OJ, Schiering C, et al. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4+ Th17 cells. The Journal of Experimental Medicine. 2012;209(9):1595–1609. doi: 10.1084/jem.20111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glatzer T, Killig M, Meisig J, et al. RORγt+ innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity. 2013;38(6):1223–1235. doi: 10.1016/j.immuni.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 37.Sciumé G, Hirahara K, Takahashi H, et al. Distinct requirements for T-bet in gut innate lymphoid cells. The Journal of Experimental Medicine. 2012;209(13):2331–2338. doi: 10.1084/jem.20122097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klose CSN, Kiss EA, Schwierzeck V, et al. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature. 2013;494(7436):261–265. doi: 10.1038/nature11813. [DOI] [PubMed] [Google Scholar]
- 39.Vonarbourg CV, Mortha A, Bui VL, et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt+ innate lymphocytes. Immunity. 2010;33(5):736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu J, Zhou L. Aryl hydrocarbon receptor promotes RORγt+ group 3 ILCs and controls intestinal immunity and inflammation. Seminars in Immunopathology. 2013;35(6):657–670. doi: 10.1007/s00281-013-0393-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu J, Guo X, Chen Z, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39(2):386–399. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiss EA, Vonarbourg C, Kopfmann S, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334(6062):1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- 43.Zelante T, Iannitti R, Cunha C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39(2):372–385. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, di Santo JP, Spits H. Regulation of cytokine secretion in human CD127+ LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity. 2010;33(5):752–764. doi: 10.1016/j.immuni.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 45.Mielke LA, Groom JR, Rankin LC, et al. TCF-1 controls ILC2 and NKp46+RORγt+ innate lymphocyte differentiation and protection in intestinal inflammation. Journal of Immunology. 2013;191(8):4383–4391. doi: 10.4049/jimmunol.1301228. [DOI] [PubMed] [Google Scholar]
- 46.Sonnenberg GF, Monticelli LA, Alenghat T, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336(6086):1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Killig M, Glatzer T, Romagnani C. Recognition strategies of group 3 innate lymphoid cells. Frontiers in Immunology. 2014;5:p. 142. doi: 10.3389/fimmu.2014.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Satoh-Takayama N, Vosshenrich CAJ, Lesjean-Pottier S, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29(6):958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 49.de Luca A, Zelante T, D’Angelo C, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunology. 2010;3(4):361–373. doi: 10.1038/mi.2010.22. [DOI] [PubMed] [Google Scholar]
- 50.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29(6):947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eken A, Singh AK, Treuting PM, Oukka M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunology. 2014;7:143–154. doi: 10.1038/mi.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuchs A, Colonna M. Innate lymphoid cells in homeostasis, infection, chronic inflammation and tumors of the gastrointestinal tract. Current Opinion in Gastroenterology. 2013;29(6):581–587. doi: 10.1097/MOG.0b013e328365d339. [DOI] [PubMed] [Google Scholar]
- 53.Garrett WS, Lord GM, Punit S, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131(1):33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Powell N, Walker AW, Stolarczyk E, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity. 2012;37(4):674–684. doi: 10.1016/j.immuni.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirchberger S, Royston DJ, Boulard O, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. Journal of Experimental Medicine. 2013;210(5):917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]