

Abstract
The carbon nanopipette (CNP) is comprised of a pulled-glass pipette terminating with a nanoscale (tens to hundreds of nm) diameter carbon pipe. The entire inner glass surface of the CNP is coated with a carbon film, providing an electrically conductive path from the carbon tip to the distal, macroscopic end of the pipette. The CNP can double as a nanoelectrode, enabling electrical measurements through its carbon lining, and as a nanoinjector, facilitating reagent injection through its hollow bore. With the aid of a lock-in amplifier, we measured, in real time and with millisecond resolution, variations in impedance as the CNP penetrated into the cytoplasm and nucleus of adherent human osteosarcoma (U20S) cells. The capacitance change associated with nucleus penetration was, on average, 1.5 times greater than the one associated with cell membrane penetration. The experimental data was compared and favorably agreed with theoretical predictions based on a simple electrical network model. As a proof of concept, the cytoplasm and nucleus were transfected with fluorescent tRNA, enabling real-time monitoring of tRNA trafficking across the nuclear membrane. The CNP provides a robust and reliable means to detect cell and nucleus penetration, and trigger injection, thereby enabling the automation of cell injection.
Keywords: carbon nanopipette, microinjection, impedance, cell, automated
Introduction
In drug discovery, vaccine development, cellular therapeutics development, basic biology, and combinatorial biochemistry, there is a need to controllably inject reagents into a large number of cells to assure statistically significant data about cellular responses. Methods like electroporation[1,2] and photoporation[3,4] are often used for bulk introduction of reagents into cells; however, these methods are difficult to optimize, lack single-cell resolution, and cannot assure that all the cells in the population are treated uniformly and that the intended composition of the reagents is preserved as they diffuse / migrate into the cells. This is significant since in many cases, one needs to control the composition of the mixture that is injected into a cell. For instance, the use of fluorescent tRNA to monitor translation (FtTM) requires high throughput, controlled injection. This recently developed technique[5] enables the identification and monitoring of active ribosome sites within live cells with submicron resolution, facilitating (i) quantitative comparison of protein synthesis among various cell types, (ii) monitoring the effects of antibiotics and stress agents on protein synthesis, and (iii) characterization of changes in spatial compartmentalization of protein synthesis upon viral infection. Despite the immense potential of FtTM for measuring translation dynamics and synthesis patterns in real time in normal and diseased cells under various physiological, pathological, and environmental conditions, its widespread adoption has been curtailed by the difficulty in introducing predetermined quantities of fl-tRNA or mRNA into large numbers of cells in an efficient and reproducible manner.
Microinjection remains the most robust method for controllably introducing precise compositions of reagents into cells. The most prohibitive obstacles to microinjection are the relatively low throughput (several hundred cells/hour for most experienced operators), the tedious manual manipulation, and the potential damage to cells. Microinjection success rates are thus highly dependent on operator skill, and it is difficult to attain statistically significant populations of injected cells.[6,7] The lack of reliable, high throughput, controllable injection techniques is the bottleneck in many significant projects.[6]
There have been many attempts to automate the cell injection process[6–20] through positioning of cells at predetermined locations in an array,[11] computer vision,[10,12,13] novel microfluidic chips,[16,20] and feedback systems.[8,9,17,18,19] While these systems have made significant advancements in microinjection rates and efficiency, they are still limited by lack of a robust feedback signal to indicate that the injector has, indeed, penetrated the cell membrane. Penetration-force measurement has been successfully used to detect large cell penetration,[8,9] but is unlikely to provide the necessary sensitivity for the smaller mammalian cells. Instead, researchers have attempted to use electrical signals.
Electrical measurements have been used with patch electrodes (micropipettes filled with a high concentration salt solution in contact with a non-polarizable electrode, often Ag/AgCl/Cl−[21–23]) to detect cellular contact and penetration in both manual[21] and automated[24] patch-clamping, and for automated single-cell electroporation.[25,26] Lukkari and co-workers[17–19] extended this technique to microinjection by placing an electrode in the injection solution. The solution in the micropipette was continuously subjected to a 10 Hz square wave, and the electric current was monitored. An impedance change was detected upon cell contact and penetration as well as upon pipette breaking/clogging. A similar technique used a DC ionic current measurement to detect cell penetration during electrokinetic injection of cells.[27] The use of the liquid inside the micropipette as the electrical conductor imposes, however, limitations on the type (typically, high salt concentration) and volume of liquids that can be used in the injection process, adversely affects cells’ viability, and limits the time resolution. Hence, it is desirable to decouple the electrical measurement indicating cell penetration from the injection liquid.
Mirkin et al.[28] detected cell penetration with solid platinum microelectrodes by introducing a redox mediator in the extracellular solution, similar to techniques used in scanning electrochemical microscopy (SECM).[23,29] The use of a redox mediator may, however, adversely impact cell viability and function,[30] and the solid microelectrodes are not suitable for introducing fluids into cells.
Recently, we have developed carbon nanopipettes (CNPs)[31–36] that consist of a pulled-quartz capillary, terminating with a nanoscale carbon pipe (Figure 1). The diameter of the carbon pipe can be adjusted from tens to hundreds of nanometers. The fabrication process[31,36] consists of pulling a quartz capillary to desired dimensions and placing it in a tube furnace in the presence of carbon precursor gases at elevated temperatures. The hydrocarbon decomposes, coating the capillary’s inner surface with a carbon film. The tip of the capillary can be chemically etched to expose the carbon. The diameter of the carbon pipe is controlled by the pulled glass template. The carbon film’s thickness is controlled by the process’ temperature and duration. The length of the exposed carbon tip is controlled by the etching time. The result of this fabrication process is a nanoscopic, hollow carbon “tube” incorporated and insulated within a quartz handle. The fabrication process does not require any assembly, overcoming one of the major challenges of nanotechnology - the interfacing between a nanostructure and a macroscopic handle. CNPs are compatible with micromanipulators, micropipette fittings, and amplifiers. Since the CNPs can be hollow, they can be used as injectors. Since the carbon tips are electrically accessible, the CNPs can also double as nano-electrodes. Figure 1 features both schematics and SEM micrographs of CNPs.
Figure 1.
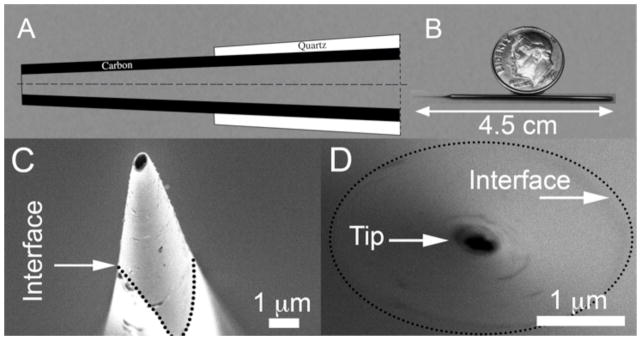
Carbon Nanopipettes (CNPs). (A) A schematic depiction of the CNP’s cross-section. (B) A photograph of a CNP fabricated from a 7.5cm-long (before pulling) capillary. (C) A SEM side micrograph of a CNP’s tip (500nm diameter, inclined at 10° off-axis). (D) A SEM axial front micrograph of a 200nm diameter CNP tip. The quartz-carbon interfaces in C and D are delineated with dotted lines.
Schrlau et al.[32–35] demonstrated that CNPs can be used for microinjection of dyes and secondary messengers into cells without affecting cell viability and function. CNPs are less intrusive and more durable than their glass counterparts. Schrlau et al.[35] also used the CNPs to measure cell membrane polarization induced by extracellular pharmacological agents. Carbon microelectrodes are not ideal, however, for potentiometry due to their large interfacial impedance, resulting in measurement instability, and requiring a high-impedance amplifier headstage.[35]
Here, we use an AC, electrical impedance measurement to detect cell and nucleus penetration with carbon nanopipettes. Using an AC signal allows us to transmit relatively large non-Faradaic (charging) currents that can be easily measured and processed with Fourier methods to yield accurate data. Much of our data is presented in terms of the equivalent capacitance, which is highly sensitive to the electrode’s local environment and not dependent on Faradaic reactions.[37] Our system has a number of advantages over previously proposed methods that utilized the injection liquid in the pipette’s bore as the conductive path to detect cell penetration. In the CNPs, the electrical signal is not dependent on the injection liquid, the injection liquid need not be conductive, and one can operate with small volumes of injection liquid (as there is no need to bring the injection liquid into contact with an electrode). Additionally, the small size of the CNP and the AC method provide much greater temporal resolution than is possible with ionic electrodes.
The manuscript is organized as follows. We first describe the experimental techniques used. Next, we introduce an analog capacitance-based network model to predict the CNPs’ response. The model assists us to interpret our experimental observations. We then report on impedance measurements of a liquid-filled CNP as a function of the CNP’s tip position and the composition of the injection solution. We examine CNPs operating in both continuous and pulse injection modes. To exemplify the relevance of the CNPs to biological research, we then describe very briefly the injection of tRNA into the cytoplasm and the nucleus and the monitoring of tRNA trafficking between the cytoplasm and nucleus.
Experimental Section
Cell Culture and Imaging
The experiments were carried out with adherent human osteosarcoma cells (U2OS, ~40 μm diameter). U2OS cells were selected for their availability and ease of culture. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (HyClone) with 10% Fetal Bovine Serum (HyClone) and 1% Penicillin/Streptomycin antibiotics (HyClone) in a standard (Fisher Isotemp) CO2 incubator (37 C, 5% CO2). The cells were then plated on Poly-L-Lysine treated glass coverslips (1 mg/mL solution) or directly grown in 35mm tissue culture dishes (Corning). The plated coverslips were transferred to petri dishes with cell culture medium for experiments.
The experiments were carried out at room temperature in standard atmosphere without any CO2 regulation. During the experiments, the cells were outside the incubator for at most two hours. The measured data did not change significantly over this time interval, and cells remained viable, as evidenced by continued proliferation several days after the experiments.
An Olympus IX-71 inverted optical microscope with long-working-distance, phase-contrast objectives, Hamamatsu CCD camera, and HCImage Software were used to image the cells and track the carbon nanopipettes during probing. The entire system was encased in a copper-mesh Faraday cage to reduce electromagnetic interference and was located on an air-damped, vibration-isolation table (TMC MICRO-g).
CNP Fabrication
CNPs were fabricated with 1mm outer diameter, 0.7 mm inner diameter, filamented quartz capillaries of 7.5 cm length (Sutter Instruments). Pipettes were pulled using a Sutter P-2000 laser-based pipette puller with the parameters: HEAT 800, FIL 4, VEL 60, DEL 128, and PULL 100. Chemical vapor deposition was performed on the pipettes in a Lindberg horizontal tube furnace, with a 1″ inner diameter quartz furnace tube at 875 C. The flow conditions were 200 sccm of methane and 300 sccm of argon (AirGas ultra-high purity) for a total deposition time of 40 minutes. Some pipettes were also fabricated in a Carbolite HVS 3-zone horizontal tube furnace with a 1.3″ inner diameter quartz tube at 905 C, with flow conditions of 400 sccm methane and 600 sccm argon, for a 3 hour duration which yielded equivalent carbon deposition. During deposition, the pipette tips were oriented against the flow of the gas, i.e., the tip pointed upstream.
The carbon deposited selectively only inside the pipette, not on the pipette’s outer surface. The carbon-coated pipettes were etched in 5:1 buffered hydrofluoric acid (Transene Buffer HF Improved) followed by a 10-minute rinse in deionized water. The pipettes were inspected under an optical microscope and imaged with a SEM (FEI Quanta 600 ESEM). The tip outer diameter ranged from 200 to 400 nm, the inner diameter ranged from 25 to 200 nm, and the exposed carbon tip length depended on the etch time, but typically was between 4 and 20 μm for etch times between 15–60s. The tip had a conical shape with a cone angle of about 2 degrees. The electrical resistance of the CNPs was on the order of tens of kΩ When in solution, the DC junction impedance was on the order of 5GΩ as measured by impedance spectroscopy. At 1 kHz in a typical cell medium, the interfacial impedance drops to 10–100 MΩ. The patch clamp amplifier headstage has a 0.5GΩ input impedance for the gain setting used.
Impedance Measurements
The CNP was connected to a HEKA EPC 10 patch clamp amplifier (HEKA Instruments Inc.) with a standard 1mm HEKA micropipette holder. A lead wire was connected at one end to the pin of the BNC connection (standard coaxial bayonet coupling). The other end of the wire was inserted into the distal end of the CNP. Slight bends in the wire endowed the wire with sufficient springiness to press it against the carbon film and form an electrical contact with the CNP’s inner carbon lining. Despite its simplicity, this electrical connection proved reliable. The pipette holder provided a hermetic seal around the CNP using a compression fitting with O-rings. Fluidic tubing with a bayonet coupler connected the hollow of the CNP to a pressure injection system (Eppendorf FemtoJet) via a simple tubing connection on the HEKA pipette holder.
The LockIn module of HEKA’s PATCHMASTER software was used to monitor the current response, complex impedance, and DC conductance. Typically, the capacitance and the real part of the impedance were monitored in real-time during probing. A voltage with amplitude of 10mV, 1 kHz frequency, and −70mV offset was used with a sampling rate of 20 kHz. The impedance and capacitance data were computed once per cycle, resulting in an effective time resolution of 1ms. We elected to operate at 1 kHz since filtering proved effective, long sweep durations (~120s) could be used, and the measurement noise was near its minimum as previously demonstrated by Chen and Gillis.[38] Examining the frequency response of the electrode impedance in a simple electrolyte solution produced a traditional Nyquist plot for a Randle’s circuit, and at 1 kHz the electrode was outside the Warburg regime.[23]
The voltage offset was selected to be approximately equal to a typical membrane potential to avoid significant polarization of the cell during probing.[37,39] The capacitance measurement was not sensitive to the offset voltage as long as the charge transfer resistance was much greater than the solution resistance, which holds true in a typical non-Faradaic system such as ours.[39] No capacitance or bridge compensation was used during the measurements. Bessel low-pass filters of 2.9 kHz and 10 kHz were used in series for the current measurement, and the resulting data (capacitance, conductance, and impedance) was further filtered in real-time using the digital filter on the PATCHMASTER oscilloscope of ~5Hz. The 1mV/pA gain setting was used. A silver/silver-chloride wire was inserted in the extracellular solution and used as a pseudo-reference and counter electrode.
Micromanipulation and Cell Experiments
The headstage of the amplifier was mounted on a piezoelectric micromanipulator (Eppendorf Transferman NK2). The CNPs were back-filled with 5–10μL of solution using Eppendorf Microloader tips and secured to the headstage using the 1.0mm pipette holder. For continuous flow injection, the control pressure was set to 10–40 hPa. For pulsed injection the control pressure was typically set to 5–10hPa. This control pressure and injection pressure were calibrated by injecting a few test cells. Pipettes were manually lowered into cells until there was a change in the impedance. The cell was either injected via the control flow, or with a burst of pressure (50–200 hPa for 0.3s) to force fluid into the cells. Cell injection was confirmed visually by cell swelling. Impedance traces and computed capacitance traces were acquired and monitored in real time. The impedance change was measured immediately after cell and nucleus penetration.
tRNA Experiments
5 nmol of Cy-5 labelled tRNA were resuspended in molecular grade, nuclease-free water to a final volume of 50μl and a concentration of 100μM. The Cy5-tRNA was back-loaded into a filamented CNP. Continuous-flow microinjection was utilized to inject the U2OS cells. A Cy5 filter was used with the Olympus IX71 scope to monitor the fluorescence as a function of time post-injection. Although the fluorescent photobleached, we had sufficient signal to confirm tRNA injection and to qualitatively monitor subcellular tRNA trafficking.
Safety Considerations
Hydrofluoric (HF) acid is highly toxic and corrosive, causing deep tissue damage, delayed burns, and systemic toxicity upon skin exposure. It should be handled with extreme caution and used with appropriate safety measures in place, including, but not limited to a fume hood, personal protective equipment, eye wash, emergency shower, and HF-exposure kit. As it dissolves glass and ceramic, any containers used with HF acid should be made of a compatible material such as polyethylene.
The chemical vapor deposition furnace is operated at high temperatures, and samples should not be loaded/unloaded until the furnace is appropriately cooled, and physical contact with the furnace should be avoided when in operation.
Results and Discussion
A Simplified Circuit Model
The basic premise of our detection scheme is that, as the CNP tip crosses the cell membrane, there is a change in the impedance due to the electrode surface contacting the intracellular solution, which differs in its characteristics from the extracellular solution, and due to the added impedance of the cell membrane. Figure 2A depicts schematically an analog electrical circuit of the CNP-cell system.[23,37,39] The symbols R and C denote, respectively, resistance and capacitance. The significance of the various subscripts and superscripts is delineated in the figure’s caption. Briefly, the electrode-liquid interface is modeled as a Stern capacitor (CS) in series with a Debye (diffuse) layer capacitor (Cd). These capacitors are connected in parallel with a Faradaic charge transfer resistor (Rt). Since, in the absence of redox species, Rt is large, we can treat the electrode as blocking (perfectly polarizable). Likewise, the membranes can be approximated as capacitors. To the first order of approximation, we can approximate the circuit model with capacitors alone, as shown in Figure 2B and 2C.
Figure 2.

A schematic depiction of a CNP penetrating an adherent cell with the equivalent circuit model overlaid. HEKA EPC 10 patch clamp amplifier shown. C and R denote, respectively, capacitors and resistors. Subscripts o, i, and j designate, respectively, extracellular, intracellular, and inner-pipette circuit components. Superscripts designate the following: S - Stern layer (capacitance), s- series (resistance), d – diffuse layer, n – nuclear membrane, m – cellular membrane, t – charge transfer. When modeling cytoplasm probing, the nuclear circuit elements (Cin and Rin) are omitted. (A) Complete circuit model. (B) Extracellular circuit approximation, only capacitors are included. (C) Intracellular (cytoplasm) circuit approximation, only capacitors are included.
When the CNP resides in the extracellular solution (Figure 2B), its equivalent capacitance is:
| (1) |
where is the combined capacitance per unit area of the Stern and Debye layers at the interface between the CNP tip’s carbon surface and the extracellular solution. Cj is the total capacitance of the surface inside the CNP’s bore. For later use, the CNP’s outer exposed surface is decomposed into area Ai and area Ao representing, respectively, the part of the CNP tip that will penetrate into the cell and the part that will remain in the extracellular solution post penetration. When the CNP is outside the cell, the entire area, Ai + Ao, is exposed to the extracellular solution.
When area Ai of the CNP tip is inside the cell, the equivalent capacitance of the CNP is:
| (2) |
where is the capacitance per unit area of the carbon surface – intracellular solution interface. Cm =Cm″ Am is the cell membrane capacitance, Cm″ is the membrane capacitance per unit area, and Am is the membrane area.
The change in capacitance upon cell penetration is thus:
| (3) |
The first term in Equation (3) represents the contribution of the CNP’s inner bore to the capacitance change. The second term is proportional to the area of the CNP tip that is submerged inside the cell. The presence of an ionic solution in the CNP’s bore greatly increases the magnitude of ΔCeq compared to the case of the empty CNP.
When a liquid-filled CNP is just slightly immersed in the cell (Ai is small) and liquid is present in the CNP’s bore, Equation (3) reduces to
| (4) |
The capacitance change upon cell penetration is dominated by the capacitance of the internal CNP’s bore and is negative.
In the absence of liquid in the CNP’s bore, Cj is negligible. The change in capacitance upon cell penetration is given by the second term in Equation (3)
| (5) |
and the change in capacitance is proportional to the area of the CNP tip submerged inside the cell. Depending on the relative magnitudes of Co″ and Ci″, the expression in Equation (5) can be either positive or negative. Under ideal conditions, when the intracellular and extracellular solutions are nearly isotonic , Equation (5) reduces to . The above equation suggests that by monitoring the capacitance of an empty CNP, in addition to detecting cell penetration, we should be able to estimate the penetration depth. While this manuscript focuses on microinjection, the ability to detect cell penetration depth may be useful to position functionalized nanoprobes at desired location within the cell. As we shall see shortly, the simplified capacitance-based theory can provide insights into many of our experimental observations presented below.
Detection of Cellular Penetration
Our primary objective of cell penetration detection is to enable microinjection automation. The bore of the CNP is filled with the solution to be injected. The pipette tip is lowered through the extracellular solution until it penetrates the cell membrane, injection takes place, and then the pipette is removed. In practice, a small backpressure is applied to the pipette as it approaches the cell to induce weak flow through the pipette’s tip. This flow serves both to prevent capillary uptake of extracellular/intracellular solution into the bore of the pipette and to minimize clogging.
The injection system operates in either continuous flow mode or pulse mode. In the continuous mode, the backpressure remains unaltered as the pipette penetrates the cell, and the infusion volume is controlled by the duration of the pipette’s penetration. In the pulse mode, upon penetration, the backpressure is increased greatly, and the infusion volume is dictated by the magnitude and duration of the pressure pulse. Below, we examine the effects of both modes of operation on the measured impedance. In all the experiments, we used filamented CNPs with ~300 nm diameter tips and ~16μm exposed tip length.
Continuous Flow Microinjection
While operating in the continuous injection mode, we lowered a liquid-filled CNP, subjected to 10–40 hPa pressure, through the extracellular solution until it penetrated into the cell. After a 1 s dwelling time, we withdrew the CNP from the cell and moved it to the next cell, repeating the process. The pressure and penetration duration were selected so as to produce a visible cell swelling, which was used to verify that infusion had, indeed, occurred. In our automated injection system, we will not rely on visual clues to determine the injected volume. Instead, we plan to use correlations that calibrate the injection volume as a function of pressure and dwelling time. The CNP’s impedance was continuously monitored throughout this process.
Figure 3A depicts the changes in the equivalent capacitance ΔCeq (lower curve) and the change in resistance, ΔRe{Z} (upper curve), upon penetrations into and withdrawals from different cells. The capacitance and resistance changes correlated with visible swelling of the cells due to injection. The background capacitance and resistance signals were very stable, with RMS noise of approximately 0.011pF. Upon cell penetration, there was a sharp drop in capacitance of 1–3 pF, consistent with Equation (4), and an increase in resistance of 200–300 kΩ. Upon removal of the CNP tip from the cell, the capacitance and the resistance returned to their extracellular, baseline values. The scatter in ΔCeq and ΔRe{Z} can be attributed to variations in the sizes and states of the cells, as well as the penetration location and depth of the pipette.
Figure 3.
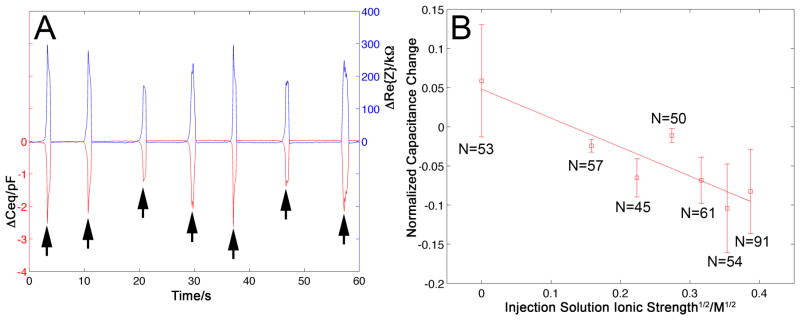
(A) The change in the CNP’s capacitance, ΔCeq (red trace, left axis), and the change in the CNP’s resistance, ΔRe{Z} (blue trace, right axis), upon penetration into and withdrawal from various cells when operating in continuous-flow microinjection mode (pressure 10–40 hPa, and 100mM KCl injection solution). (B) The normalized change in capacitance ΔCeq/C0 upon cell penetration as a function of the square root of the injection solution’s (KCl) ionic strength. C0 is the CNP capacitance when in the extracellular solution. The symbols represent the average of N measurements and the vertical bars represent one standard deviation. The solid line is a linear best-fit. R2=0.80. Each data point corresponds to a distinct CNP. All the data was acquired from the same cell culture on the same day, with the exception of the data point to the far right, which was obtained with two different CNPs and two different cell petri dishes.
Effect of Solution’s Ionic Strength on ΔCeq
Although normally it is preferred to inject the cell with a solution of similar osmotic strength to that of the cytoplasm to prevent stressing the cell, occasionally, it is necessary to use other solution concentrations. Thus, it is of interest to examine the effect of the injection solution’s ionic strength on ΔCeq. Equation (4) suggests that the capacitance change ΔCeq is proportional to the capacitance of the CNP’s carbon film – injection solution interface (Cj). Cj is composed, in part, of the electric double layer capacitance next to the inner carbon layer[23] (ε/λD), where ε and λD are, respectively, the solution permittivity and the Debye screening length. Since the Debye screening length is inversely proportional to the square root of the ionic strength, one would expect ΔCeq to decrease linearly with the square root of the injection solution’s ionic strength. Figure 3B depicts ΔCeq/C0 (C0 being the extracellular baseline capacitance) as a function of the square root of the injection solution’s ionic strength. Each data point represents the average of N measurements. The error bars correspond to one standard deviation. The far right data point includes results from the pulsed microinjection experiment, which was also carried out with a 150mM KCl solution. In the aggregate, the figure summarizes results of 411 events. The solid line is a linear best fit with R2=0.8. Consistent with expectations, the experimental data decreases nearly linearly as the square root of the ionic strength increases.
When the injection solution had very low ionic strength, the capacitance increased upon cell penetration. We hypothesize that the low concentration solution effusing out of the CNP mixed with the solution enveloping the tip, reducing its concentration and the magnitude of the electric double layer capacitance Co″ in the absence of effusion. Upon cell penetration, the extracellular interface re-equilibrated with the surrounding buffer, Co″ increased, while the effusion kept Ci″< Co″. Since under these circumstance Cj is also small, the net effect is that ΔCeq>0, consistent with experiments.
Pulsed Microinjection
In pulsed microinjection, the background pressure of the microinjection pipette is kept relatively low. The majority of the injection is achieved through a controlled burst of pressure after the CNP’s insertion into the cell. To test the penetration detection when operating in pulse mode and to examine the effect of the injection pulse on the measured impedance, we injected 150 mM KCl solution into U2OS cells (plated on tissue culture dishes with cell media) while concurrently recording Ceq and Re{Z}. This particular salt concentration was selected to approximately match the ionic strength of the cytoplasm.[40] A control pressure of 10 hPa was applied continuously to the CNP prior to and during cell penetration. Subsequent to cell penetration, we applied a pressure pulse of 125 hPa for 0.3 s to inject the KCl solution into the cell. We verified the injection by visually monitoring cell swelling. The amount of cell swelling in our experiments was consistent with that observed by others[6] during microinjection. The injected volume was small enough (<10% of the cell volume) as not to impair cell viability. Although we have not done so, the injection volume could be quantified by including fluorescent dye in the injected solution, monitoring the dye emission intensity, and using calibration table to correlate the emission intensity with the injected volume.[41–43]
Figure 4 illustrates pulse injections into the cytoplasm and the nucleus. In applications, the pipette would be withdrawn from the cell promptly after the injection. However, in the experiments of Figure 4, we left the CNPs’ tip inside the cell for ~80s to monitor signal stability. Figure 4A and 4C are, respectively, micrographs of cellular and nuclear penetration and the subsequent injection-induced swelling. The edges of the cell (Figure 4A) and nucleus (Figure 4C) are outlined with dotted lines for better visibility. As the cell swelled, various organelles were seen more clearly than prior to swelling due to the phase contrast filter on the microscope. For example, the nucleus becomes more distinct as the cell membrane is displaced by the fluid injected into the cytoplasm (Figure 4A).
Figure 4.

Concurrent penetration and microinjection detection. Control pressure: 10hPa. Injection pulse: 125hPa for 0.3s. (A) Micrographs of the cell: CNP in extracellular solution (i); CNP’s tip inside the cytoplasm (ii); injection of 150mM KCl into the cytoplasm (iii–iv); CNP tip withdrawn from the cell (v). (B) ΔCeq (lower red trace) and ΔRe{Z} (upper blue trace) as functions of time during cytoplasmic penetration and microinjection events. (C) Micrographs of the CNP tip position relative to the cell nucleus: CNP outside (above) the cell (i); CNP’s tip in the nucleus (ii); nucleus is injected with 150mM KCl solution (iii–iv); CNP withdrawn from the cell (v). (D) ΔCeq (lower red trace) and ΔRe{Z} (upper blue trace) as functions of time during the nuclear penetration and injection. The cell (A) and the nucleus (C) are outlined with dotted lines for better visibility.
Figure 4B and 4D depict the measured ΔCeq (lower red trace and LHS ordinate) and ΔRe{Z} (upper blue trace and RHS ordinate) as functions of time during penetration, injection, and post injection into the cytoplasm (Figure 4B) and the nucleus (Figure 4D). There was a slight upward drift in the extracellular capacitance signal prior to cell penetration. No such drift was observed in Re{Z}. We used the extracellular values after the CNP was withdrawn from the cell as the reference. Upon CNP penetration into the cytoplasm and the nucleus, the capacitances dropped, respectively, by −3.2pF and −4.5pF relative to the CNP’s capacitance when in the extracellular solution. Concurrently, Re{Z} increased, respectively, by 540kΩ and 750kΩ upon cytoplasm and nucleus penetration relative to the extracellular values. After the cell and nucleus penetration, we observed a gradual swelling of the cell and nucleus, which was most likely caused by slow ejection of liquid from the CNP, induced by the CNP’s control pressure. In our setup, during the nucleus penetration experiments, due to the close proximity of the nucleus to the cell membrane for adherent cells, it was not possible to detect the intermediary state after penetration of the cell membrane, but prior to penetration of the nuclear membrane.
The femtoinjector applied a pressure pulse to the CNP to inject solution into the cell and nucleus (two different cells) at time t~30s (Figure 4B) and time t~20s (Figure 4D), respectively. The injection caused a rapid swelling of the cell and nucleus. The injection into the cytoplasm resulted in a +0.6 pF pulse in ΔCeq and a −120kΩ pulse in ΔRe(Z). The injection into the nucleus resulted in a +0.9 pF pulse in ΔCeq and a −130kΩ pulse in ΔRe(Z). The pulses in ΔCeq and ΔRe(Z) likely may have been caused by alterations in the CNP tip’s local ionic environment, membrane swelling, and physiological changes in the cell triggered by the injection event. An interesting question, which we defer to future work, is whether the magnitude of the “injection pulse” correlates with the injection volume. ΔCeq and ΔRe{Z} assumed steady values post injection. These corresponded, respectively, to ΔCeq = −0.5pF and −0.7pF for cytoplasm and nucleus, relative to the extracellular values after CNP removal. The corresponding ΔRe(Z) were 98kΩ and 120kΩ. Steady state was reached faster for the nuclear injection, presumably due to the smaller volume of the nucleus. Upon removal of the CNP from the cell, both Ceq and Re{Z} resumed stable extracellular baseline values.
The steadiness of the impedance measurements when the CNP tips were embedded in the cytoplasm and the nucleus suggests that the CNP penetration, injection, and dwelling in the cell and nucleus did not significantly compromise the cell and nuclear membranes. This is consistent with our prior work,[32–35] which demonstrated that probing cells with CNPs and injecting secondary messengers into the cytoplasm did not harm cells. Furthermore, the CNPs can be used to record electrically significant events in the cell and nucleus for prolonged periods of time.
Cytoplasm vs. Nucleus Penetration
When measuring CNP impedance change upon cell and nucleus penetration we found a significant difference between the ΔCeq associated with cytoplasm and the ΔCeq associated with nuclear penetration (P<0.0001). When the liquid-filled CNP (150mM KCl) penetrated the cell (subscript ‘CP’) and the nuclear membrane (subscript ‘N’), ΔCeq,CP (± one standard deviation) = −5.8±2.7pF (N=28) and ΔCeq,N =−9.1±3.9pF (N=31). The corresponding changes in the real part of the impedance were ΔRe{Z}CP = 670±387kΩ and ΔRe{Z}N = 1020±591kΩ. The ratios ΔCeq,N /ΔCeq,CP ~ 1.56 and ΔRe{Z}N/ΔRe{Z}CP ~1.52.
The differences in ΔCeq,CP and ΔCeq,N are due to the nuclear membrane’s impedance. If one were to measure the CNP’s impedance as the CNP’s tip transverses from the cytoplasm into the nucleus, one should be able to identify nuclear penetration from the capacitance data. Variations in the biological state of various cells, penetration depth of the CNP, and tolerances of the CNPs make it difficult to determine whether the tip is in the cytoplasm or nucleus based on a single impedance measurement, but with calibration and test-injections it was typically possible to distinguish between the two based on magnitude.
Fluorescent tRNA (fl-tRNA) Transfection with CNPs
To demonstrate the CNPs’ utility for biological studies, we transfected fluorescent tRNA into the cytoplasm and nucleus and monitored tRNA trafficking between the cytoplasm and nucleus. tRNA is involved in protein synthesis and serves to assemble specific sequences of amino acids at active sites of the ribosome, corresponding to complementary codons on mRNA.[44] Lipofection is typically used to introduce fl-tRNA into cells. However, lipofection requires one to develop specific protocols for different cell lines, a process that sometimes consumes months of trial and error. Microinjection offers many advantages over lipofection as it is readily adaptable to various cell lines and allows better control of the injection process, enabling, among other things, the study of dose-dependent behavior.[42] In addition, CNPs enable one to selectively introduce tRNA into the nucleus and the cytoplasm to monitor subcellular dynamics of tRNA transport across the nuclear membrane in response to cell stresses, tRNA modifications, and other factors. The transport is known to be bidirectional as tRNAs travel from the cytoplasm to the nucleus for modifications, regulation of protein synthesis, and tRNA quality control.[45]
CNPs loaded with Cy5 fl-tRNA (100 μM labeled bulk yeast tRNA) suspended in molecular-grade, nuclease-free water penetrated U2OS cells, and the ΔCeq associated with the cytoplasm penetration was monitored. We measured an increase in capacitance of 2.9 ± 2 pF (N=83) upon cell penetration (Figure 3). As we discussed earlier in the paper, we attribute the increase in capacitance to the use of a very low ionic strength suspending medium. Concurrently with the electrical measurements, we monitored florescence emission from the cells.
Figure 5A–5C and 5G–5I document tRNA injection into the cytoplasm while Figure 5D–5F shows injection into the nucleus. Within the monitoring time interval, the tRNA injected into the cytoplasm remained in the cytoplasm (Figure 5B–5C) and the tRNA injected into the nucleus (Figure 5E–5F) remained in the nucleus. Figure 5H–5I exhibits, however, a different behavior. tRNA injected into the cytoplasm migrated and accumulated in the nucleus. We hypothesize that this tRNA trafficking resulted from stresses imposed on the cell by environmental conditions such as depletion of extracellular media. This hypothesis is consistent with reports that stressors drive tRNA into the nucleus for the modifications needed to regulate protein synthesis. Although the studies of tRNA trafficking are at their infancy and beyond the scope of this paper, this section demonstrates the utility of the CNPs for important biological studies.
Figure 5.

Penetration detection and transfection of fl-tRNA with a CNP. The brightness and contrast of the various images have been adjusted for better visibility. (A–C) Micrographs of a single cell injected with Cy5 fl-tRNA into the cytoplasm. (A) Phase contrast image prior to injection. (B) Fluorescence image immediately after injection. (C) Fluorescence image about 2 min post injection, no visible change in the cell. (D–F) Micrographs of a single cell injected with Cy5 fl-tRNA into the nucleus. (D) Phase contrast image prior to injection. (E) Fluorescence image immediately post injection. (F) Fluorescence image about 2min after injection, no visible change in the cell. (G–I) Micrographs of a single cell injected with Cy5 fl-tRNA into the cytoplasm. (G) Phase contrast image prior to injection. (H) Fluorescence image immediately after injection. (I) Fluorescence image about 1 min after injection. Witness the tRNA migration from the cytoplasm into the nucleus.
Conclusions
Reliable, controllable, high throughput methods for cell injection are critical for, and are the bottleneck in, many important projects in biomedical research. To enable high throughput automated injection, it is desirable to detect cell penetration to trigger the injector. The electrical monitoring of carbon nanopipettes’ (CNPs’) impedance provides a relatively simple means to detect cell penetration. Since CNPs have independent paths for electrical signal monitoring through their carbon lining and for injection through their hollow bore, they are uniquely suitable for automated injection. Unlike other proposed electrical detection methods for cell penetration, the CNPs do not rely on the injection liquid itself to provide the conductive path. Additionally, the CNPs provide smaller dimensions, improved biocompatibility, and better optical contrast for visual feedback during micromanipulation than traditional pulled glass pipettes and much greater controllability than cell transfusion techniques based on cell membrane poration and vectors.
We have demonstrated that CNPs can robustly detect cellular and nuclear penetration through an impedance measurement. By applying a kHz-frequency potential difference between the CNP and a counter electrode submerged in the extracellular solution, we attain stable, low-noise impedance measurements with high time resolution (<1 ms) without a need to rely on a redox mediator in the extracellular solution or high concentration ionic electrolyte in the injection pipette. Trends in the experimental observations are predicted well with a simple, equivalent circuit model. Although a redox mediator is not necessary for our measurements, one could be employed as an alternative means for penetration detection if desired.
Data collected from many microinjection events demonstrates that there is a statistically significant difference between capacitance signal magnitudes when probing the cytoplasm of a cell versus the nucleus, with an average difference of 3.3pF for 150mM KCl solution-filled CNPs.
The proposed measurement technique is reliable and stable, can be used with any composition or volume of injection solution, and can be applied for most, if not all, mammalian cell types. To demonstrate the utility of the CNPs in biological research, we injected the cytoplasm and nucleus with fluorescently labeled tRNA and monitored tRNA trafficking between the cytoplasm and nucleus, a simple real-time method that can help aid in understanding the subcellular dynamics of tRNA. The CNPs allow one to selectively introduce tRNA into the cytoplasm or nucleus, the composition of tRNA mixture can be precisely controlled and the time history of the tRNA can be observed in real time immediately post-injection, none of which is possible with current transfection and fluorescence in situ hybridization techniques.[45]
Another advanced biotechnology procedure that would benefit from microinjection automation is transcriptome induced phenotype remodeling (TIPeR), whereby a transcriptome from one cell type (i.e., A) is transferred into a distinct cell type B with the result that the host cell phenotype B converts to a destination cell-like phenotype A.[46] The CNPs enable introducing the RNA populations into the host cell with the desired composition. Current methods use phototransfection require a skilled operator, are tedious, and do not guarantee that the intended RNA composition enters the cell. Automation would increase the success and throughput of the TIPeR technique allowing for significant improvements in experimental throughput and statistical validity for cellular therapeutic studies.
Acknowledgments
The work was supported, in part, by NIH grant 1R21EB016343-01 to the University of Pennsylvania, by the Commonwealth of Pennsylvania under the Ben Franklin Technology Development Authority and the Nanotechnology Institute, Department of Education GAANN program, Grant number P200A120237, and the NSF DMR08-32802, NSEC - Nano/Bio Interface Center. E. Brailoiu (Temple University) supplied the U2OS cell line used in our experiments and provided advice on cell culturing. J. Griepenberg, C. Lanci, and I. Dmochowski (University of Pennsylvania) provided access to their cell culture facilities. R. Singhal and Y. Gao (Drexel University) assisted with the CNP fabrication. B. Cooperman and I. Farrell (University of Pennsylvania) supplied fl-tRNA for transfection.
Footnotes
Author Contributions
The research presented in this paper was performed by Sean E. Anderson, under the direct supervision of Prof. Haim H. Bau. SEA and HHB designed the experiments, discussed the results, and wrote the paper. SEA carried out the experiments.
References
- 1.Potter H, Heller R. Curr Protoc in Molec Biol. 2010;Chapter 9(Unit 9.3) doi: 10.1002/0471142727.mb0903s92. [DOI] [PubMed] [Google Scholar]
- 2.Gehl J. ACTA Physiologica Scandinavica. 2003;177:437–447. doi: 10.1046/j.1365-201X.2003.01093.x. [DOI] [PubMed] [Google Scholar]
- 3.Paterson L, Agate B, Comrie M, Ferguson R, Lake T, Morris J, Carruthers A, Brown CT, Sibbett W, Bryant P, Gunn-Moore F, Riches AC, Dholakia K. Optics Express. 2005;13:595–600. doi: 10.1364/opex.13.000595. [DOI] [PubMed] [Google Scholar]
- 4.Stevenson DJ, Gunn-Moore FJ, Campbell P, Dholakia K. Journal of the Royal Society Interface. 2010;7:863–871. doi: 10.1098/rsif.2009.0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barhoom S, Kaur J, Cooperman BS, Smorodinsky NI, Smilansky Z, Ehrlich M, Elroy-Stein O. Nucleic Acids Research. 2011:1–13. [Google Scholar]
- 6.Wang W, Sun Y, Zhang M, Anderson R, Langille L, Chan W. Review of Scientific Instruments. 2008;79:104302. doi: 10.1063/1.3006000. [DOI] [PubMed] [Google Scholar]
- 7.Ansorge W, Pepperkok R. Journal of Biochemical and Biophysical Methods. 1988;16:283–292. doi: 10.1016/0165-022x(88)90062-0. [DOI] [PubMed] [Google Scholar]
- 8.Pillarisetti A, Pekarev M, Brooks AD, Desai JP. Symposium on Haptic Interfaces for Virtual Environment and Teleoperator Systems; 2006; Alexandria, VA. [Google Scholar]
- 9.Xie Y, Sun D, Liu C, Tse HY, Cheng SH. The International Journal of Robotics Research. 2009;29:1222–1232. [Google Scholar]
- 10.Zappe S, Fish M, Scott MP, Solgaard O. Lab on a Chip. 2006;6:1012–1019. doi: 10.1039/b600238b. [DOI] [PubMed] [Google Scholar]
- 11.Wang W, Liu X, Gelinas D, Ciruna B, Sun Y. PLoS ONE. 2007;9:e862. doi: 10.1371/journal.pone.0000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan KK, Huang S, Tang KZ. The International Journal of Medical Robotics and Computer Assisted Surgery. 2009;5:85–98. doi: 10.1002/rcs.239. [DOI] [PubMed] [Google Scholar]
- 13.Mattos LS, Grant E, Thresher R, Kluckman K. IEEE Transactions on Information Technology in Biomedicine. 2009;13:822–831. doi: 10.1109/TITB.2009.2023664. [DOI] [PubMed] [Google Scholar]
- 14.Youoku S, Suto Y, Ando M, Ito A. Proc Of SPIE-OSA Biomedical Optics. 2007;6633:66330S. [Google Scholar]
- 15.Matusoka H, Komazaki T, Mukai Y, Shibusawa M, Akane H, Chaki A, Uetake N, Saito M. Journal of Biotechnology. 2005;116:185–194. doi: 10.1016/j.jbiotec.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Adamo A, Jensen KF. Lab on a Chip. 2008;8:1258–1261. doi: 10.1039/b803212b. [DOI] [PubMed] [Google Scholar]
- 17.Lukkari M, Kallio P. Proceedings of the IEEE International Symposium on Computational Intelligence in Robotics and Automation; 2005. [Google Scholar]
- 18.Kallio P, Ritala T, Lukkari M, Kuikka S. The International Journal of Robotics Research. 2007;26:1303. [Google Scholar]
- 19.Lukkari MJ, Karjalainen MI, Sarkanen R, Linne ML, Jalonen TO, Kallio PJ. Proceedings of the 26th Annual International Conference of the IEEE EMBS; 2004. [DOI] [PubMed] [Google Scholar]
- 20.Sharei A, Zoldan J, Adamo A, Sim WY, Cho N, Jackson E, Mao S, Schneider S, Han MJ, Lytton-Jean A, Bastoe PA, Jhunjhunwalab S, Leef J, Hellerb DA, Kangh JW, Hartoularosa GC, Kimd KS, Andersona DG, Langera R, Jensen KF. PNAS. 2013;110:2082–2087. doi: 10.1073/pnas.1218705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown KT, Flaming DG. Advanced Micropipette Techniques for Cell Physiology. John Wiley & Sons Inc; 1986. [Google Scholar]
- 22.Geddes LA. ELECTRODES AND THE MEASUREMENT OF BIOELECTRIC EVENTS. WILEY-INTERSCIENCE, a Division of John Wiley & Sons, Inc; 1972. [Google Scholar]
- 23.Bard AJ, Faulkner LR. ELECTROCHEMICAL METHODS: Fundamentals and Applications. 2. John Wiley & Sons Inc; 2001. [Google Scholar]
- 24.Kodandaramaiah SB, Franzesi GT, Chow BY, Boyden ES, Forest CR. Nature Methods. 2012;9:585–590. doi: 10.1038/nmeth.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakaki K, Esmaeilsabzali H, Dechev N, Burke RD, Park EJ. IEEE Trans Autom Sci Eng. 2012;9:226– 236. [Google Scholar]
- 26.Esmaeilsabzali H, Sakaki K, Dechev N, Burke RD, Park EJ. Med Biol Eng Comput. 2012;50:11–21. doi: 10.1007/s11517-011-0831-2. [DOI] [PubMed] [Google Scholar]
- 27.Nagai M, Torimoto T, Miyamoto T, Kawashima T, Shibata T. Japanese Journal of Applied Physics. 2013;52:047002. [Google Scholar]
- 28.Sun P, Laforge FO, Abeyweera TP, Rotenberg SA, Carpino J, Mirkin MV. PNAS. 2008;105:443–448. doi: 10.1073/pnas.0711075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun P, Laforge FO, Mirkin MV. Phys Chem Chem Phys. 2007;9:802–823. doi: 10.1039/b612259k. [DOI] [PubMed] [Google Scholar]
- 30.Liu C, Sun T, Zhai Y, Dong S. Talanta. 2009;78:613–617. doi: 10.1016/j.talanta.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 31.Kim BM, Murray T, Bau HH. Nanotechnology. 2005;16:1317–1320. [Google Scholar]
- 32.Schrlau MG. PhD Thesis. University of Pennsylvania; Philadelphia, PA: 2009. [Google Scholar]
- 33.Schrlau MG, Falls EM, Ziober BL, Bau HH. Nanotechnology. 2008;19:01501. doi: 10.1088/0957-4484/19/01/015101. [DOI] [PubMed] [Google Scholar]
- 34.Schrlau MG, Brailoiu E, Patel S, Gogotsi Y, Dun NJ, Bau HH. Nanotechnology. 2008;19:325102. doi: 10.1088/0957-4484/19/32/325102. [DOI] [PubMed] [Google Scholar]
- 35.Schrlau MG, Dun NJ, Bau HH. ACS Nano. 2009;3:563–568. doi: 10.1021/nn800851d. [DOI] [PubMed] [Google Scholar]
- 36.Singhal R, Bhattacharyya S, Orynbayeva Z, Vitol E, Friedman G, Gogotsi Y. Nanotechnology. 2010;21:015304. doi: 10.1088/0957-4484/21/1/015304. [DOI] [PubMed] [Google Scholar]
- 37.Katz E, Willner I. Electroanalysis. 2003;15:913–947. [Google Scholar]
- 38.Chen P, Gillis KD. Biophysical Journal. 2000;79:2162–2170. doi: 10.1016/S0006-3495(00)76464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gillis KD. Eur J Physiol. 2000;439:655–664. doi: 10.1007/s004249900173. [DOI] [PubMed] [Google Scholar]
- 40.Alberts B, Bray D, Hopkin K, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Essential Cell Biology. 3. Garland Science; 2010. [Google Scholar]
- 41.Minaschek G, Bereiter-Hahn J, Bertholdt G. Experimental Cell Research. 1989;183:434–444. doi: 10.1016/0014-4827(89)90402-3. [DOI] [PubMed] [Google Scholar]
- 42.Shan JW, Lang DB, Dimotakis PE. Experiments in Fluids. 2004;36:268–273. [Google Scholar]
- 43.Ilegems E, Pick HM, Vogel H. Nucleic Acids Research. 2002;30:e128. doi: 10.1093/nar/gnf128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barhoom S, Kaur B, Cooperman BS, Smorodinsy NI, Smilansky Z. Nucleic Acids Res. 2011;39:e129. doi: 10.1093/nar/gkr601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hopper AK. Genetics. 2013;194:43–67. doi: 10.1534/genetics.112.147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sul JY, Wu CK, Zeng F, Jochems J, Lee MT, Kim TK, Peritz T, Buckley P, Cappelleri D, Maronski M, Kim M, Kumar V, Meaney D, Kim J, Eberwine J. PNAS. 2009 doi: 10.1073/pnas.0902161106. [DOI] [PMC free article] [PubMed] [Google Scholar]