

Abstract
Oculocutaneous albinism (OCA) is an autosomal recessive disorder caused by either complete lack of or a reduction of melanin biosynthesis in the melanocytes. The OCA1A is the most severe type with a complete lack of melanin production throughout life, while the milder forms OCA1B, OCA2, OCA3, and OCA4 show some pigment accumulation over time. Mutations in TYR, OCA2, TYRP1, and SLC45A2 are mainly responsible for causing oculocutaneous albinism. Recently, two new genes SLC24A5 and C10orf11 are identified that are responsible to cause OCA6 and OCA7, respectively. Also a locus has been mapped to the human chromosome 4q24 region which is responsible for genetic cause of OCA5. In this paper, we summarized the clinical and molecular features of OCA genes. Further, we reviewed the screening of pathological mutations of OCA genes and its molecular mechanism of the protein upon mutation by in silico approach. We also reviewed TYR (T373K, N371Y, M370T, and P313R), OCA2 (R305W), TYRP1 (R326H and R356Q) mutations and their structural consequences at molecular level. It is observed that the pathological genetic mutations and their structural and functional significance of OCA genes will aid in development of personalized medicine for albinism patients.
1. Background
Albinism is a group of disorders caused by reduction of polymeric pigment melanin [1]. Melanin production is closely regulated in the body and occurs in specialized ectodermal derived cells called melanocytes. Generally, melanocytes are possibly cutaneous (hair, skin) or extracutaneous (eye, cochlea) which derived from different ectodermal lineages. The terms albinism, oculocutaneous albinism (OCA), and ocular albinism (OA) can be used both as a phenotypic descriptions and as references to specific syndromes. Sir Arcibald Garrod initially states albinism as an inborn error of metabolism [2, 3], but it is now believed to be a heterogeneous genetic disorder caused by mutations in several different genes. Population studies have shown a genetic heterogeneity with evidence pointing to several loci [3].
Oculocutaneous albinism (OCA) is a heterogeneous and autosomal recessive disorder. Based on occurrence of mutation, OCA is identified as nonsyndromic OCA genes (TYR, OCA2, TYRP1, and SLC45A2) and syndromic OCA genes (HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, PLDN, LYST, MYO5A, RAB27A, and MLPH) [4, 5]. The genes TYR, OCA2, TYRP1, and SLC45A2 are mainly responsible for OCA. Apart from these four genes, other genes are also involved to cause OCA. Several genes encoding melanosomal proteins including SILV, RAB7, TYRP2, SLC24A5, and RAB38 have been considered as good candidates for OCA. However, until recently, no pathological mutations of these genes are reported in human OCA patients [6–9]. Very recently, two new OCA genes are found. Mutations of C10orf11 gene are identified in a family from the Faroe Islands and in a Lithuanian patient [10]. Mutations of SLC24A5 gene are found in a Chinese patient presenting with nonsyndromic OCA [11]. In addition, an OCA locus is mapped to 4q24 in a consanguineous Pakistani family, but the gene has not yet been described [12].
Many polymorphisms have not been experimentally illustrated in terms of their possible disease association. Also, the underlying mechanisms by which a genetic mutation has a deleterious functional effect on its gene product thus causes disease are not yet fully understood. In this study we elucidate the molecular basis of OCA disorder caused by disease-associated mutations. Further we have highlighted a better understanding of the relationships between genetic and phenotypic variation as well as protein structure and function. Our review data further helps in the field of pharmacogenomics to develop a personalized medicine for OCA-associated disorders.
2. Prevalence
Albinism affects one in 20,000 individuals worldwide, but the prevalence of individual subtypes varies among different ethnic backgrounds [13]. OCA-1 is the most common subtype found in Caucasians and accounts for about 50% of cases worldwide [7, 14]. OCA-2, or brown OCA (BOCA), accounts for 30% of cases worldwide and is most common in Africa, where it is estimated to affect one in 10,000 and as many as one in 1,000 in certain populations [15, 16]. This is primarily due to an OCA2 found deletion seen at high frequencies within this population [16–19]. OCA3, or rufous OCA (ROCA), is virtually unseen in Caucasians but affects approximately one in 8,500 individuals from southern Africa or 3% of cases worldwide [14]. OCA-4 is also rare among Caucasians as well as Africans, but worldwide it accounts for 17% of cases and in Japan it is diagnosed in one of four persons affected with OCA [14, 20].
3. Symptoms
In general all types of albinism have some lack of pigmentation, but the amount is different depending on the type. OCA1 causes complete absence of pigment in the skin, hair, and eyes, but some individuals may have some degree of pigmentation. OCA1 also affects reduced visual acuity, photophobia (sensitivity to light), and nystagmus (involuntary eye movement). OCA2 causes a minimal to moderate degree of pigmentation in the hair, skin, and eyes. OCA3 has been difficult to identify based on appearance alone. It has been clearly noticeable when a very light-skinned child is born to dark-skinned parents. Ocular albinism affects only the eyes, causing minimal pigmentation. Difficulty controlling eye movements, reduced visual acuity, and nystagmus may occur [21]. OCA6 and OCA7 genes associated with albinism shows the classic visual symptoms and signs but without an obvious change in the pigmentation patterns [10, 11].
4. Genes Associated with Oculocutaneous Albinism
4.1. TYR
OCA1 (MIM 203100) is caused by mutations in the tyrosinase gene (TYR, MIM 606933) on chromosome 11q14.3 [22]. TYR gene consists of 5 exons spanning about 65 kb of genomic DNA which encodes a protein called tyrosinase and consist of 529 amino acids [23]. TYR (EC 1.14.18.1) is a copper-containing enzyme catalysing the first two steps in the melanin biosynthesis pathway, converting tyrosine to L-dihydroxy-phenylalanine (DOPA) and subsequently to DOPAquinone [24]. Mutations completely abolishing tyrosinase activity result in OCA1A, while mutations rendering some enzyme activity result in OCA1B allowing some accumulation of melanin pigment over time. Almost 303 mutations in TYR are known [25] (Table 1).
Table 1.
Mutations detected for albinism associated genes.
| Albinism | Gene | Chromosome location | Mutations |
|---|---|---|---|
| OCA1 | TYR | 11q14-q21 | 303 |
| OCA2 | OCA2 | 15q11.2-q12 | 154 |
| OCA3 | TYRP1 | 9p23 | 16 |
| OCA4 | SLC45A2 | 5p13.3 | 78 |
| OCA5 | ND | 4q24 | 1 |
| OCA6 | SLC24A5 | 15q21.1 | 2 |
| OCA7 | C10ORF11 | 10q22.2-q22.3 | 1 |
| OA1 | GPR143 | Xp22.3 | 114 |
| LYST | CHS1 | 1q42.1-q42.2 | 53 |
| HPS1 | HPS1 | 10q23.1-q23.3 | 31 |
| AP3B1 | HPS2 | 5q14.1 | 20 |
| HPS3 | HPS3 | 3q24 | 7 |
| HPS4 | HPS4 | 22cen-q12.3 | 13 |
| HPS5 | HPS5 | 11p14 | 11 |
| HPS6 | HPS6 | 10q24.32 | 9 |
| HPS7 | DTNBP1 | 6p22.3 | 2 |
| HPS8 | BLOC1S3 | 19q13.32 | 2 |
| HPS9 | BLOC1S6 | 15q21.1 | 1 |
Source: human gene mutation database, 05 Dec, 2013; ND: not determined.
Type 1 temperature sensitive oculocutaneous albinism (OCA1-TS) is an extremely rare form of OCA1 characterized by the production of temperature sensitive tyrosinase proteins leading to dark hair on the legs, arms, and chest (cooler body areas) and white hair on the scalp, axilla, and pubic area (warmer body areas). Mutation in temperature sensitive tyrosinase protein is inactivated at 37°C.
4.2. OCA2
Mutations in the OCA2 gene (also known as P-gene) (MIM 203200) cause the OCA2 phenotype (MIM 203200) [26]. The OCA2 gene consists of 24 exons (23 coding), spanning almost 345 kb of genomic DNA in the region of 15q11.2-q12. It is highly polymorphic [27] and is suspected to play an important role in human pigmentation [28–30]. This gene encodes P protein, a 110-kDa integral melanosomal protein with 12 predicted transmembrane domains and consists of 838 amino acids [27, 31, 32]. The P protein acts as a precursor to melanin synthesis, within the melanocyte, and serves as a key control point at which ethnic skin colour variation is determined. Moreover, it may stabilize or traffic the melanosomal protein such as tyrosinase which regulate melanosomal pH or serve as a melanosomal tyrosine transporter [26, 33–36]. Currently, in human gene mutation database (HGMD) (http://www.hgmd.org/), 154 mutations in OCA2 gene are listed to cause OCA2 (Table 1). Due to mutation, P protein might disturb the pigmentation characteristics via altering the melanosomal tyrosine or tyrosinase bioavailability or function.
4.3. TYRP1
OCA3 is mostly caused by the genetic mutation in TYRP1 (MIM 115501) gene. OCA3 is also known as Rufous oculocutaneous albinism. The human TYRP1 gene consists of 8 exons and 7 introns, spanning almost 15–18 kb of genomic DNA in the region of 9p23 [37–40]. This gene that encodes a protein called tyrosinase-related protein 1 (Tyrp1) has a molecular weight of ~75 kDa and appears to be the most abundant melanosomal protein of the melanocyte [41, 42]. Tyrp1 contains of 537 amino acid residues and shares 40–52% of amino acid homology to tyrosinase protein. Tyrp1 is involved in the maintenance of melanosome structure and affects melanocyte proliferation and cell death [43–46]. Tyrp1 shows tyrosine hydroxylase activity, albeit under low substrate (L-tyrosine) concentration, but no DOPA oxidase activity [47, 48]. Human Tyrp1 also is involved in conversion of L-tyrosine to DOPA with low turnover rates, by the generation of low amounts of DOPA. It is an essential cofactor for tyrosinase activity [49]. Tyrp1 has also been attributed with various other catalytic functions including dopachrome tautomerase (Dct), dihydroxyindole (DHI) oxidase [50], and 5,6-dihydroxyindole-2-carboxylic acid (DHICA) [51]. To date, 16 mutations were found in TYRP1 gene which is responsible for OCA3 (Table 1).
4.4. SLC45A2
Mutations in the membrane-associated transporter protein gene (MATP, also known as SLC45A2 and MIM 606202) cause OCA4 (MIM 606574) [52]. MATP consists of 7 exons spanning approximately 40 kb of genomic DNA, mapping to chromosomal position 5p13.3. The SLC45A2/MATP protein consists of 530 amino acids which contains 12 putative transmembrane domains and shows sequence and structural similarity to plant sucrose transporters. It is expressed in melanosomal cell lines [53, 54]. The function of SLC45A2 is still unknown, but studies from Medaka fish show that the SLC45A2 protein plays an important role in pigmentation and probably functions as a membrane transporter in melanosomes [53]. Mutations in SLC45A2 were found for the first time in a Turkish OCA patient [52] and have since been found in German, Japanese, and Korean OCA patients [20, 55–57]. Mutations in SLC45A2 cause misrouting of tyrosinase similar to the cellular phenotype of OCA-2 [58, 59]. To date, 78 mutations were predicted in SLC45A2 gene which is responsible for OCA4 (Table 1).
4.5. SLC24A5 and C10orf11
Mutations in SLC24A5 encode another solute carrier protein. It is a well-known gene in the pigment cell arena and is associated with a new form of OCA, named as OCA6 (MIM 609802). SLC24A5 gene was located in the chromosomal position of 15q21.1 [11]. SLC24A5 mutations were detected in patients of diverse ethnic origins, thus indicating that OCA6 is not restricted to the Chinese population. It is evident that the cutaneous phenotype was heterogeneous with hair colour changing from white to blond and dark brown [60]. A SNP in SLC24A5 (rs1426654) encoding an alanine or threonine at position 111 was detected. Prominently, Thr111 is present in almost all individuals of European American origin, while Ala111 is present in African Asian populations. Thr111 is associated with lighter pigmented skin, thus suggesting an important role of this SNP in the establishment of human pigmentation [61]. Recent results indicate a role of SLC24A5 in the maturation of melanosomes. The assembly of SLC24A5 into melanosomes showed an important role for the melanosomal architecture and to ensure that melanin is synthesized properly. Hence, the lack of SLC24A5 may impair or disrupt melanosomal maturation and melanin biosynthesis [11].
Mutations in the C10orf11 gene were associated with new form of OCA, designated as OCA7 (MIM 615179). C10orf11 was located in the chromosomal position of 10q22.2-q22.3 [10]. C10orf11 encodes a 198 amino acid protein containing three leucine-rich repeats (LRRs) and one LRR C-terminal (LRRCT) domain. The family of LRRs-containing proteins encompasses members with a variety of functions, including cell adhesion and signalling, extracellular-matrix assembly, neuronal development, and RNA processing [62]. The updated mutation list of these genes was shown in Table 1.
5. Other Partial Albinism Disorders and Their Genes
Ocular albinism (OA1) (MIM: 300500) is a form of albinism that affects only the eyes. This disease is caused by mutation in OA1/GPR143 gene which is located on the X chromosome [63] and mutations lists were shown in Table 1. Pigment of the skin and hair is normal or only slightly diluted. Human eyes are severely affected with reduced visual acuity and photophobia. Strabismus or nystagmus is frequently affected with OA1. The fundus and irides are depigmented.
Hermansky-Pudlak syndrome (HPS) (MIM: 203300) is a rare autosomal recessive [64] disorder which results in oculocutaneous albinism (decreased pigmentation), bleeding problems due to storage of an abnormal fat-protein compound (lysosomal accumulation of ceroid lipofuscin), and platelet abnormality (platelet storage pool defect). HPS type 1–9 is caused by mutations in following genes: HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, and BLOC1S6, respectively, and their updated mutations lists were shown in Table 1. The disease can affect the dysfunctions of lungs, intestine, kidneys, or heart. The major severe form of disorder is pulmonary fibrosis, which routinely exhibits in patient's ages of 40–50 years [65]. The disorder is more common in Puerto Rico [66], where it affects approximately 1 in 1,800 individuals [67].
Chédiak-Higashi syndrome (CHS1) (MIM: 214500) is a rare autosomal recessive disorder caused by the mutation of a lysosomal trafficking regulator protein and leads to a decrease in phagocytosis [68]. The decrease in phagocytosis results in recurrent pyogenic infections, peripheral neuropathy, and partial albinism. The eye, skin, and hair pigment is reduced or diluted in CHS [69, 70]. Mutations in the CHS1 gene (also known as LYST) have been found to be connected with Chédiak-Higashi syndrome (Table 1). CHS1 gene provides instructions for making a protein known as the lysosomal trafficking regulator. Scientists believe that this protein plays a major role in the transport (trafficking) of materials into structures called lysosomes. Lysosomes act as recycling centers within cells. Digestive enzymes are used to break down toxic substances, digest bacteria that invade the cell, and recycle worn-out cell components.
6. Computational Screening of Pathological Mutation and Their Molecular Mechanism
Due to the presence of huge amount of variations data, experimental study of each variant cannot be achieved in a reasonable timescale. Therefore, predictive analysis of the effects of polymorphisms on gene function is needed in order to prioritize the cases that require further study, to elucidate the molecular basis of albinism disorders caused by nonsynonymous mutations. Further, it helps to observe the structural and functional changes of protein upon mutation at atomic level. Screening of nsSNPs of OCA 1–4 genes were analyzed by following computational methods SIFT (deleterious = ≥0.05; tolerated = ≤0.05), PolyPhen (damaging = ≥1.5; benign = ≤1.5), PolyPhen 2.0 (damaging = ≤0.5; benign = ≥0.51), I-Mutant 2.0 (decrease stability = (DDG < 0; increase stability = DDG > 0), I-Mutant 3.0 (decrease = ≤0.5 kcal/moL; Increase = ≥0.5 kcal/moL), and PANTHER (deleterious (>−3); tolerated <−3)). The nsSNPs which were commonly predicted by the above servers were further applied in PHD-SNP, SNP&GO, Pmut, and Mutpred servers to screen the most deleterious and disease-associated mutations in OCA 1–4 genes. The strategy of our investigation is depicted in Figure 1.
Figure 1.
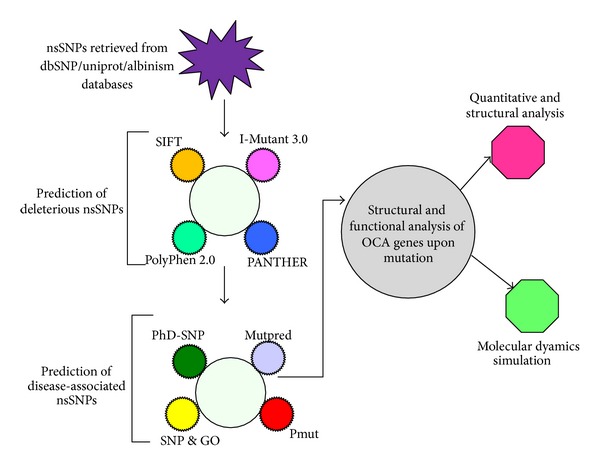
Flow chart of OCA 1–4 gene analysis.
The predicted disease-associated mutations could be endorsed with the observed experimental data [71, 72]. The SNP information of OCA 1–4 genes was retrieved from dbSNP/Swiss-prot/albinism database. To understand the atomic arrangement in 3D space, the native and mutant structures of OCA (Tyr, P, Tyrp1, and SLC45A2) proteins were modelled. Molecular dynamics simulation approach was applied to observe the structural and functional behaviour of OCA proteins upon mutation at atomic level. In this review we highlighted the structural and functional consequence of TYR, OCA2, and TYRP1 genes, respectively.
6.1. OCA1A
In this study, we rationalized the structural and functional behaviour of tyrosinase protein upon mutation. Based on our investigations we reported the potential candidate SNPs for advanced studies. Out of 57 nsSNPs, four nsSNPs T373K, N371Y, M370T, and P313R in tyrosinase protein were predicted to be functionally significant from our datasets (Table 2).
Table 2.
Gene names, chromosome location, protein name, and predicted mutations of OCA1–4.
| Disease types | Gene | Chromosome location | Protein | Predicted mutations |
|---|---|---|---|---|
| OCA1A | TYR | 11q14.3 | Tyrosinase | T373K, N371Y, M370T, P313R |
| OCA2 | OCA2 | 15q11.2-q12 | P protein/melanocyte-specific transporter protein | R305W |
| OCA3 | TYRP1 | 9p23 | Tyrosinase related protein-1 | R326H, R356Q |
| OCA4 | SLC45A2/MATP | 5p13.3 | Membrane-associated transporter protein | Y317C |
Threading based approach was applied to predict the model structure of native and mutant TYR proteins. Molecular dynamics simulation approach was applied for refinement of predicted structure of native and mutant TYR proteins. Quantitative and structural approach was performed to rationalized deleterious mutation in TYR gene and molecular mechanism in OCA1A. Due to mutation, TYR structural orientation was altered and became more rigid in nature. This structural disturbance might affect the function of protein and thus the reason to cause OCA1A [73]. The superimposed structure of native and mutant tyrosinase protein and its mutation was displayed in Figures 2(a)–2(d).
Figure 2.
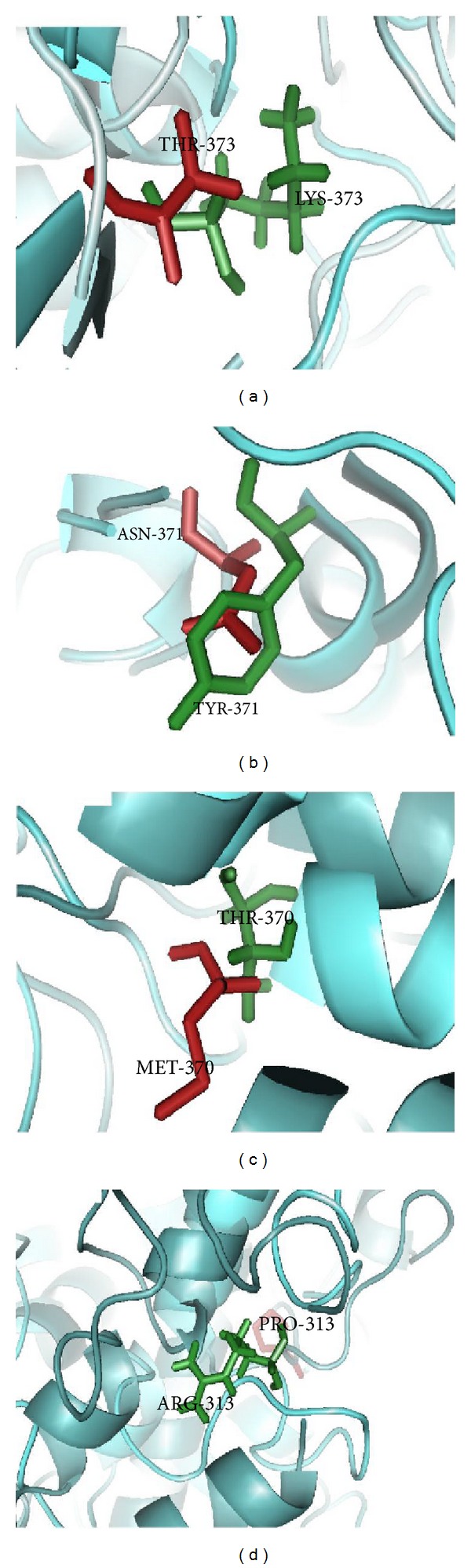
The superimposed structure of native and mutant TYR proteins and mutant residues in stick model: (a) T373K, (b) N371Y, (c) M370T, and (d) P313R.
6.2. OCA2
Using computational approach we screened the most deleterious and disease-associated mutation (R305W) on OCA2 gene from 95 input variants datasets (Table 2). In this analysis, we applied molecular modelling and molecular dynamics simulation approach to examine the structural and functional behaviour of P protein upon mutation. From the molecular dynamics simulation analysis, we confirmed that due to occurrence of mutation the structure loss stability and became more flexible in conformation [74]. Due to flexibility, P protein losses the catalytic function in melanin biosynthesis and might also play a significant role in inducing OCA2 [74]. The superimposed structure of native and mutant P protein and its mutation was shown in Figure 3(b).
Figure 3.
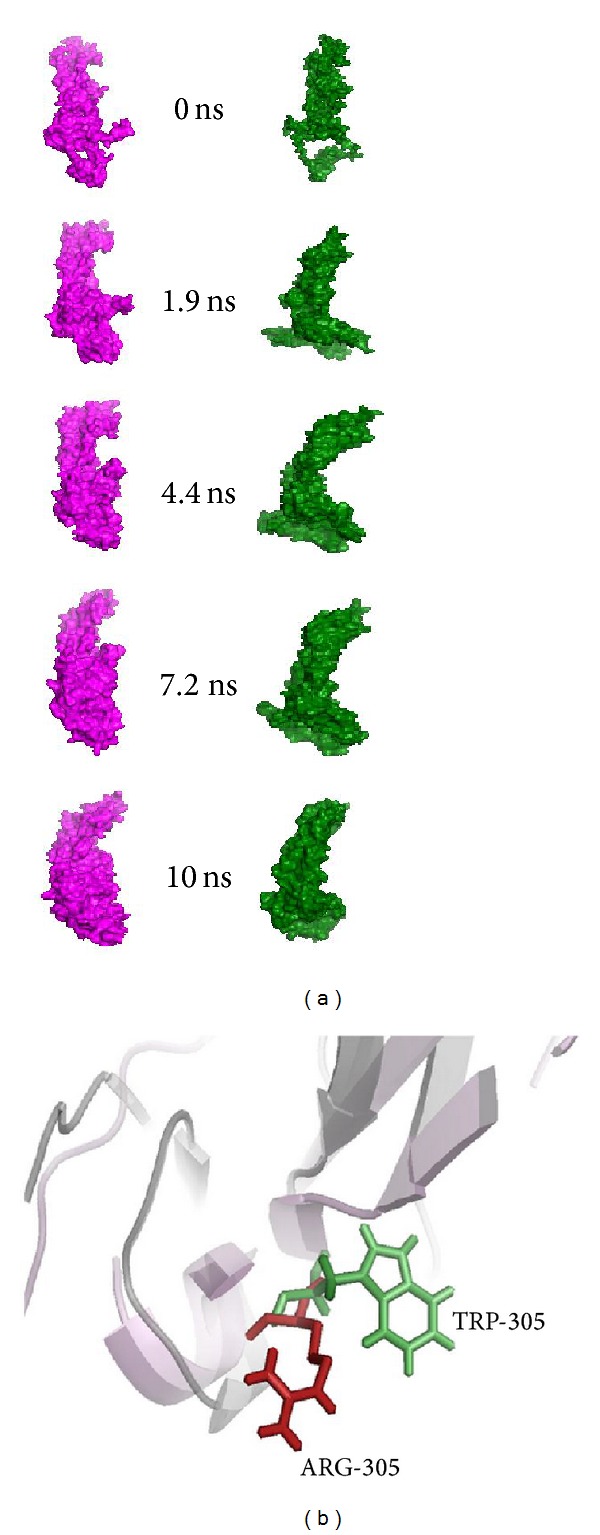
(a) Snapshots of native and mutant P protein conformation at different simulation time steps [74]. (b) The superimposed structure of native and mutant (R305W) P proteins and mutant residues in a stick model.
6.3. OCA3
In this analysis, we applied in silico approach to screen the most disease-associated mutation on TYRP1 gene. Out of 63 nsSNPs, we screened the most disease-associated (R326H and R356Q) mutations on TYRP1 gene (Table 2). The structural analyses of native and mutant Tyrp1 proteins were scrutinized by molecular modelling and molecular dynamics simulation (MDS) approach. From this analysis we confirmed that, TYRP1 protein alter the structural conformation and loss the flexibility behaviour upon mutation [75]. The structural alteration of TYRP1 protein upon mutation was clearly discussed and shown in our previous studies (Figure 4) [75].
Figure 4.
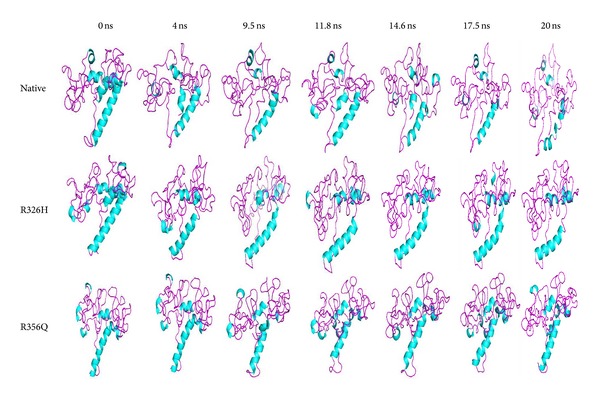
Snapshots of native and mutant (R326H and R356Q) TYRP1 protein conformation at different simulation time steps [75].
In Figure 4, both mutant (R326H and R356Q) structures showed an increase in helical content which leads to became more rigid on conformation. Due to rigidity, Tyrp1 protein may lose stability of the tyrosinase protein and modulate its catalytic activity in eumelanin synthesis. It may also disturb the maintenance of melanosome structure and may lead to affect melanocyte proliferation and cell death [75]. The superimposed structure of native and mutant Tyrp1 protein and its mutation residues was shown in Figure 5(a).
Figure 5.

(a) The superimposed structure of native and mutant structures (R326H and R356Q) TYRP1 proteins and mutant residues in a stick model. (b) The superimposed structure of native and mutant (Y317C) SLC45A2 proteins and mutant residue as a stick model.
6.4. OCA4
In this investigation, we implemented multiple computational approaches to identity the most disease-associated mutations in SLC45A2 gene. Based on SIFT, PolyPhen 2.0, I-Mutant 3.0, PANTHER, SNP&GO, PhD-SNP, Pmut, and MutPred servers, we screened the most disease-associated mutation (Y317C) on SLC45A2 gene (Table 2). It may lead to affect the structural conformation and function of the SLC45A2/MATP protein upon mutation. The superimposed structure of native and mutant (Y317C) SLC45A2 protein and its mutation was shown in Figure 5(b). The predicted mutations on TYR, OCA2, and TYRP1 genes and their molecular mechanism can further help wet lab scientist to develop a potent drug target for OCA type 1–3.
7. Preclinical Attempts for Albinisms
To date, three drugs were found for albinism related disorders. L-DOPA [76–79] and Adeno associated viral vectors (AAV) [80, 81] helps to treat the OCA1 and OA1. A third therapeutic approved drug, nitisinone, originally devised to treat patients with hereditary tyrosinemia type 1 [82]. This drug has a useful side effect, because it triggers the anomalous accumulation of tyrosine in the blood, thus increasing the concentration of the reagent used by the enzyme tyrosinase (TYR) and resulting in an increased oxidation and improved pigmentation in eyes and skin of mouse models of OCA1B, with some residual tyrosinase activity. Also, the group of Fukai (Osaka, Japan) has suggested the use of aminoglycosides, known to allow the read-through effect over certain nonsense mutations, as a potential therapeutic intervention for some mutations commonly found in albinism [83]. These very promising results soon will be also investigated in humans, in tentative clinical trials. Some people are working to develop a potent drug targets for skin (reduced pigmentation) disorders [84–87]. Additional studies and validations are still required before transferring any of these therapeutic proposals, here listed, to routine treatment for human subjects.
8. Concluding Remarks
Albinisms are studied for many years as a congenital rare disease without cure, where a person born with albinism will ultimately die with the same disease. This situation has now changed and will continue improving in the near future. Oculocutaneous albinism is caused by all the ethnic backgrounds of humans. It is caused by the mutation in OCA genes. Due to mutation, the melanin biosynthesis pathway inhibits the action of melanin synthesis, which leads to albinism. Our predictions suggest a significant computational approach to detect the OCA 1–3 (TYR, TYRP1, OCA2, and SLC45A2 genes) associated SNPs from the large SNP dataset and reduce the expenses in experimental depiction of pathological mutations. Most of this pathological evidence identified in OCA has been reported and still we lack in understanding their associated molecular mechanism. To develop an efficient therapeutic approach, it is highly necessary to understand the detailed knowledge of their molecular mechanisms, to illustrate their structural and functional behaviours in detail. The SLC45A2, SLC24A2, and C10orf11 genes structural and functional behaviours are still unknown. We must also continue to elucidate the high resolution 3D structures and their localization patterns of OCA genes, as these data will aid in developing a potent drug target for albinism related disorders.
Acknowledgments
The authors gratefully acknowledge the management of Vellore Institute of Technology University for providing the facilities to carry out this work. They thank the anonymous reviewers for their helpful comments and critical reading of the paper.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Garrod AE. Inborn Errors of Metabolism. London, UK: Frowde and Hodder & Stoughton; 1923. [Google Scholar]
- 2.Wildsoet CF, Oswald PJ, Clark S. Albinism: its implications for refractive development. Investigative Ophthalmology & Visual Science. 2000;41(1):1–7. [PubMed] [Google Scholar]
- 3.Kasmann-Kellner B, Hille K, Pfau B. Eye and general illnesses in the public school for the blind and usually handicapped students in Saarland development in the last 20 years. Opthamologe. 1998;95(1):51–54. doi: 10.1007/s003470050235. [DOI] [PubMed] [Google Scholar]
- 4.Li W, He M, Zhou H, Bourne JW, Liang P. Mutational data integration in gene-oriented files of the Hermansky-Pudlak Syndrome database. Human Mutation. 2006;27(5):402–407. doi: 10.1002/humu.20309. [DOI] [PubMed] [Google Scholar]
- 5.Wei A, Li W. Hermansky-Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell and Melanoma Research. 2013;26(2):176–192. doi: 10.1111/pcmr.12051. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki T, Miyamura Y, Inagaki K, Tomita Y. Characterization of the human RAB38 and RAB7 genes: exclusion of new major pathological loci for Japanese OCA. Journal of Dermatological Science. 2003;32(2):131–136. doi: 10.1016/s0923-1811(03)00071-9. [DOI] [PubMed] [Google Scholar]
- 7.Hutton SM, Spritz RA. Comprehensive analysis of oculocutaneous albinism among non-hispanic caucasians shows that OCA1 Is the most prevalent OCA type. Journal of Investigative Dermatology. 2008;128(10):2442–2450. doi: 10.1038/jid.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grønskov K, Ek J, Sand A, et al. Birth prevalence and mutation spectrum in Danish patients with autosomal recessive albinism. Investigative Ophthalmology & Visual Science. 2009;50(3):1058–1064. doi: 10.1167/iovs.08-2639. [DOI] [PubMed] [Google Scholar]
- 9.Mondal M, Sengupta M, Samanta S, Sil A, Ray K. Molecular basis of albinism in India: Evaluation of seven potential candidate genes and some new findings. Gene. 2012;511(2):470–474. doi: 10.1016/j.gene.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 10.Grønskov K, Dooley CM, Østergaard E, et al. Mutations in C10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism. The American Journal of Human Genetics. 2013;92(3):415–421. doi: 10.1016/j.ajhg.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei AH, Zang DJ, Zhang Z, et al. Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic Oculocutaneous albinism . Journal of Investigative Dermatology. 2013;133(7):1834–1840. doi: 10.1038/jid.2013.49. [DOI] [PubMed] [Google Scholar]
- 12.Kausar T, Bhatti MA, Ali M, Shaikh RS, Ahmed ZM. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clinical Genetics. 2013;84(1):91–93. doi: 10.1111/cge.12019. [DOI] [PubMed] [Google Scholar]
- 13.Gargiulo A, Testa F, Rossi S, et al. Molecular and clinical characterization of albinism in a large cohort of Italian patients. Investigative Ophthalmology and Visual Science. 2011;52(3):1281–1289. doi: 10.1167/iovs.10-6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rooryck C, Morice-Picard F, Elçioglu NH, Lacombe D, Taieb A, Arveiler B. Molecular diagnosis of oculocutaneous albinism: new mutations in the OCA1-4 genes and practical aspects. Pigment Cell & Melanoma Research. 2008;21(5):583–587. doi: 10.1111/j.1755-148X.2008.00496.x. [DOI] [PubMed] [Google Scholar]
- 15.Okoro AN. Albinism in Nigeria. A clinical and social study. British Journal of Dermatology. 1975;92(5):485–492. [PubMed] [Google Scholar]
- 16.Puri N, Durham-Pierre D, Aquaron R, Lund PM, King RA, Brilliant MH. Type 2 oculocutaneous albinism (OCA2) in Zimbabwe and Cameroon: distribution of the 2.7-kb deletion allele of the P gene. Human Genetics. 1997;100(5-6):651–656. doi: 10.1007/s004390050568. [DOI] [PubMed] [Google Scholar]
- 17.Durham-Pierre D, Gardner JM, Nakatsu Y, et al. African origin of an intragenic deletion of the human P gene in tyrosinase positive oculocutaneous albinism. Nature Genetics. 1994;7(2):176–179. doi: 10.1038/ng0694-176. [DOI] [PubMed] [Google Scholar]
- 18.Stevens G, van Beukering J, Jenkins T, Ramsay M. An intragenic deletion of the P gene is the common mutation causing tyrosinase-positive oculocutaneous albinism in Southern African Negroids. American Journal of Human Genetics. 1995;56(3):586–591. [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens G, Ramsay M, Jenkins T. Oculocutaneous albinism (OCA2) in sub-Saharan Africa: Distribution of the common 2.7-kb P gene deletion mutation. Human Genetics. 1997;99(4):523–527. doi: 10.1007/s004390050400. [DOI] [PubMed] [Google Scholar]
- 20.Inagaki K, Suzuki T, Shimizu H, et al. Oculocutaneous albinism type 4 is one of the most common types of albinism in Japan. The American Journal of Human Genetics. 2004;74(3):466–471. doi: 10.1086/382195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boissy RE. Albinism. eMedicine. 2003, http://emedicine.medscape.com/article/1068184-overview.
- 22.Tomita Y, Takeda A, Okinaga S, Tagami H, Shibahara S. Human oculocutaneous albinism caused by single base insertion in the tyrosinase gene. Biochemical and Biophysical Research Communications. 1989;164(3):990–996. doi: 10.1016/0006-291x(89)91767-1. [DOI] [PubMed] [Google Scholar]
- 23.Kwon BS, Haq AK, Pomerantz SH, Halaban R. Isolation and sequence of a cDNA clone for human tyrosinase that maps at the mouse c-albino locus. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(21):7473–7477. doi: 10.1073/pnas.84.21.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooksey CJ, Garratt PJ, Land EJ, et al. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. The Journal of Biological Chemistry. 1997;272(42):26226–26235. doi: 10.1074/jbc.272.42.26226. [DOI] [PubMed] [Google Scholar]
- 25.HGMD. The Human Gene Mutation Database at the Institute Medical Genetics in Cardiff. 2013. http://www.hgmd.org/ [Google Scholar]
- 26.Rinchik EM, Bultman SJ, Horsthemke B, et al. A gene for the mouse pink-eyed dilution locus and for human type II oculocutaneous albinism. Nature. 1993;361(6407):72–76. doi: 10.1038/361072a0. [DOI] [PubMed] [Google Scholar]
- 27.Lee S-T, Nicholls RD, Jong MTC, Fukai K, Spritz RA. Organization and sequence of the human P gene and identification of a new family of transport proteins. Genomics. 1995;26(2):354–363. doi: 10.1016/0888-7543(95)80220-g. [DOI] [PubMed] [Google Scholar]
- 28.Ancans J, Flanagan N, Hoogduijn MJ, Thody AJ. P-locus is a target for the melanogenic effects of MC-1R signaling: a possible control point for facultative pigmentation. Annals of the New York Academy of Sciences. 2003;994:373–377. doi: 10.1111/j.1749-6632.2003.tb03202.x. [DOI] [PubMed] [Google Scholar]
- 29.Sturm RA, Duffy DL, Box NF, et al. The role of melanocortin-1 receptor polymorphism in skin cancer risk phenotypes. Pigment Cell Research. 2003;16(3):266–272. doi: 10.1034/j.1600-0749.2003.00041.x. [DOI] [PubMed] [Google Scholar]
- 30.Jannot A, Meziani R, Bertrand G, et al. Allele variations in the OCA2 gene (pink-eyed-dilution locus) are associated with genetic susceptibility to melanoma. European Journal of Human Genetics. 2005;13(8):913–920. doi: 10.1038/sj.ejhg.5201415. [DOI] [PubMed] [Google Scholar]
- 31.Grønskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet Journal of Rare Diseases. 2007;2(1, article 43) doi: 10.1186/1750-1172-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosemblat S, Durham-Pierre D, Gardner JM, Nakatsu Y, Brilliant MH, Orlow SJ. Identification of a melanosomal membrane protein encoded by the pink-eyed dilution (type II oculocutaneous albinism) gene. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(25):12071–12075. doi: 10.1073/pnas.91.25.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rebbeck TR, Kanetsky PA, Walker AH, et al. P gene as an inherited biomarker of human eye color. Cancer Epidemiology Biomarkers and Prevention. 2002;11(8):782–784. [PubMed] [Google Scholar]
- 34.Korner A, Pawelek J. Mammalian tyrosinase catalyzes three reactions in the biosynthesis of melanin. Science. 1982;217(4565):1163–1165. doi: 10.1126/science.6810464. [DOI] [PubMed] [Google Scholar]
- 35.Manga P, Boissy RE, Pifko-Hirst S, Zhou B-K, Orlow SJ. Mislocalization of melanosomal proteins in melanocytes from mice with oculocutaneous albinism type 2. Experimental Eye Research. 2001;72(6):695–710. doi: 10.1006/exer.2001.1006. [DOI] [PubMed] [Google Scholar]
- 36.Brilliant MH. The mouse p (pink-eyed dilution) and human P genes, oculocutaneous albinism type 2 (OCA2), and melanosomal pH. Pigment Cell Research. 2001;14(2):86–93. doi: 10.1034/j.1600-0749.2001.140203.x. [DOI] [PubMed] [Google Scholar]
- 37.Murty VVVS, Bouchard B, Mathew S, Vijayasaradhi S, Houghton AN. Assignment of the human TYRP (brown) locus to chromosome region 9p23 by nonradioactive in situ hybridization. Genomics. 1992;13(1):227–229. doi: 10.1016/0888-7543(92)90228-k. [DOI] [PubMed] [Google Scholar]
- 38.Shibahara S, Taguchi H, Muller RM, et al. Structural organization of the pigment cell-specific gene located at the brown locus in mouse: its promoter activity and alternatively spliced transcripts. Journal of Biological Chemistry. 1991;266(24):15895–15901. [PubMed] [Google Scholar]
- 39.Sturm RA, O'Sullivan BJ, Box NF, et al. Chromosomal structure of the human TYRP1 and TYRP2 loci and comparison of the tyrosinase-related protein gene family. Genomics. 1995;29(1):24–34. doi: 10.1006/geno.1995.1211. [DOI] [PubMed] [Google Scholar]
- 40.Box NF, Wyeth JR, Mayne CJ, O'Gorman LE, Martin NG, Sturm RA. Complete sequence and polymorphism study of the human TYRP1 gene encoding tyrosinase-related protein 1. Mammalian Genome. 1998;9(1):50–53. doi: 10.1007/s003359900678. [DOI] [PubMed] [Google Scholar]
- 41.Vijayasaradhi S, Doskoch PM, Houghton AN. Biosynthesis and intracellular movement of the melanosomal membrane glycoprotein gp75, the human b (Brown) locus product. Experimental Cell Research. 1991;196(2):233–240. doi: 10.1016/0014-4827(91)90256-t. [DOI] [PubMed] [Google Scholar]
- 42.Halaban R, Moellmann G. Murine and human b locus pigmentation genes encode a glycoprotein (gp75) with catalase activity. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(12):4809–4813. doi: 10.1073/pnas.87.12.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi T, Imokawa G, Bennett DC, Hearing VJ. Tyrosinase stabilization by Tyrp1 (the brown locus protein) The Journal of Biological Chemistry. 1999;273(48):31801–31805. doi: 10.1074/jbc.273.48.31801. [DOI] [PubMed] [Google Scholar]
- 44.Hirobe T, Abe H. Genetic and epigenetic control of the proliferation and differentiation of mouse epidermal melanocytes in culture. Pigment Cell Research. 1999;12(3):147–163. doi: 10.1111/j.1600-0749.1999.tb00508.x. [DOI] [PubMed] [Google Scholar]
- 45.Johnson R, Jackson IJ. Light is a dominant mouse mutation resulting in premature cell death. Nature Genetics. 1992;1(3):226–229. doi: 10.1038/ng0692-226. [DOI] [PubMed] [Google Scholar]
- 46.Fang D, Tsuji Y, Setaluri V. Selective down-regulation of tyrosinase family gene TYRP1 by inhibition of the activity of melanocyte transcription factor, MITF. Nucleic Acids Research. 2002;30(14):3096–3106. doi: 10.1093/nar/gkf424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarangarajan R, Boissy RE. Tyrp1 and oculocutaneous albinism type 3. Pigment Cell Research. 2001;14(6):437–444. doi: 10.1034/j.1600-0749.2001.140603.x. [DOI] [PubMed] [Google Scholar]
- 48.Boissy RE, Sakai C, Zhao H, Kobayashi T, Hearing VJ. Human tyrosinase related protein-1 (TRP-1) does not function as a DHICA oxidase activity in contrast to murine TRP-1. Experimental Dermatology. 1998;7(4):198–204. doi: 10.1111/j.1600-0625.1998.tb00324.x. [DOI] [PubMed] [Google Scholar]
- 49.Zhao H, Zhao Y, Nordlund JJ, Boissy RE. Human TRP-1 has tyrosine hydroxylase but no dopa oxidase activity. Pigment Cell Research. 1994;7(3):131–140. doi: 10.1111/j.1600-0749.1994.tb00040.x. [DOI] [PubMed] [Google Scholar]
- 50.Urabe K, Aroca P, Hearing VJ. From gene to protein: determination of melanin synthesis. Pigment Cell Research. 1993;6(4):186–192. doi: 10.1111/j.1600-0749.1993.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 51.Olivares C, Jiménez-Cervantes C, Lozano JA, Solano F, García-Borrón JC. The 5,6-dihydroxyindole-2-carboxylic acid (DHICA) oxidase activity of human tyrosinase. Biochemical Journal. 2001;354(1):131–139. doi: 10.1042/0264-6021:3540131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newton JM, Cohen-Barak O, Hagiwara N, et al. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. American Journal of Human Genetics. 2001;69(5):981–988. doi: 10.1086/324340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fukamachi S, Shimada A, Shima A. Mutations in the gene encoding B, a novel transporter protein, reduce melanin content in medaka. Nature Genetics. 2001;28(4):381–385. doi: 10.1038/ng584. [DOI] [PubMed] [Google Scholar]
- 54.Harada M, Li YF, El-Gamil M, Rosenberg SA, Robbins PF. Use of an in vitro immunoselected tumor line to identify shared melanoma antigens recognized by HLA-A*0201-restricted T cells. Cancer Research. 2001;61(3):1089–1094. [PubMed] [Google Scholar]
- 55.Rundshagen U, Zühlke C, Opitz S, Schwinger E, Käsmann-Kellner B. Mutations in the MATP gene in five German patients affected by oculocutaneous albinism type 4. Human Mutation. 2004;23(2):106–110. doi: 10.1002/humu.10311. [DOI] [PubMed] [Google Scholar]
- 56.Inagaki K, Suzuki T, Ito S, et al. Oculocutaneous albinism type 4: six novel mutations in the membrane-associated transporter protein gene and their phenotypes. Pigment Cell Research. 2006;19(5):451–453. doi: 10.1111/j.1600-0749.2006.00332.x. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki T, Inagaki K, Fukai K, et al. A Korean case of oculocutaneous albinism type IV caused by a D157N mutation in the MATP gene. British Journal of Dermatology. 2005;152(1):174–175. doi: 10.1111/j.1365-2133.2005.06403.x. [DOI] [PubMed] [Google Scholar]
- 58.Costin GE, Valencia JC, Vieira WD, Lamoreux ML, Hearing VJ. Tyrosinase processing and intracellular trafficking is disrupted in mouse primary melanocytes carrying the underwhite (uw) mutation. A model for oculocutaneous albinism (OCA) type 4. Journal of Cell Science. 2003;116(15):3203–3212. doi: 10.1242/jcs.00598. [DOI] [PubMed] [Google Scholar]
- 59.Cullinane AR, Vilboux T, O'Brien K, et al. Homozygosity mapping and whole-exome sequencing to detect SLC45A2 and G6PC3 mutations in a single patient with oculocutaneous albinism and neutropenia. Journal of Investigative Dermatology. 2011;131(10):2017–2025. doi: 10.1038/jid.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fanny MP, Eulalie L, Stéphane F, et al. SLC24A5 mutations are associated with non-syndromic oculocutaneous albinism. Journal of Investigative Dermatology. 2013 doi: 10.1038/jid.2013.360. [DOI] [PubMed] [Google Scholar]
- 61.Lamason RL, Mohideen MPK, Mest JR, et al. Genetics: SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005;310(5755):1782–1786. doi: 10.1126/science.1116238. [DOI] [PubMed] [Google Scholar]
- 62.Bella J, Hindle KL, McEwan PA, Lovell SC. The leucine-rich repeat structure. Cellular and Molecular Life Sciences. 2008;65(15):2307–2333. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bassi MT, Schiaffino MV, Renieri A, et al. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nature Genetics. 1995;10(1):13–19. doi: 10.1038/ng0595-13. [DOI] [PubMed] [Google Scholar]
- 64.Oh J, Ho L, Ala-Mello S, et al. Mutation analysis of patients with Hermansky-Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity. The American Journal of Human Genetics. 1998;62(3):593–598. doi: 10.1086/301757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Depinho RA, Kaplan KL. The Hermansky-Pudlak syndrome. Report of three cases and review of pathophysiology and management considerations. Medicine. 1985;64(3):192–202. [PubMed] [Google Scholar]
- 66.Santiago Borrero PJ, Rodríguez-Pérez Y, Renta JY, et al. Genetic testing for oculocutaneous albinism type 1 and 2 and Hermansky-Pudlak syndrome type 1 and 3 mutations in Puerto Rico. Journal of Investigative Dermatology. 2006;126(1):85–90. doi: 10.1038/sj.jid.5700034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Witkop CJ, Rao GH, Gaudier F, et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Boletin de la Asociacion Medica de Puerto Rico. 1990;82(8):333–339. [PubMed] [Google Scholar]
- 68.Jorizzo J, Bolognia J. Dermatology 2-Volume Set. St. Louis, Mo, USA: Mosby; 2007. [Google Scholar]
- 69.Chediak MM. New leukocyte anomaly of constitutional and familial character. Revue d'Hématologie. 1952;7(3, article 362) [PubMed] [Google Scholar]
- 70.Fukai K, Ishii M, Kadoya A, Chanoki M, Hamada T. Chediak-Higashi syndrome: report of a case and review of the Japanese literature. The Journal of Dermatology. 1993;20(4):231–237. doi: 10.1111/j.1346-8138.1993.tb03867.x. [DOI] [PubMed] [Google Scholar]
- 71.Montoliu L, Grønskov K, Wei AH, et al. Increasing the complexity: new genes and new types of albinism. Pigment Cell & Melanoma Research. 2014;27(1):11–18. doi: 10.1111/pcmr.12167. [DOI] [PubMed] [Google Scholar]
- 72.Simeonov DR, Wang X, Wang C, et al. DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Human Mutation. 2013;34(6):827–835. doi: 10.1002/humu.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Balu K, Purohit R. Mutational analysis of TYR gene and its structural consequences in OCA1A. Gene. 2013;513(1):184–195. doi: 10.1016/j.gene.2012.09.128. [DOI] [PubMed] [Google Scholar]
- 74.Kamaraj B, Purohit R. Computational screening of disease-associated mutations in oca2 gene. Cell Biochemistry and Biophysics. 2014;68(1):97–109. doi: 10.1007/s12013-013-9697-2. [DOI] [PubMed] [Google Scholar]
- 75.Kamaraj B, Purohit R. In silico screening and molecular dynamics simulation of disease-associated nsSNP in TYRP1 gene and its structural consequences in OCA3. BioMed Research International. 2013;2013:13 pages. doi: 10.1155/2013/697051.697051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lopez VM, Decatur CL, Stamer WD, Lynch RM, McKay BS. L-DOPA is an endogenous ligand for OA1. PLoS Biology. 2008;6(9, article e236) doi: 10.1371/journal.pbio.0060236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lavado A, Jeffery G, Tovar V, de la Villa P, Montoliu L. Ectopic expression of tyrosine hydroxylase in the pigmented epithelium rescues the retinal abnormalities and visual function common in albinos in the absence of melanin. Journal of Neurochemistry. 2006;96(4):1201–1211. doi: 10.1111/j.1471-4159.2006.03657.x. [DOI] [PubMed] [Google Scholar]
- 78.Murillo-Cuesta S, Contreras J, Zurita E, et al. Melanin precursors prevent premature age-related and noise-induced hearing loss in albino mice. Pigment Cell & Melanoma Research. 2010;23(1):72–83. doi: 10.1111/j.1755-148X.2009.00646.x. [DOI] [PubMed] [Google Scholar]
- 79.Slominski A, Zmijewski MA, Pawelek J. L-tyrosine and L-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment Cell and Melanoma Research. 2012;25(1):14–27. doi: 10.1111/j.1755-148X.2011.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gargiulo A, Bonetti C, Montefusco S, et al. AAV-mediated tyrosinase gene transfer restores melanogenesis and retinal function in a model of oculo-cutaneous albinism type 1 (OCA1) Molecular Therapy. 2009;17(8):1347–1354. doi: 10.1038/mt.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Surace EM, Domenici L, Cortese K, et al. Amelioration of both functional and morphological abnormalities in the retina of a mouse model of ocular albinism following AAV-mediated gene transfer. Molecular Therapy. 2005;12(4):652–658. doi: 10.1016/j.ymthe.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Onojafe IF, Adams DR, Simeonov DR, et al. Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism. The Journal of Clinical Investigation. 2011;121(10):3914–3923. doi: 10.1172/JCI59372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fukai K, Kunimoto H, Nakajima K, et al. In vitro analysis of read-through effect of aminoglycosides to tyrosinase R278X nonsense mutation in melan-c cells. Proceedings of the 24th Annual Meeting of the Japanese Society for Pigment Cell Research (JSPCR '12); 2012; pp. 904–922. [DOI] [PubMed] [Google Scholar]
- 84.Sivamani RK, Goodarzi H, Garcia MS, et al. Biologic therapies in the treatment of psoriasis: a comprehensive evidence-based basic science and clinical review and a practical guide to tuberculosis monitoring. Clinical Reviews in Allergy and Immunology. 2013;44(2):121–140. doi: 10.1007/s12016-012-8301-7. [DOI] [PubMed] [Google Scholar]
- 85.Fitzmaurice SD, Sivamani RK, Isseroff RR. Antioxidant therapies for wound healing: a clinical guide to currently commercially available products. Skin Pharmacology and Physiology. 2011;24(3):113–126. doi: 10.1159/000322643. [DOI] [PubMed] [Google Scholar]
- 86.Sivamani RK, Ghiya M, Maibach HI. Shedding light on sunscreens and their labels. Testing policies need to match actual use. The American Journal of Preventive Medicine. 2010;38(6):679–681. doi: 10.1016/j.amepre.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 87.Sivamani RK, Crane LA, Dellavalle RP. The benefits and risks to ultraviolet (UV) tanning and it alternatives: the role of prudent sun exposure. Dermatologic Clinics. 2009;27(2):149–154. doi: 10.1016/j.det.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]