

Abstract
Microtubule depolymerization can provide force to transport different protein complexes and protein-coated beads in vitro. The underlying mechanisms are thought to play a vital role in the microtubule-dependent chromosome motions during cell division, but the relevant proteins and their exact roles are ill-defined. Thus, there is a growing need to develop assays with which to study such motility in vitro using purified components and defined biochemical milieu. Microtubules, however, are inherently unstable polymers; their switching between growth and shortening is stochastic and difficult to control. The protocols we describe here take advantage of the segmented microtubules that are made with the photoablatable stabilizing caps. Depolymerization of such segmented microtubules can be triggered with high temporal and spatial resolution, thereby assisting studies of motility at the disassembling microtubule ends. This technique can be used to carry out a quantitative analysis of the number of molecules in the fluorescently-labeled protein complexes, which move processively with dynamic microtubule ends. To optimize a signal-to-noise ratio in this and other quantitative fluorescent assays, coverslips should be treated to reduce nonspecific absorption of soluble fluorescently-labeled proteins. Detailed protocols are provided to take into account the unevenness of fluorescent illumination, and determine the intensity of a single fluorophore using equidistant Gaussian fit. Finally, we describe the use of segmented microtubules to study microtubule-dependent motions of the protein-coated microbeads, providing insights into the ability of different motor and nonmotor proteins to couple microtubule depolymerization to processive cargo motion.
Keywords: Basic Protocol, Issue 85, microscopy flow chamber, single-molecule fluorescence, laser trap, microtubule-binding protein, microtubule-dependent motor, microtubule tip-tracking
Introduction
Microtubules are highly conserved cytoskeletal structures that are important for cellular architecture, cell motility, cell division, and intracellular transport1. These dynamic polymers assemble from tubulin in the presence of GTP, and they switch spontaneously between growth and shortening2. Microtubules are very thin (only 25 nm in diameter) therefore special optical techniques to enhance contrast should be used to observe microtubules with a light microscope. Previous work with these polymers examined their dynamic behavior using differential interference contrast (DIC)3. This and similar studies in vitro revealed that under typical experimental conditions, the microtubules undergo catastrophe and switch to the depolymerization only rarely, once every 5-15 min (this frequency is for 7-15 mM soluble tubulin concentration examined at 28-32 °C)4. Different techniques have thus been proposed to induce microtubule depolymerization in a controlled manner. Microtubule shortening can be triggered by washing away soluble tubulin5,6, cutting microtubules with a laser beam7, or using segmented microtubules8, as described here. Previous work using segmented microtubules, as well as stochastically switching polymers, has found that small intracellular cargos, such as chromosomes, vesicles, and protein-coated beads, can move at the ends of the shortening microtubules9-13. This phenomenon is thought to have a direct implication for chromosome motions in mitotic cells, and the underlying mechanisms are currently under active investigation14-16.
Recently, fluorescent-based techniques, including the total internal reflection fluorescence (TIRF) microscopy, have been employed to study motility with dynamic microtubule ends17-24. The advantage of this approach is that it allows examination of interactions between microtubules and microtubule-binding proteins in real time using proteins labeled with different fluorophores. Several protein complexes were found to move processively with elongating and/or shortening microtubule ends. They include the microtubule-associated proteins Dam110,12,18, Ska119, and XMAP21520, as well as kinesin motors Kif18A21,22, MCAK23 and CENP-E24. These proteins exhibit processive tip-tracking, which is fundamentally different from that of the classic tip-tracking proteins like EB125. Although EB1 molecules and the associated partners appear to remain stably associated with dynamic microtubule ends, the individual molecules remain bound to the microtubule tip for only ~0.8 sec, rapidly exchanging with the soluble pool26. In contrast, processive tip-trackers, like Dam1, travel with microtubule ends for many microns, and their association with microtubule tips can last for many seconds. The tip association time, as well as the resulting rate of tracking, depends strongly on the number of molecules that form the tip-tracking complex27. Larger protein ensembles are usually much better tip-trackers. For example, such complex assemblies as the isolated yeast kinetochores can remain coupled to microtubule ends for hours28. Some microtubule-binding proteins, e.g. Ndc80 kinetochore protein complex, have been found to be unable to track with microtubule ends at a single molecule level, yet Ndc80 is very efficient in coupling the motion of bead cargo19,29-31. Thus, to understand the mechanism of tip-tracking by different protein complexes, as well as their biological roles, it is important to examine tip-tracking as a function of the number of molecules in the tip-tracking complex, as well as to determine the ability of these complexes to exhibit collective motility on the surface of bead cargo.
Below we provide detailed protocols to prepare and conduct experiments with segmented microtubules (Figure 1A). First, the commercially available glass slides are modified to attach short polyethylene tubing (Protocol 1). The reusable microscopy flow chamber is then assembled from such a slide and the chemically or plasma-cleaned and silanized coverslip (protocol 2)32-34. The resulting chamber volume is only 20-25 μl (or as small as 15 μl, see Note 3 in Protocol 1), including the volume of the inlet tubing. Commercially available flow chambers can also be used, but their volume is usually larger, leading to the unnecessary waste of proteins. If a larger chamber is employed, the volume of all solutions in the protocols below should be scaled proportionally. Microtubule seeds are then prepared, for example using slowly hydrolysable GTP analog, GMPCPP (Guanosine-5’-[(α,β)-methyleno]triphosphate) (protocol 3, see also Hyman et al.35). The seeds are immobilized on a cleaned coverslip and the surface is subsequently blocked to prevent nonspecific absorption of other proteins32 (protocol 4 describes seeds immobilization using digoxigenin). The segmented microtubules can then be prepared using Protocol 5. The main rationale for this approach is that dynamic microtubule polymers, which form in the presence of GTP, can be stabilized temporarily by adding the short “caps” of stable tubulin segments, which contain GMPCPP. These caps also contain Rhodamine-labeled tubulin, so they can be removed simply by illuminating the field of view with a 530-550 nm laser or mercury arc lamp (Protocol 6)36. Fluorescence intensity of the tip-tracking signal can then be used to estimate the number of molecules that travel with the disassembling microtubule ends, taking into account the unevenness of the microscope field illumination (Protocol 7). A similar approach can be used to study interactions between depolymerizing microtubules and protein-coated beads, prepared as described in27 (Protocol 8). Some proteins will readily bind to the walls of segmented microtubules, but laser tweezers can also be used to hold the bead near the microtubule wall, thereby promoting its binding.
Protocol
Required equipment: The experiments described below require a light microscope equipped for DIC and fluorescence imaging (Table 1). Bright field LED illumination can be used to significantly improve the detection of the coverslip-attached microtubule seeds37, which are difficult to observe with a regular Halogen lamp. To control liquid flow in microscopy chambers, the solutions should be exchanged with a peristaltic pump capable of flow speeds from 10-100 μl/min. A syringe pump can also be used but care should be taken to avoid air bubbles that may form when the flow speed is changed abruptly. For handling protein-coated beads, for example to bring them close to the segmented microtubule wall, a 1,064 nm continuous wave laser beam can be introduced into the microscope's optical axis and focused with a high numerical aperture objective (1.3 or higher) to produce a trap. For quantitative analysis of the fluorescent intensity of single molecules the excitation light should be provided by a laser-base source since the intensity of this light source is more stable than that generated by a mercury lamp. To minimize mechanical vibrations, the microscope should be placed on an optical table. More sophisticated equipment is required to study the movement of the beads with the depolymerizing microtubule ends under a constant force, and to measure the single-shot force signals11,38,39, these methods will be described elsewhere.
1. Manufacturing Reusable Flow Chambers
Glass slides for reusable flow chambers can be ordered from a local glass manufacturing facility using schematics in Figure 1B (see Table 2 for details about our supplier). With ultrasonic milling modify regular microscope slides (75 mm x 25 mm, 1.0 mm thick) to make two grooves 15±1 mm long, 1.0±0.1 mm wide and 0.8±0.05 mm deep. Distance between the closest ends should be 14±1 mm; this distance is optimal for a chamber assembled with 22 mm x 22 mm coverslip. See Table 2 for a list of other materials.
Place a 100 mm long polyethylene tube (O.D. 0.61 mm, Table 2) in each groove in the slide, leaving ~5 mm overhangs at the inner ends of the grooves. Fix the tubes inside the grooves with cyanoacrylate adhesive, embedding the tubes completely inside the grooves.
Fill the grooves with epoxy glue, while avoiding spilling the glue inside the tubes. Let the glue dry for ~1 day.
With a sharp razor blade cut the solidified glue mass 3-4 mm from the distal end of each attachment site, removing the parts proximal to the center of the slide. The tubes should remain inside their grooves. Removing the proximal parts will also cut and remove the inner overhangs, creating a flat surface with two tube openings.
Fill a syringe with water and test whether the tubes are working properly. If the liquid flows freely, put a drop of epoxy glue (~5 mm in diameter) at the outer ends of the groves, dry for 1 day (Figure 1D). This will make chambers more durable, so they can be used repeatedly for many months. Note 1: To make a chamber for an inverted microscope, the slides should be modified additionally to make two small holes at the opposite ends of the grooves (Figure 1C). Insert the tubes through the holes in the slide, bend the tubes and fit them tightly inside the grooves (Figure 1E). Follow steps 1.2-1.4, but remove the epoxy glue from the entire surface, which will be used to make a flow chamber. Note 2: To reduce chamber volume, use milling to make two indentations 0.050±0.005 mm deep, leaving the central part of the slide 5.0±0.5 mm wide and slightly elevated (see “etched areas” on Figures 1B and 1C). When the flow chamber is assembled (as described below), place the double-sided tape inside these indentations. Note 3: To reuse these modified slides, after finishing the experiments remove the coverslip and double-sided tape using a razor blade. Remove the sealant by peeling it off and by wiping the slide with 70% ethanol. Place the slide in a container with 1-2% of a lab dishwashing detergent, attach tubing to a peristaltic pump and perfuse 50-70 ml, follow with equal volume of deionized water, dry and store in a dust-free compartment.
2. Preparation of Coverslips
This protocol takes 6-8 hr and will help to prepare 12 coverslips. You will need one ceramic coverslip holder and 3 coverslip staining jars with lids; jar volume should be 15 ml, so each will hold 4 coverslips stacked together. A glass jar with a lid (250 ml) should be used to incubate coverslips with silane. Use regular No.1 glass coverslips (22 mm x 22 mm or 22 mm x 30 mm, see Tables 2 and 3 for a list of materials). All steps should be carried out in a fume hood, while wearing gloves.
Put the coverslips into the glass coverslip staining jars and fill the jars with acetone. Incubate for 1 hr, wash 10x with deionized water.
Incubate the coverslips 10 min with ethanol and wash again 10x with deionized water.
Prepare “piranha” solution. Put 60 ml of hydrogen peroxide solution (30% in water) in a heat-resistant glass vessel and slowly add 100 ml of sulfuric acid (final ratio of acid to hydrogen peroxide solution is 5:3). Solution will heat up, this is normal but use caution. Piranha solution is extremely corrosive! Use thick lab coat, gloves and goggles!
Fill the coverslip staining jars with “piranha” solution, close the lids and place the jars in a water bath preheated to 90 °C for 1 hr.
Pour off the “piranha” solution and discard as instructed by the safety regulations at your workplace. Wash coverslips 10x with deionized water.
Fill the coverslip staining jars with 0.1 M KOH, incubate 10 min, and wash 10x with deionized water. This will neutralize any acid residues left on the coverslips after “piranha” treatment.
Dry coverslips one at a time by holding each coverslip with Teflon coated flat-edged tweezers (to minimize damage to a glass surface) and while blowing compressed dry nitrogen. Make sure that coverslips are dried completely, because silane solution is highly reactive with water.
Stack the dried coverslips in ceramic holders (12 coverslips per holder), which should be thoroughly predried with nitrogen. Keep the ceramic holders covered to avoid dust from sticking to the coverslip surface.
Cover the bottom of 250 ml glass jar (6 cm in diameter) with Molecular Sieves, Grade 564, for water absorption.
Fill the jar with 200 ml of PlusOne Repel Silane solution and slowly immerse a ceramic holder with coverslips in a jar, close the lid and incubate for 5 min at room temperature. This will create hydrophobic coating on the coverslip surface.
Slowly remove the holder with coverslips from the jar and transfer coverslips one at a time into the coverslip staining jars filled with methanol.
Place a metal or glass pedestal into the water reservoir of a sonic bath, such that the coverslip staining jar is immersed for 2/3 of its height. Sonicate at 70 W for 20 min, changing methanol solution every 5 min, then rinse 10x with deionized water. If the silanization worked properly, the coverslips will appear dry when removed from water.
Thoroughly remove any residual water using nitrogen, as above.
Interlay the coverslips with Kimwipes to avoid surface-to-surface contact between the coverslips. Coverslips can be stored in a sealed container for several weeks at room temperature. Note 1: Steps 2.1-2.6 can be replaced by cleaning the coverslips with Plasma Cleaner for 15 min at 30 W, greatly reducing the total preparation time. Pressure inside the cleaning chamber is set at 100-200 mTorr. Both atmospheric and compressed oxygen can be used. Stack the plasma-cleaned coverslips in ceramic holders and proceed to step 2.7.
3. Preparation of GMPCPP-stabilized Microtubule Seeds
This procedure will take ~1 hr and the resulting microtubule seeds are stable for 1-2 days at room temperature. See Table 4 for a list of reagents.
- Mix on ice:
- 10 μl unlabeled tubulin (100 μM, Table 4) in BRB-80 buffer (80 mM Pipes, 1 mM EGTA, 4 mM MgCl2, pH 6.9 with KOH; supplement with 1-2 mM DTT using fresh aliquot for each experiment).
- 2.6 μl digoxigenin-labeled tubulin (Table 4). Adjust volume depending on preparation, such that the final ratio of labeled to unlabeled tubulin is ~1:10. Mix well by pipetting.
- 1.4 μl 10 mM GMPCPP (final concentration 1 mM)
Incubate 15 min at 35 °C, the seeds will grow 2-3 μm long. Adjust time if different microtubule length is desired.
Add 35 μl BRB-80 (prewarmed to 35 °C), mix by pipetting, and centrifuge for 15 min at 25,000 x g to pellet the seeds at room temperature.
Discard supernatant, wash the pellet by gently adding and removing 50 μl of warm BRB-80.
Resuspend pellet well in 25 μl BRB-80.
4. Attachment of Microtubule Seeds to the Coverslips
Protocols 4 and 5 will require 2-3 hr, so two flow chambers are used per day.
- Assemble flow chamber as per manufacturer's instructions using silanized coverslips and proceed to step 4.2. If using custom-made coverslips (Protocol 1), follow the steps below.
- Attach two pieces of double-sided tape (5 mm x 30 mm) along the central ~5 mm wide area, put silanized coverslip atop the tape, press firmly.
- Fill the chamber with BRB-80 through one of the tubes and plug both tubes with round toothpicks.
- Squeeze a small drop of two-colored Kwik Cast sealant on top of a small plastic Petri dish, and mix quickly but thoroughly using a toothpick. The sealant will turn green; apply immediately, carefully sealing all edges of the coverslip. If the sealant penetrates too deeply under the coverslip, open one of the tubes by removing the toothpick plug and apply gentle pressure to prevent sealant from leaking inside the tubes.
- Let the chamber dry for 10 min and confirm that the flow is not restricted before proceeding further.
Place the chamber on a microscope stage prewarmed to 32 °C and attach one of the tubes to a pump, which will pump the liquid out. The length of the inlet tube should be minimized to avoid the unnecessary loss of reagents: the recommended length is 5-7 cm. Immerse this end in a 0.5 ml vial with BRB-80 buffer. This and all solutions below should be prewarmed to 32-35 °C.
Apply a gentle pressure with a pump or simply lift the end of the outlet tube to squeeze out the air bubbles, which may form occasionally when the inlet tube plug is removed.
Set the pump rate at 100 μl/min. Wash with 2 chamber volumes of anti-digoxigenin antibodies diluted 1:30 in BRB-80, incubate 15 min to allow antibody adsorption.
Wash with 5-10 chamber volumes of warm BRB-80, incubate 10 min with 1% Pluronic F-127 in BRB-80 to block the hydrophobic surface of silanized coverslip.
Wash with 5-10 chamber volumes of motility buffer (BRB-80 supplemented with 0.4 mg/ml of casein).
Reduce the pump speed to 10 μl/min and perfuse microtubule seeds diluted 1:200-1:600 in 30-40 μl motility buffer. Incubate 15 min to promote binding of the seeds to the coverslip-adsorbed antibodies.
Wash the chamber at 100 μl/min with 400 μl of motility buffer to remove any unbound material. Note 1: The resulting density of seeds should be 10-30 per microscope field (Figure 2A). To troubleshoot, use fluorescently labeled tubulin during polymerization (step 3.1) for easier detection of the coverslip-attached seeds. Note 2: Axonemes prepared from Chlamydomonas40 or other biological sources, as well as the pellicles of lysed and deciliated Tetrahymena cells41 can also be used as microtubule nucleators. These nucleators are useful for creating small microtubule arrays, and are preferred when microtubules with specific number of protofilaments are desired (GMPCPP seed nucleates one microtubule that contains ≥14 protofilaments42). These structures can be attached to the cleaned coverslips by nonspecific absorption, but the attachment is usually less stable compared with antibody-based attachment, especially when using the silanized coverslips.
5. Preparation of Segmented Microtubules
All solution volumes below are for chamber volume 15-20 μl; increase proportionally if larger chamber is used.
Prewarm unlabeled tubulin mix (45 μl motility buffer supplemented with 1 mM Mg-GTP and with 10-15 μM unlabeled tubulin) for 30 sec at 35 °C. Perfuse at 30 μl/min.
Monitor microtubule growth with DIC optics (Figure 2B, Video 1). During 5-7 min incubation the microtubules usually grow ~10 μm long.
Prepare Rhodamine-tubulin mix (65 μl motility buffer supplemented with 0.5 mM GMPCPP and 2-5 μM of Rhodamine-labeled tubulin with 0.5-1 molar ratio of Rhodamine to tubulin) and warm the solution at 35 °C for 30 sec.
Perfuse immediately at 30 μl/min. Incubate for 8-10 min to promote formation of stable fluorescent caps at the microtubule tips. Stable microtubule segments will also nucleate spontaneously and will be visible with DIC optics.
Wash the chamber well with 100 μl of motility buffer at 20 μl/min to remove tubulins and nucleotides, as well as soluble microtubule fragments.
With DIC, confirm that the microtubules are visible (Figure 2D); their number, however, should decrease because many microtubules disassemble during capping (Video 2 shows a typical field with segmented microtubules). Note 1: Segmented microtubules are very stable and can be used for at least 2 hr. However, the lifetime of these microtubules decreases with excessive solution exchange, or if 2-mercaptoethanol is used in the imaging buffer.
6. Experimental Observation of the Protein Tracking with Depolymerizing Microtubule Ends
Introduce 30-50 μl of fluorescent protein (0.1-20 nM) into the chamber at 10 μl/min. If protein sticking to the coverslip is evident, supplement the motility buffer with 4-8 mg/ml BSA. Alexa488-Dam1 tip-tracking additionally requires 10 mM DTT or 0.5-1% βME10.
Limit the illumination field using a microscope field diaphragm to avoid the unnecessary bleaching and disassembling of the microtubules.
Start video acquisition using GFP filter cube, then switch to Rhodamine filter cube without interrupting the image recording. The red segments at the microtubule ends should be clearly visible; they will begin to fade and disintegrate quickly (Video 3).
Continue to illuminate until the caps almost disappear (usually for 10-20 sec but this time will be longer for the caps grown with a lower Rhodamine labeling ratio), and switch back to the GFP channel to record protein tracking with microtubule disassembly.
Analyze the resulting sequences by constructing kymographs, i.e. two-dimensional images that show fluorescence intensity along microtubule axis for various times during observation) using MetaMorph, freely available ImageJ or other image processing software (Figure 2E). Note 1: Acquisition rate should be adjusted depending on the timing of the observed events. The recommended rate is 2-3 frames per second (fps) for the slow-moving, ring-sized Dam1 complexes27 but acquisition time for single molecules should be >20 fps19. Note 2: A highly sensitive EMCCD, e.g. ANDOR iXon3, is required for the fast recording of tip-tracking events with depolymerizing microtubules. The recommended settings for Andor iXon3 camera are: gain 5x, EM gain 200, 1 MHz readout speed, 16-bit sensor mode, 80 msec exposure time. Note 3: Using TIRF microscopy will improve signal-to-noise ratio, however, shorter microtubules should be used, such that the fluorescent stabilizing caps remain within the reach of evanescent field.
7. Quantitative Analysis of the Molecular Size of Microtubule Tip-tracking Complexes
The rationale for this approach is to determine the number of molecules in a tip-tracking complex by finding the ratio of total fluorescent intensity of the tip-tracking complex to the intensity of a single fluorophore. This approach can be applied to GFP-protein fusions and proteins labeled with fluorescent dyes, but it may underestimate the number of molecules in the tip-tracking complexes if some protein molecules in the preparations are not fluorescent.
- Record photobleaching kinetics for the fluorescently labeled protein molecules.
- Assemble regular microscopy chamber using a nonmodified glass slide, two strips of double-sided tape, and a clean coverslip, which can be prepared using the entire Protocol 2, or only steps 2.1-2.6 of this protocol.
- Add approximately 50 nM protein in motility buffer, wash briefly with motility buffer and seal the chamber with VALAP (Table 4). Optimize protein concentration to obtain the field with evenly dispersed spots (Figure 3A), representing single molecules and their small aggregates (trimers and tetramers, which may form spontaneously in the solution or may appear when several single molecules are close together and cannot be resolved). This step is very important for obtaining a multi-peak distribution of photobleaching steps and accurate determination of a step size (see below).
- Minimize the illumination laser intensity at which the individual fluorescent spots are still visible; with lower illumination the photobleaching time is extended, so longer photobleaching traces can be obtained. Also minimize the exposure time to reduce the probability of more than one fluorophore bleaching during one frame. The recommended setting for Andor iXon3 camera: gain 5.0x, EM gain 999, 10 MHz readout speed, 50-100 msec exposure time.
- Focus at the surface of the coverslip, close the illumination shutter, move to a fresh field, open the illumination shutter and acquire images until all complexes have bleached (thereafter referred to as img(x,y)).
- Correct the acquired images for unevenness of illumination (Figure 3B).
- Prepare solution of any fluorophore, e.g. 1 µM Fluorescein isothiocyanate (FITC) in BRB-80. Such solution can be prepared in advance, aliquoted and stored at -20 °C.
- Assemble a chamber as in Section 7.1.1 but use a regular coverslip. Add fluorophore solution and seal the chamber using VALAP.
- Collect >50 images of the entire microscope field: move the stage to a new unbleached area while the illumination shutter is closed, and acquire the images immediately after opening the shutter.
- Create average projection of this stack and filter with Gaussian blur with 5 pixel radius using ImageJ or other software (Figure 3C). The resulting image represents the distribution of the illumination intensity of the field (Illum(x,y), where x and y correspond to pixel's coordinates).
- Determine the maximum pixel brightness of this image (Max(Illum)).
- With the closed illumination shutter and using same camera settings as in Section 7.2.3 acquire one image, determine the average pixel intensity of this image; this value corresponds to CN, camera noise.
- Use the above values and image (Illum(x,y)) to normalize the experimental image (img(x,y)) using the following expression:
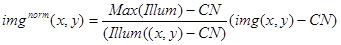 Use the resulting image imgnorm(x,y) for the quantitative analysis of the brightness of the stationary fluorescent complexes, and also to normalize the images with tip-tacking complexes (Figure 3D).
Use the resulting image imgnorm(x,y) for the quantitative analysis of the brightness of the stationary fluorescent complexes, and also to normalize the images with tip-tacking complexes (Figure 3D).
- Determine intensity of a single fluorophore.
- Using normalized images imgnorm(x,y) and any image processing software select a fluorescent spot with a circular region (5-6 pixels in diameter) and determine its integral intensity for all frames, generating the photobleaching traces. Avoid very bright dots (> 5-fold brighter than the dimmer ones).
- Using the same circular region tool, select at least 3 spot-free areas, generate the corresponding photobleaching traces, average and fit with exponential decay function.
- Tabulate this background intensity curve to match the experimental time points and subtract it from the photobleaching curves.
- Smooth the photobleaching curves (average with the sliding window of 3-5 points). Visually inspect the resulting curves and discard any curve that shows an abrupt increase in fluorescence or lack of obvious bleaching (Figure 3E).
- For each of the remaining curves (usually 50-70% of the total number of curves), visually select the final plateau, when the fluorescent spot has bleached. Shorten this segment to leave only ~100 points and average these intensities. Subtract this value from the shortened photobleaching curve to minimize small variations is the background levels and to reduce the size of the background peak (below).
- Plot a histogram of the intensities for all time points from 20 or more photobleaching curves (>1,000 time points). The histogram should exhibit at least 4 distinct peaks (see Note 2).
- Fit the histogram with equidistant Gaussian distribution10,43 using MATLAB, Mathematica or similar software (Figure 3F):
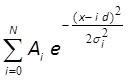 where Ai and σi , d and N are fitting parameters. Parameters Ai and σi correspond to amplitude and width of i-th peak; d is distance between peaks; N is integer number, which corresponds to the total number of peaks in the distribution. If centers of the first 3 or more peaks show a visually good match to the fitted line, the distance between these peaks (parameter d) corresponds to a fluorescent intensity of a single fluorophore. Note 1: Number of examined dots (Section 7.3.6) should be increased if the microscopy system exhibits significant vibrations or there is another source of noise (e.g. unstable laser beam). Note 2: It is essential to obtain >3 peaks for accurate analysis with equidistant Gaussian fit. If fewer peaks are obtained, the false (e.g. double) step size could be obtained when the illumination conditions are not optimal, e.g. when the dots bleach too fast and single steps are not resolved well.
where Ai and σi , d and N are fitting parameters. Parameters Ai and σi correspond to amplitude and width of i-th peak; d is distance between peaks; N is integer number, which corresponds to the total number of peaks in the distribution. If centers of the first 3 or more peaks show a visually good match to the fitted line, the distance between these peaks (parameter d) corresponds to a fluorescent intensity of a single fluorophore. Note 1: Number of examined dots (Section 7.3.6) should be increased if the microscopy system exhibits significant vibrations or there is another source of noise (e.g. unstable laser beam). Note 2: It is essential to obtain >3 peaks for accurate analysis with equidistant Gaussian fit. If fewer peaks are obtained, the false (e.g. double) step size could be obtained when the illumination conditions are not optimal, e.g. when the dots bleach too fast and single steps are not resolved well.
- Determine the molecular size of the tip-tracking complex.
- Use images collected with Protocol 6 and select the first 2-4 frames, which were acquired immediately after opening the shutter. If the field was illuminated for some time before the tip-tracking was observed, estimate the original intensity from the kinetics of photobleaching under the same experimental conditions.
- Average the selected frames and normalize the resulting image as in Sections 7.2.5-7.2.7.
- Measure integral intensity of the fluorescent tip-tracking complex using the same region size as in Section 7.3.1.
- Measure integral intensity of 3 background areas located near the tip-tracking complex and using the same region, average these values and subtract from the intensity of the tip-tracking complex from Section 7.4.3.
- Calculate the number of fluorophore molecules in the complex by dividing the fluorescent intensity obtained in Section 7.4.4 by the intensity of the single fluorophore obtained in Section 7.3.5. Note 1: It is desirable that the illumination and acquisition settings for protocol 7.4 are the same as in protocol 7.1. If either the exposure time or laser intensity was adjusted during these steps, the resulting fluorescent values should be scaled accordingly. However, the accuracy of such scaling should be verified by imaging the same sample (e.g. fluorescent time) under these different conditions and calculating the ratio of the resulting intensities.
8. Microtubule Tip-tracking by the Protein Coated Beads
Carry out experiments with tip-tracking beads by triggering MT disassembly as in protocol 6. Intensity of DIC light source should be reduced to allow viewing Rhodamine fluorescence simultaneously with DIC imaging.
Prepare the beads as in Grishchuk et al.10 and Asbury et al.11 Introduce 30-50 μl of bead suspension into the chamber at 10 μl/min. The suggested bead concentration is 10-16-10-17 M.
If using upright microscope, remove the chamber from the microscope stage and invert it for 5-10 min to allow beads to sediment at the coverslip. This promotes a better binding of the bead to the microtubules tethered to the coverslip, but this procedure is not successful with 0.5 μm polystyrene beads, which show little gravity-based sedimentation during such a short time.
Select a bead that is attached to the coverslip-tethered microtubule; the bead should move in a clear arc44 (Figure 4A). The tethered bead should be located 1-3 μm away from the coverslip surface, which is clearly visible owing to the occasional coverslip-attached beads that remain motionless.
Switch to Rhodamine filter cube and begin collecting images while using DIC illumination.
Open shutter to illuminate the imaging field (restricted with a field diaphragm) with a mercury lamp or 530-550 nm laser. Continue recording until the bead detaches or moves with the disassembling microtubule end (Figures 4D, 4F, and 4G). Note 1: Optical trap can be used to promote interaction between the microtubule wall and protein-coated bead. This is especially useful when working with beads coated with kinesin motors (Figures 4E-G). Follow same protocols as above but supplement motility buffer with 2 mM Mg-ATP. In step 8.3, capture a free floating bead with the 1,064 nm laser beam, move the stage to bring the trapped bead closer to the segmented microtubule wall. Begin imaging with low light DIC and via the Rhodamine filter cube and wait for the bead to begin walking toward the capped microtubule end. After the directed bead motion is observed, close the shutter for trapping beam and open the shutter for fluorescent illumination. Continue recording until the bead detaches or tracks with the disassembling microtubule end.
Representative Results
Protein tracking with depolymerizing microtubule ends. Yeast kinetochore component Dam1 is by far the best tip-tracker of the depolymerizing microtubule ends14. This 10-subunit complex labeled with GFP can be readily expressed and purified from bacterial cells18,38, so we recommend using it as a positive control for the tip-tracking assay. A fluorescent protein that tracks with the depolymerizing end of a microtubule is seen as a bright fluorescent spot steadily moving towards the coverslip-attached seed (Video 3). It will also appear as an oblique line on a corresponding kymograph (Figure 2E). In contrast, if the protein fails to tip-track, the microtubule shortens without showing enrichment in fluorescent signal at the microtubule end, such as seen for human Ndc80-GFP complex19 (Figure 2F). When the processive tracking is observed, the rate of microtubule shortening can be determined by tracking the moving dot or by measuring the slope of the oblique line on the corresponding kymograph. This can give information about the strength of protein-microtubule end binding45. Proteins that bind strongly to the microtubule, like the Dam1 ring complex, slow down the rate of microtubule depolymerization. This effect, however, depends strongly on the size of the tip-tracking complex and the small complexes may exert little or no effect on the rate of microtubule disassembly27. Thus, the change in the rate of microtubule shortening should be interpreted in the context of the size of the tip-tracking complex. Under some conditions, the tracking may be accompanied by the increase in brightness of the tip-tracking complex, as the depolymerizing end “collects” the protein that was bound to the microtubule lattice. In this case the rate of depolymerization often slows down concomitantly with the increasing size of the tip-tracking complex. In other cases the relationship is more complex. For example, many subunits of the Dam1 protein complex, in which GFP was conjugated at the N-terminus of Dam1 subunit, can be collected at the end of the shortening microtubule (Figure 2G). Microtubule disassembly eventually stalls; presumably because the force of microtubule depolymerization is not strong enough to move complexes that are too big. The disassembly often resumes after a decrease in tip brightness, which indicates dissociation of some of the tip-associated Dam1 subunits10. Careful examination of these relationships is important because the microtubule-binding proteins that do not track processively can also slow down the rate of microtubule disassembly19,31,46.
Bead tracking with depolymerizing microtubule ends. Many microtubule-binding proteins have already been identified as couplers for the microbead motion with dynamic microtubule ends5,11,27,39,47. Strikingly, even the Ndc80 protein complex can sustain the microtubule end tip-tracking by the beads29,30. Usually, binding between protein-coated beads and microtubules is strong, so when beads are added to the microscopy chamber they bind readily to the segmented, coverslip-attached microtubules (Figure 4A). It is not always possible to definitively see with DIC whether the bead became attached to only one microtubule. We have developed several criteria to avoid recording the beads that have formed attachments to several microtubules. First, a bead that is attached to one microtubule should show an arc-like motion, as the microtubule pivots slightly around the site of its growth from the cover-slip attached seed (Figure 4B). Second, when the stabilizing cap is illuminated, only one red segment should be seen distal from the seed and bead (Figure 4C). The cap can often be seen following the bead’s arc-like motion until the cap bleaches (Video 4). Third, the bead motion (or its detachment) should be observed shortly after the cap has bleached and the direction of bead motion should be consistent with microtubule orientation, which was deduced based on the above observations. Fourth, as the microtubule shortens and the bead moves towards the seed, the amplitude of the arc-like oscillations should decrease (Figure 4D).
When the tracking by the bead is very processive, as shown in Figure 4D, these criteria are often satisfied. However, if the bead tracking is not processive, after some initial directed motion the bead detaches and begins diffusing randomly. With larger beads, e.g. 1 μm glass beads, this thermal motion is slow enough that it could be misinterpreted as an arc-containing motion with the shortening microtubule end. A formal criterion based on the magnitude of bead’s deviation from the linear track can be used to discriminate such events29. In our previous work we calculated average bead excursions across the microtubule axis and estimated that if this value exceeded 0.4 μm/1 μm of the microtubule-parallel bead motion, the bead was likely to diffuse randomly with 95% confidence. Using this measure we found that even in “control” bead preparations, e.g. beads coated with nonspecific antibody or streptavidin-coated beads used in conjunction with biotinylated microtubules, 5% of beads were judged to move processively. If greater accuracy is desired to monitor motions of beads with low processivity, we recommend using a laser trap, where the directed bead motions are easier to identify.
Laser trap should also be used if the beads fail to form lasting attachments to stable microtubules. We used a laser beam to capture beads coated with different protein constructs of plus-end-directed kinesin CENP-E and to “launch” such beads on the segmented microtubule walls (Figure 4E)24. In the presence of ATP these beads move rapidly (20-25 μm/min) towards the capped microtubule plus ends; on a kymograph, such motion results in the oblique line directed toward the cap (Figures 4F and 4G). After the cap is destroyed, a bead coated with the truncated CENP-E protein detaches from the microtubule (green arrow on Figure 4F). However, the full length CENP-E protein can sustain bead motion in the reverse direction, i.e. toward the minus microtubule end (Video 5). As a result, the kymograph for such beads shows a zigzag pattern, indicating the presence of both plus-end and minus-end directed motions by the beads coated with full length, and not truncated CENP-E. We expect that other microtubule-dependent motors may exhibit similar motility5,13, and hope that the assays we describe will help to advance the studies of microtubule tip-tracking by these and other microtubule-binding proteins.
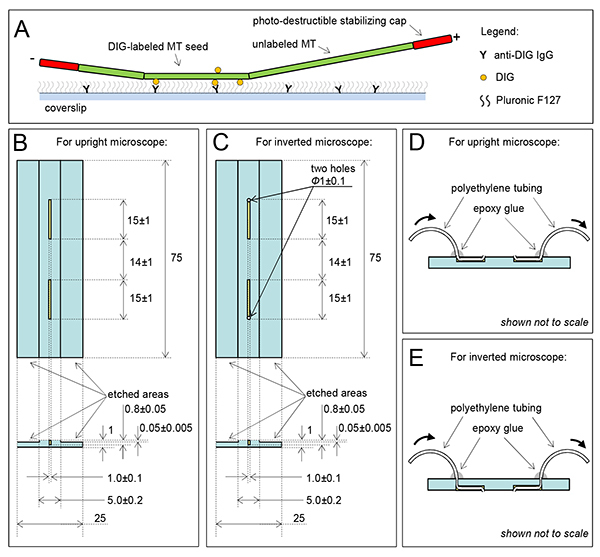 Figure 1. Flow chamber for in vitro studies with segmented or dynamic microtubules.
A. Diagram of a segmented microtubule (MT). Stable MT seeds are prepared with digoxigenin (DIG)-labeled tubulin and GMPCPP, attached to coverslip via anti-DIG antibodies. Nonspecific binding of tubulin and other proteins is blocked with Pluronic F127. Microtubules are extended from the seeds using unlabeled tubulin and GTP, and these extensions are capped with short segments containing Rhodamine labeled tubulin and GMPCPP (in red). MT Plus end (labeled with +) can usually be distinguished from a much shorter minus end (-). B and C. Detailed schemes of the modified flow chamber slides to use with upright and inverted microscopes. Sonic slots are shown in yellow. Numbers are in millimeters. D and E. Side views of the modified slides (not to scale) with attached tubes; thick arrows indicate liquid flow through the tubes after the chambers are fully assembled (not shown). Click here to view larger image.
Figure 1. Flow chamber for in vitro studies with segmented or dynamic microtubules.
A. Diagram of a segmented microtubule (MT). Stable MT seeds are prepared with digoxigenin (DIG)-labeled tubulin and GMPCPP, attached to coverslip via anti-DIG antibodies. Nonspecific binding of tubulin and other proteins is blocked with Pluronic F127. Microtubules are extended from the seeds using unlabeled tubulin and GTP, and these extensions are capped with short segments containing Rhodamine labeled tubulin and GMPCPP (in red). MT Plus end (labeled with +) can usually be distinguished from a much shorter minus end (-). B and C. Detailed schemes of the modified flow chamber slides to use with upright and inverted microscopes. Sonic slots are shown in yellow. Numbers are in millimeters. D and E. Side views of the modified slides (not to scale) with attached tubes; thick arrows indicate liquid flow through the tubes after the chambers are fully assembled (not shown). Click here to view larger image.
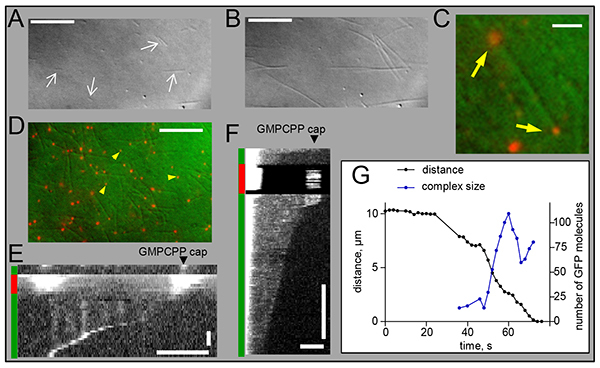 Figure 2. Preparation of segmented microtubules and typical protein tip-tracking results.
A. Coverslip-attached microtubule seeds visualized with DIC optics (step 4.8 of the protocol); arrows point to some of the seeds. The image is an average of 10 frames acquired sequentially (100 msec exposure each). B. Same microscope field after tubulin and GTP were incubated for 8 min at 32 °C, microtubules are seen emanating from the seeds (step 5.2 of the protocol). C. A pseudo-colored image showing a segmented microtubule viewed in DIC (green) and the Rhodamine-labeled stabilizing caps (arrows), viewed with epifluorescence (red). D. Same image as on panel C but with a larger field of view. Some Rhodamine-containing microtubule fragments, which nucleate spontaneously in the solution during step 5.2 of the protocol, are also seen (arrowheads). E. Kymograph of the Dam1–GFP protein tracking the disassembling microtubule end. Color-coded bars on the left show a fluorescent channel used for acquisition of the corresponding portion of the kymograph. Vertical fluorescent lines are created by the Dam1-GFP complexes that bind to the microtubule wall and show little motion until the microtubule end comes by. F. Kymograph as on E but with GFP labeled Ndc80 complex19. Initially, microtubule is decorated uniformly, although increased GFP fluorescence is observed at the capped end after Rhodamine excitation. This increase depends on the soluble pool of Ndc80-GFP protein, but the underlying mechanism is not known. After the cap disintegrates, microtubule shortening becomes evident but there is little enrichment of fluorescence at the microtubule tip, indicating a lack of tip-tracking. G. Distance from the tip-tracking Dam1 spot to the axoneme, which was used for microtubule nucleation in this experiment10, was measured using MetaMorph Track Points drop-in. The initial brightness of the moving complex corresponds to about 15 subunits: enough to form a single Dam1 ring. The brightness of the moving complex was normalized to the intensity of coverslip-attached, motionless fluorescent spots to correct for bleaching. Horizontal scale bars: panels A, B, D (10 μm), panels C, F, E (3 μm). Vertical scale bars: 10 sec. Click here to view larger image.
Figure 2. Preparation of segmented microtubules and typical protein tip-tracking results.
A. Coverslip-attached microtubule seeds visualized with DIC optics (step 4.8 of the protocol); arrows point to some of the seeds. The image is an average of 10 frames acquired sequentially (100 msec exposure each). B. Same microscope field after tubulin and GTP were incubated for 8 min at 32 °C, microtubules are seen emanating from the seeds (step 5.2 of the protocol). C. A pseudo-colored image showing a segmented microtubule viewed in DIC (green) and the Rhodamine-labeled stabilizing caps (arrows), viewed with epifluorescence (red). D. Same image as on panel C but with a larger field of view. Some Rhodamine-containing microtubule fragments, which nucleate spontaneously in the solution during step 5.2 of the protocol, are also seen (arrowheads). E. Kymograph of the Dam1–GFP protein tracking the disassembling microtubule end. Color-coded bars on the left show a fluorescent channel used for acquisition of the corresponding portion of the kymograph. Vertical fluorescent lines are created by the Dam1-GFP complexes that bind to the microtubule wall and show little motion until the microtubule end comes by. F. Kymograph as on E but with GFP labeled Ndc80 complex19. Initially, microtubule is decorated uniformly, although increased GFP fluorescence is observed at the capped end after Rhodamine excitation. This increase depends on the soluble pool of Ndc80-GFP protein, but the underlying mechanism is not known. After the cap disintegrates, microtubule shortening becomes evident but there is little enrichment of fluorescence at the microtubule tip, indicating a lack of tip-tracking. G. Distance from the tip-tracking Dam1 spot to the axoneme, which was used for microtubule nucleation in this experiment10, was measured using MetaMorph Track Points drop-in. The initial brightness of the moving complex corresponds to about 15 subunits: enough to form a single Dam1 ring. The brightness of the moving complex was normalized to the intensity of coverslip-attached, motionless fluorescent spots to correct for bleaching. Horizontal scale bars: panels A, B, D (10 μm), panels C, F, E (3 μm). Vertical scale bars: 10 sec. Click here to view larger image.
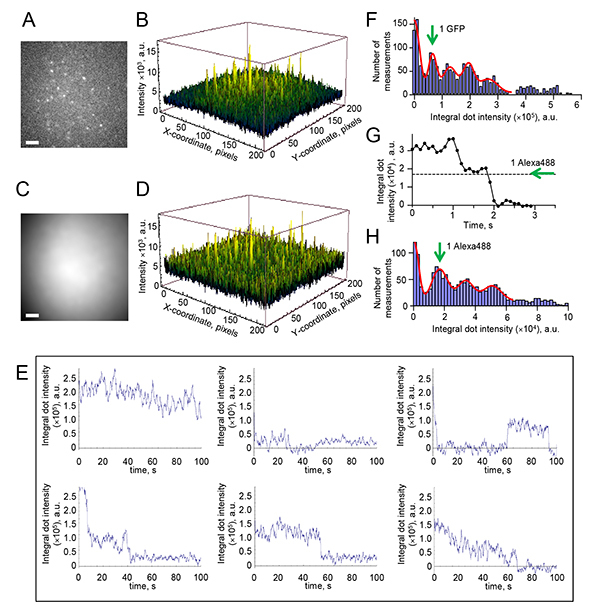 Figure 3. Quantitative analysis of fluorescent signals.A. Example image of the microscope field with Dam1-Alexa488 complexes bound nonspecifically to the coverslip surface. Note heterogeneity in dots brightness and unevenness of the illumination. Scale bar is 2 µm. B. Quantitative representation of the same image as in panel A. C. Image of soluble fluorescent dye is used to quantify the two-dimensional laser intensity profile. Image was generated by averaging 50 images, and using Gaussian filter, as described in Section 7.2.4. Scale bar is 2 µm. D. Normalized image from panel A plotted as intensity surface using Mathematica 9 (Wolfram Research). Note that this surface is more flat than on B and the peaks are less heterogeneous in height. E. Examples of the photobleaching curves obtained with CENP-E-GFP protein. The integral intensity of each dot was collected, normalized to take into account unevenness of illumination and the curves were smoothed (average with the sliding window of 5 points). Signals in the upper row were excluded from further analysis for reasons described in Section 7.3.4. Curves like those shown in the lower row were further processed and used to build a histogram on panel F, as described in Sections 7.3.4-7.3.5. F. Histogram of integral intensities collected from 22 bleaching CENP-E-GFP dots (total 1,900 data points). Red line is fitting with equidistant Gaussian function; 5 distinct peaks are seen. The integral intensity of a single GFP fluorophore determined from this histogram was (64.7±1.1) x 104 a.u. G. Typical photobleaching curve for Dam1-Alexa488, processed as described in protocol 7.3.5. H. Histogram of integral intensities collected from 48 photobleaching Dam1-Alexa488 dots (total 1,548 data points); 4 distinct peaks are seen. The integral intensity of one Alexa488 molecule under these conditions was (17.4±0.8) x 103 a.u. Click here to view larger image.
Figure 3. Quantitative analysis of fluorescent signals.A. Example image of the microscope field with Dam1-Alexa488 complexes bound nonspecifically to the coverslip surface. Note heterogeneity in dots brightness and unevenness of the illumination. Scale bar is 2 µm. B. Quantitative representation of the same image as in panel A. C. Image of soluble fluorescent dye is used to quantify the two-dimensional laser intensity profile. Image was generated by averaging 50 images, and using Gaussian filter, as described in Section 7.2.4. Scale bar is 2 µm. D. Normalized image from panel A plotted as intensity surface using Mathematica 9 (Wolfram Research). Note that this surface is more flat than on B and the peaks are less heterogeneous in height. E. Examples of the photobleaching curves obtained with CENP-E-GFP protein. The integral intensity of each dot was collected, normalized to take into account unevenness of illumination and the curves were smoothed (average with the sliding window of 5 points). Signals in the upper row were excluded from further analysis for reasons described in Section 7.3.4. Curves like those shown in the lower row were further processed and used to build a histogram on panel F, as described in Sections 7.3.4-7.3.5. F. Histogram of integral intensities collected from 22 bleaching CENP-E-GFP dots (total 1,900 data points). Red line is fitting with equidistant Gaussian function; 5 distinct peaks are seen. The integral intensity of a single GFP fluorophore determined from this histogram was (64.7±1.1) x 104 a.u. G. Typical photobleaching curve for Dam1-Alexa488, processed as described in protocol 7.3.5. H. Histogram of integral intensities collected from 48 photobleaching Dam1-Alexa488 dots (total 1,548 data points); 4 distinct peaks are seen. The integral intensity of one Alexa488 molecule under these conditions was (17.4±0.8) x 103 a.u. Click here to view larger image.
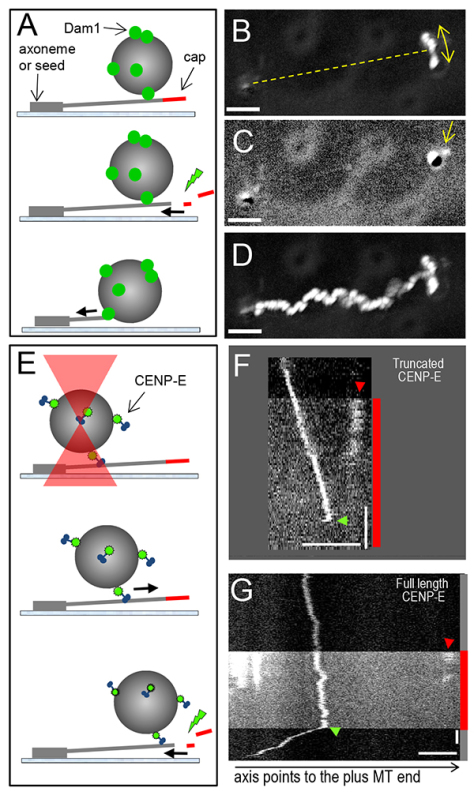 Figure 4. Illustration of the motions of microtubule-associated beads. A. Schematic of the experiment with beads coated with microtubule-binding proteins. Thick arrows indicate direction of microtubule disassembly. B. Maximum intensity projection of the DIC images of one GFP-Dam1-coated bead, which was stably attached to a segmented microtubule (based on 26 images). Bead showed the arc-like motion (arrow), suggesting that it was attached to a microtubule oriented as shown with a broken line. C. Single image of the same bead as in panel B, but taken during step 8.6 of the protocol immediately after the fluorescent shutter was opened. Arrow points to the fluorescent cap located near the bead, but on the opposite side from the microtubule attachment site. D. A maximum intensity projection shows the trajectory of the bead’s motion with the disassembling microtubule. Image was created from 115 frames acquired sequentially from the start of imaging (note the arc-like projection) and until bead’s detachment (Video 4). E. Schematic of the experiment with beads coated with plus-end-directed kinesin CENP-E. The bead is brought in contact with microtubule using laser trap. The trap is then switched off and the bead starts moving towards microtubule plus end. After the microtubule-stabilizing cap is remove and microtubule disassembled, the bead reverses the direction of its motion. F. Kymograph shows a 0.5 μm bead coated with truncated X. laevis CENP-E kinesin moving towards microtubule plus end24. After the bead traveled about 1 μm, fluorescent shutter was opened (red bar), and the stabilizing cap became visible (red arrowhead). The bead detached suddenly (green arrowhead), presumably when the motor encountered the shortening microtubule end. G. Same experiment but when the bead was coated with full length CENP-E. Note that this bead suddenly changed the direction of its motion (green arrowhead). Horizontal scale bars: 3 μm. Vertical scale bars: 10 sec. Click here to view larger image.
Figure 4. Illustration of the motions of microtubule-associated beads. A. Schematic of the experiment with beads coated with microtubule-binding proteins. Thick arrows indicate direction of microtubule disassembly. B. Maximum intensity projection of the DIC images of one GFP-Dam1-coated bead, which was stably attached to a segmented microtubule (based on 26 images). Bead showed the arc-like motion (arrow), suggesting that it was attached to a microtubule oriented as shown with a broken line. C. Single image of the same bead as in panel B, but taken during step 8.6 of the protocol immediately after the fluorescent shutter was opened. Arrow points to the fluorescent cap located near the bead, but on the opposite side from the microtubule attachment site. D. A maximum intensity projection shows the trajectory of the bead’s motion with the disassembling microtubule. Image was created from 115 frames acquired sequentially from the start of imaging (note the arc-like projection) and until bead’s detachment (Video 4). E. Schematic of the experiment with beads coated with plus-end-directed kinesin CENP-E. The bead is brought in contact with microtubule using laser trap. The trap is then switched off and the bead starts moving towards microtubule plus end. After the microtubule-stabilizing cap is remove and microtubule disassembled, the bead reverses the direction of its motion. F. Kymograph shows a 0.5 μm bead coated with truncated X. laevis CENP-E kinesin moving towards microtubule plus end24. After the bead traveled about 1 μm, fluorescent shutter was opened (red bar), and the stabilizing cap became visible (red arrowhead). The bead detached suddenly (green arrowhead), presumably when the motor encountered the shortening microtubule end. G. Same experiment but when the bead was coated with full length CENP-E. Note that this bead suddenly changed the direction of its motion (green arrowhead). Horizontal scale bars: 3 μm. Vertical scale bars: 10 sec. Click here to view larger image.
Video 1. Preparation of segmented microtubules. Video shows DIC images of a microscope field during preparation of segmented microtubules. Imaging starts after the microtubule seeds have already attached to the coverslip. Small round dots are from particulates found in our preparation of anti-digoxigenin antibodies. After tubulin and GTP are added, microtubules begin to elongate from the seeds (step 5.2). After they reach the desired length (8-15 μm), the solution is exchanged quickly to introduce Rhodaminated tubulin and GMPCPP. During the exchange, microtubule polymers bend in the direction of flow but after the pump is stopped they assume the unstrained orientation. During the following “capping” stage, some tubulin begins to polymerize spontaneously and small microtubule fragments are seen floating in the chamber. They are removed during the following wash (step 5.5), which also removes all soluble tubulin and nucleotides. During these stages some microtubules fail to assemble the robust caps, and when soluble tubulin is washed away they disassemble quickly. The capped microtubules, however, are stable for several hours unless they are illuminated; when Rhodamine is excited, the caps become fragmented, and the uncapped microtubule segments become exposed. Images were acquired every second, played at 30 fps.
Video 2. Uncapping of segmented microtubules. Video shows a fully assembled segmented microtubule via DIC, followed by fluorescent images of the same microtubule via the Rhodamine filter cube. A bright red segment at the end of the microtubule (arrow) moves in arc and fades quickly due to photo-bleaching. Images acquired at 6 fps and played at 10 fps.
Video 3. Protein tracking with depolymerizing microtubule end. A segmented microtubule (MT) is visible thanks to the decoration by GFP-labeled Dam1, which forms distinct dots (green images). Rhodamine fluorescence is then excited and the red cap becomes visible, moving in a gentle arc at the end of the microtubule (red images); the rest of the microtubule is not visible because the labeled tubulin became incorporated only at the polymer’s end. The cap bleaches fast, begins to crumble and the fluorescent cube is switched back to GFP, just in time to see the start of shortening of the microtubule segment that does not have the Rhodaminated tubulin and GMPCPP. When depolymerizing end reaches the Dam1 dots, the microtubule end becomes bright green. Subsequently, a green fluorescent spot is seen moving with the disassembling microtubule, illustrating the processive tip-tracking. Concentration of Dam1 was 3 nM, images were acquired and played at 0.4 fps. Scale bar 5 μm.
Video 4. Bead tracking with depolymerizing microtubule ends. Glass bead coated with Dam1 is seen moving in an arc, indicating that it is tethered to the coverslip by a microtubule. When Rhodamine fluorescence is excited, the red cap is seen distally from the bead and moving in synchrony with the bead’s motion (images in red). Soon, the bead starts moving directionally, while its arc-like motion decreases in amplitude (Figure 4D). DIC images were acquired via Rhodamine filter cube every 300 msec and played 6 fps; bead is 1 μm, scale bar 5 μm.
Video 5. Kinesin-coated bead moving bi-directionally on the segmented microtubule. Imaging starts after the bead coated with full length CENP-E kinesin was placed on the microtubule (MT) wall. Arrow points to the initial bead position. The bead is seen moving away from the arrow, as the motor walks toward the plus microtubule end. Soon after the plus-end cap is illuminated (arrowhead, red images), the bead begins to move in the opposite direction. Only full length CENP-E kinesin can couple motion of the bead to the shortening microtubule end. Beads coated with truncated CENP-E can move toward the plus-MT end but they detach soon after the microtubule depolymerization is triggered (compare Figures 4F and 4G). DIC images were acquired every 1 sec and played at 16 fps; bead is 0.5 μm.
Discussion
Many single molecule assays nowadays routinely use specially treated coverslips to drastically reduce nonspecific protein sticking. The procedure we describe here is a modification of the original protocol developed in Howard lab32, and we find that silanizing the coverslips is well worth the effort even with DIC-based bead assays, which do not use fluorescence. Chambers assembled with such coverslips show much cleaner surfaces, and the results obtained in the presence of soluble microtubule-binding protein are more reproducible, especially if a very low protein concentration is used.
The most critical steps for successful preparation of segmented MTs are:
optimization of tubulin concentration and incubation times (steps 5.1-5.2). The times and concentrations given in the protocol are approximate and may vary due to differences in tubulin preparations, chamber volume, etc.
specific and tight attachment of the microtubule nucleators to the coverslip, and
the ability to control flow speeds accurately. In our hands, the best results are achieved when experiments are conducted in the sealed flow chambers.
The main advantage of this protocol is that microtubule depolymerization can be triggered with high temporal and spatial resolution, thereby assisting analysis of motions at the disassembling microtubule ends. With this technique, buffers can be changed rapidly and microtubule-binding proteins and beads can be added in a variety of buffers after the microtubules have formed. This allows for the study of the effects of a protein on microtubule depolymerization separately from its possible roles in microtubule assembly. This is useful if the protein can also interact with soluble tubulin, which may mask its interactions with the microtubules. However, since free, unpolymerized tubulin is present in the cell, caution should be used when interpreting physiological significance of such results.
The main limitation of the segmented microtubule approach is that it is not suitable to study motions during microtubule assembly, or the effects of tracking proteins on microtubule catastrophe and rescue. For these questions, assays with dynamic microtubules grown in the presence of soluble tubulin are more appropriate. Another constraint of this method is that motility buffers should be free from oxygen-scavenging enzymes. Such enzyme mixtures, e.g. based on catalase and glucose-oxidase48, can significantly improve the lifetime of fluorescent molecules. However, if these reagents are used in the segmented microtubule assay, the photo-induction of disassembly is infrequent because microtubule caps bleach without disintegration. In quantitative fluorescence assays, the rate of bleaching should always be taken into account, as described above or for example in reference23, especially if anti-bleaching agents are not used.
Several microtubule-binding proteins tested in the segmented microtubule assay showed preferential binding to the Rhodamine-labeled stabilizing caps vs. the unlabeled microtubule segments. Binding to the caps may reduce the fraction of processive beads, since the cap-associated beads often detach from the microtubule when the cap disintegrates. Finally, we note that if the motor-coated beads walk toward the plus microtubule end very fast, they frequently reach the red caps before microtubule depolymerization is triggered, in which case they will also dissociate from the microtubule and such an experiment will not be productive.
In summary, the ability of microtubule-binding proteins with no inherent motor activity to follow the shortening end of a microtubule is an interesting, yet poorly understood phenomenon. The protocols described here should assist studies of such proteins and their properties in vitro. Microtubule depolymerization-dependent transport plays an important role in chromosome segregation during mitosis. Thus, we hope that the techniques described here will ultimately help to gain more insight into the mechanisms of cell division.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank F. I. Ataullakhanov for helping to design and manufacture reusable flow chambers, N. Dashkevich, N. Gudimchuk and A. Korbalev for providing images for figures, N. Gudimchuk and P. Zakharov for developing a protocol and providing reagents to prepare digoxigenin-labeled microtubule seeds, A. Potapenko for help with text editing and other members of Grishchuk lab for tips and discussions. This work was supported in part by NIH grant GM R01-098389 and a pilot grant from Pennsylvania Muscle Institute to E.L.G., who is a Kimmel Scholar, by RFBR grants 12-04-00111-a, 13-04-40190-H and 13-04-40188-H, Russian Academy of Sciences Presidium Grants (Mechanisms of the Molecular Systems Integration and Molecular and Molecular Cell Biology programs) to F. I. Ataullakhanov, NIH grant GM R01 GM033787 to J.R. McIntosh, and a Dmitry Zimin Dynasty Foundation postdoctoral fellowship to V.A.V.
References
- Desai A, Mitchison TJ. Microtubule polymerization dynamics. Ann. Rev. Cell Dev. Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- Mitchison TM, Kirschner MW. Dynamic instability of microtubule growth. Nature. 1984;312(15):237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- Walker RA, Brien O, et al. Dynamic Instability of Individual Microtubules Analyzed by Video Light Microscopy: Rate Constants and Transition Frequencies. J. Cell Biol. 1988;107:1437–1448. doi: 10.1083/jcb.107.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner MK, Zanic M, Gell C, Bormuth V, Howard J. Depolymerizing Kinesins Kip3 and MCAK Shape Cellular Microtubule Architecture by Differential Control of Catastrophe. Cell. 2011;147(5):1092–1103. doi: 10.1016/j.cell.2011.10.037. [DOI] [PubMed] [Google Scholar]
- Lombillo VA, Stewart RJ, McIntosh JR. Minus-end-directed motion of kinesin-coated microspheres driven by microtubule depolymerization. Nature. 1995;373:161–164. doi: 10.1038/373161a0. [DOI] [PubMed] [Google Scholar]
- Franck AD, Powers AF, Gestaut DR, Gonen T, Davis TN, Asbury CL. Tension applied through the Dam1 complex promotes microtubule elongation providing a direct mechanism for length control in mitosis. Nat. Cell Biol. 2007;9(7):832–837. doi: 10.1038/ncb1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PT, Walker RA, Salmon ED. A metastable intermediate state of microtubule dynamic instability that differs significantly between plus and minus ends. J. Cell Biol. 1997;138(1):105–117. doi: 10.1083/jcb.138.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk EL, Molodtsov MI, Ataullakhanov FI, McIntosh JR. Force production by disassembling microtubules. Nature. 438:384–388. doi: 10.1038/nature04132. [DOI] [PubMed] [Google Scholar]
- Coue M, Lombillo A, Richard J. Microtubule Depolymerization Promotes Particle and Chromosome Movement In Vitro. J. Cell Biol. 1991;112(6):1165–1175. doi: 10.1083/jcb.112.6.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk EL, Spiridonov IS, et al. Different assemblies of the DAM1 complex follow shortening microtubules by distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 2008;105(19):6918–6923. doi: 10.1073/pnas.0801811105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asbury CL, Gestaut DR, Powers AF, Franck AD, Davis TN. The Dam1 kinetochore complex harnesses microtubule dynamics to produce force and movement. Proc. Natl. Acad. Sci. U.S.A. 2006;103(26):9873–9878. doi: 10.1073/pnas.0602249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann S, Wang H-W, Avila-Sakar A, Drubin DG, Nogales E, Barnes G. The Dam1 kinetochore ring complex moves processively on depolymerizing microtubule ends. Nature. 2006;440(7083):565–569. doi: 10.1038/nature04409. [DOI] [PubMed] [Google Scholar]
- Grissom PM, Fiedler T, Grishchuk EL, Nicastro D, West RR, Mcintosh JR. Kinesin-8 from Fission Yeast A Heterodimeric , Plus-End – directed Motor that Can Couple Microtubule Depolymerization to Cargo Movement. Mol. Biol. Cell. 2009;20:963–972. doi: 10.1091/mbc.E08-09-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JR, Volkov V, Ataullakhanov FI, Grishchuk EL. Tubulin depolymerization may be an ancient biological motor. J. Sci. 2010;123:3425–3434. doi: 10.1242/jcs.067611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk EL, McIntosh JR, Molodtsov MI, Ataullakhanov FI. Force generation by dynamic microtubule polymers. Compr. Biophys. 2012;4:93–117. [Google Scholar]
- Asbury CL, Tien JF, Davis TN. Kinetochores' gripping feat: conformational wave or biased diffusion. Trends Cell Biol. 2011. pp. 38–46. [DOI] [PMC free article] [PubMed]
- Tien JF, Umbreit NT, et al. Cooperation of the Dam1 and Ndc80 kinetochore complexes enhances microtubule coupling and is regulated by aurora B. Cell Biol. 2010;189(4):713–723. doi: 10.1083/jcb.200910142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestaut DR, Graczyk B, et al. Phosphoregulation and depolymerization-driven movement of the Dam1 complex do not require ring formation. Nat. Biol. 2008;10(4):407–414. doi: 10.1038/ncb1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, Arthanari H, et al. The Kinetochore-Bound Ska1 Complex Tracks Depolymerizing Microtubules and Binds to Curved Protofilaments. Dev. Cell. 2012;23(5):968–980. doi: 10.1016/j.devcel.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouhard GJ, Stear JH, et al. XMAP215 is a processive microtubule polymerase. Cell. 2008;132(1):79–88. doi: 10.1016/j.cell.2007.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpff J, Du Y, et al. A Tethering Mechanism Controls the Processivity and Kinetochore-Microtubule Plus-End Enrichment of the Kinesin-8 Kif18A. Mol. Cell. 2011;43(5):764–775. doi: 10.1016/j.molcel.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Qui W, Gupta M, Pereira-Leal J, Reck-Peterson SL, Pellman D. Mechanisms underlying the dual-mode regulation of microtubule dynamics by Kip3/kinesin-8. Mol. Cell. 2011;43(5):751–763. doi: 10.1016/j.molcel.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius J, Brouhard G, Kalaidzidis Y, Diez S, Howard J. The depolymerizing kinesin MCAK uses lattice diffusion to rapidly target microtubule ends. Nature. 2006;441(7089):115–119. doi: 10.1038/nature04736. [DOI] [PubMed] [Google Scholar]
- Gudimchuk N, Vitre B, et al. Kinetochore kinesin CENP-E is a processive bi-directional tracker of dynamic microtubule tips. Nat. Cell Biol. 2013;15(9):1079–1088. doi: 10.1038/ncb2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz M. Microtubule +TIPs at a glance. J. Sci. 2010;20(Pt 20):3415–3419. doi: 10.1242/jcs.062414. [DOI] [PubMed] [Google Scholar]
- Dixit R, Barnett B, Lazarus J, Tokito M, Goldman Y, Holzbaur E. Microtubule plus-end tracking by CLIP-170 requires EB1. Proc. Natl. Acad. Sci. U.S.A. 2009;106(2):492–497. doi: 10.1073/pnas.0807614106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk EL, Efremov AK, et al. The Dam1 ring binds microtubules strongly enough to be a processive as well as energy-efficient coupler for chromosome motion. Proc. Natl. Acad. Sci. U.S.A. 2008;105(40):15423–15428. doi: 10.1073/pnas.0807859105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyoshi B, Sarangapani KK, et al. Tension directly stabilizes reconstituted kinetochore-microtubule attachments. Nature. 2010;468(7323):576–579. doi: 10.1038/nature09594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JR, Grishchuk EL, et al. Fibrils Connect Microtubule Tips with Kinetochores A Mechanism to Couple Tubulin Dynamics to Chromosome Motion. Cell. 2008;135(2):322–333. doi: 10.1016/j.cell.2008.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers AF, Franck AD, et al. The Ndc80 kinetochore complex forms load-bearing attachments to dynamic microtubule tips via biased diffusion. Cell. 2009;136(5):865–875. doi: 10.1016/j.cell.2008.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbreit NT, Gestaut DR, et al. The Ndc80 kinetochore complex directly modulates microtubule dynamics. Proc. Natl. Acad. Sci. U.S.A. 2012;109(40):16113–16118. doi: 10.1073/pnas.1209615109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gell C, Bormuth V, et al. Microtubule Dynamics Reconstituted In Vitro and Imaged by Single-Molecule Fluorescence Microscopy. Methods Biol. 2010;95:221–245. doi: 10.1016/S0091-679X(10)95013-9. [DOI] [PubMed] [Google Scholar]
- Dixit R, Ross JL. Studying Plus-End Tracking at Single Molecule Resolution Using TIRF Microscopy. Methods Cell Biol. 2010;95:543–554. doi: 10.1016/S0091-679X(10)95027-9. [DOI] [PubMed] [Google Scholar]
- Beausang FJ, Sun Y, Quinlan EM, Forkey NJ, Goldman Y. Construction of Flow Chambers for Polarized Total Internal Reflection Fluorescence Microscopy (polTIRFM) Cold Spring Harbour Protoc. 2012;6:712–715. doi: 10.1101/pdb.prot069385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Salser S, Drechsel DN, Unwin N, Mitchison TJ. Role of GTP Hydrolysis in Microtubule Dynamics: Information from a Slowly Hydrolyzable Analogue, GMPCPP. Mol. Biol. Cell. 1992;3:1155–1167. doi: 10.1091/mbc.3.10.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk EL, Ataullakhanov FI. In Vitro Assays to Study the Tracking of Shortening Microtubule Ends and to Measure Associated Forces. Methods Cell Biol. 2010;95:657–676. doi: 10.1016/S0091-679X(10)95033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Medina B, Block SM. Visualizing individual microtubules by bright field microscopy. Am. J. Phys. 2010;78(11):1152–1159. [Google Scholar]
- Volkov VA, Zaytsev AV, et al. Long tethers provide high-force coupling of the Dam1 ring to shortening microtubules. Proc. Natl. Acad. Sci. U.S.A. 2013;110(19):7708–7713. doi: 10.1073/pnas.1305821110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laan L, Pavin N, et al. Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell. 2012;148(3):502–514. doi: 10.1016/j.cell.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myster SH, Knott JA, O'Toole E, Porter ME. The Chlamydomonas Dhc1 gene encodes a dynein heavy chain subunit required for assembly of the I1 inner arm complex. Mol. Biol. Cell. 1997;8:607–620. doi: 10.1091/mbc.8.4.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombillo VA, Coue M, McIntosh JR. In vitro motility assays using microtubules tethered to Tetrahymena pellicles. Methods Cell Biol. 1993;39:149–165. doi: 10.1016/s0091-679x(08)60168-5. [DOI] [PubMed] [Google Scholar]
- Hyman A, Chrétien D, Arnal I, Wade R. Structural changes accompanying GTP hydrolysis in microtubules: information from a slowly hydrolyzable analogue guanylyl-(alpha,beta)-methylene-diphosphonate. J. Cell Biol. 1995;128(1-2):117–125. doi: 10.1083/jcb.128.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Kim H, Kim D, Song NW. Counting the Number of Fluorophores Labeled in Biomolecules by Observing the Fluorescence-Intensity Transient of a Single Molecule. Bull. Chem. Soc. Jap. 2005;78:1612–1618. [Google Scholar]
- Welburn JPI, Grishchuk EL, Backer CB, Wilson-Kubalek EM, Yates JR, Cheeseman IM. The human kinetochore Ska1 complex facilitates microtubule depolymerization-coupled motility. Dev. Cell. 2009;16(3):374–385. doi: 10.1016/j.devcel.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov A, Grishchuk EL, Mcintosh JR, Ataullakhanov FI. In search of an optimal ring to couple microtubule depolymerization to processive chromosome motions. Natl. Acad. Sci. 2007;U.S.A.(48):19017–19022. doi: 10.1073/pnas.0709524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Hisanaga S, Hosoi T, Kishimoto T, Hotani H. Phosphorylation states of microtubule-associated protein 2 (MAP2) determine the regulatory role of MAP2 in microtubule dynamics. Biochemistry. 1997;36(41):12574–12582. doi: 10.1021/bi962606z. [DOI] [PubMed] [Google Scholar]
- Oguchi Y, Uchimura S, Ohki T, Mikhailenko SV, Ishiwata S. The bidirectional depolymerizer MCAK generates force by disassembling both microtubule ends. Nat. 2011;Biol.(6):1–8. doi: 10.1038/ncb2256. [DOI] [PubMed] [Google Scholar]
- Kishino A, Yanagida T. Force measurements by micromanipulation of a single actin filament by glass needles. Nature. 1988;334:74–76. doi: 10.1038/334074a0. [DOI] [PubMed] [Google Scholar]
- Borisy GG, Marcum JM, Olmsted JB, Murphy DB, Johnson KA. Purification of tubulin and associated high molecular weight proteins from porcine brain and characterization of microtubule assembly in vitro. Ann. NY Acad. Sci. 1975;253:107–132. doi: 10.1111/j.1749-6632.1975.tb19196.x. [DOI] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo S, Kirschner MW. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. U.S.A. 1975;72(5):1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlund PO, Podolski M, et al. One-step purification of assembly-competent tubulin from diverse eukaryotic sources. Mol. Biol. Cell. 2012;23(22):4393–4401. doi: 10.1091/mbc.E12-06-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A, Drechsel D, et al. Preparation of Modified Tubulins. Methods Enzymol. 1991;196:478–485. doi: 10.1016/0076-6879(91)96041-o. [DOI] [PubMed] [Google Scholar]