

Abstract
Even genetically distant prokaryotes can exchange genes between them, and these horizontal gene transfer events play a central role in adaptation and evolution. While this was long thought to be restricted to prokaryotes, certain eukaryotes have acquired genes of bacterial origin. However, gene acquisitions in eukaryotes are thought to be much less important in magnitude than in prokaryotes. Here, we describe the complex evolutionary history of a bacterial catabolic gene that has been transferred repeatedly from different bacterial phyla to stramenopiles and fungi. Indeed, phylogenomic analysis pointed to multiple acquisitions of the gene in these filamentous eukaryotes—as many as 15 different events for 65 microeukaryotes. Furthermore, once transferred, this gene acquired introns and was found expressed in mRNA databases for most recipients. Our results show that effective inter-domain transfers and subsequent adaptation of a prokaryotic gene in eukaryotic cells can happen at an unprecedented magnitude.
Keywords: inter-domain transfer, lateral gene transfer, adaptive horizontal transfer
1. Introduction
In nature, species need to constantly adapt to changing environments, and this can be achieved by modifying their genetic repertoire to acquire new functions. Indeed, gene duplications (followed by evolution of new functions) and other genomic rearrangements have shaped eukaryotic genomes [1]. However, genetic innovation can also result from the acquisition of exogenous genes by horizontal gene transfer (HGT). Prokaryotes adapt largely by HGT, and strains of a particular species can differ by large fractions of their genome [2–6].
Long thought to be a prokaryote specialty, HGT is now recognized as a mechanism of genetic innovation in eukaryotes as well [7–10]. Genome analysis of eukaryotes revealed that several genes had been horizontally transferred [11,12], with important implications for environmental adaptation [13–15]. Indubitably, HGT can enable acquisition of entirely novel functions, which is more drastic than the gradual evolutionary processes that rely on modification of pre-existing genes [16–18] and may enhance ecological opportunities. In this context, the prokaryotic gene pool can serve as a large reservoir of potential functions for eukaryotes [9]. Indeed, it appears that prokaryote-to-eukaryote inter-domain HGT events are more prevalent than eukaryote-to-eukaryote ones [19]. As described for the insect Hypothenemus hampei, where inter-domain HGT of a mannase-encoding gene from a Firmicute enabled the insect to parasitize coffee berries [7], the acquisition of a single gene can lead to enhanced competitiveness and ecological specialization. However, inter-domain HGT can involve more than one gene. Thus, previous studies reported that significant parts of genome, up to 10% of the gene repertoire of the multicellular rotifer Adineta ricciae [13], had been horizontally acquired. Although these acquisitions resulted from several transfers, the extent to which a given bacterial gene may undergo inter-domain transfers to eukaryotes remains unclear.
The bacterial gene acdS has been evidenced not only in taxonomically contrasted bacteria [20], notably in strains with plant growth-promotion activity, but also in a few fungi [21–23]. The AcdS enzyme 1-aminocyclopropane-1-carboxylate (ACC) deaminase (EC 4.1.99.4) can transform the plant's ethylene precursor ACC to α-ketobutyrate and ammonia. By degrading ACC in exudates, plant-interacting bacteria can indirectly lower ethylene level in roots, thus stimulating root growth and modulating plant stress resistance [24,25]. In addition to the acdS gene itself, AcdS catalytic activity was also found in fungi, namely the Ascomycota Trichoderma asperellum [22], Cyberlindnera (formerly Hansenula) saturnus [23], Penicillium citrinum [21] and Magnaporthe oryzae (M.B., C.P.-C., P.L., Y.M.-L. & D.M. 2013, unpublished data), raising the question of the evolutionary origin of this gene in eukaryotes.
In this study, we show that the bacterial gene acdS has been repeatedly transferred to a wide range of eukaryotic recipients (i.e. fungi and stramenopiles). Phylogenetic analysis pointed to multiple acdS acquisitions, from different bacterial phyla to different eukaryotes. Ancestral state character reconstruction confirmed past occurrence of multiple, independent transfers of acdS from each of these bacterial phyla to different types of eukaryotes. Moreover, transferred acdS genes were effectively transcribed and occasionally acquired introns in eukaryotic recipients. Altogether, these results show that prokaryote-to-eukaryote transfer of a single gene can happen at high frequency, with adaptation of the transferred gene to its new host cell machinery.
2. Results
(a). acdS prevalence in eukaryotes
We found as many as 65 acdS homologues in eukaryotes—four in stramenopiles (all oomycetes) and 61 in fungi—after analysis of 149 sequenced genomes based on the AcdS protein sequence of the proteobacterium Pseudomonas fluorescens F113. Significant sequence identity (at least 38% amino acid identity) was evidenced between bacterial and eukaryotic AcdS proteins. Moreover, conservation of nucleotide sequences between bacterial and eukaryotic acdS genes was also high (at least 40% identity). It reached as much as 65% between the actinobacterium Streptomyces violaceusniger Tu4113 and the ascomycotan M. oryzae 70–15, and 78% between the proteobacterium Acidovorax radicis N35 and the stramenopile Phytophthora infestans T30-4. Such gene sequence conservation across two different domains of life suggests both a common evolutionary origin and genetic transfer(s) between them.
(b). Distribution of acdS homologues in eukaryotes
Phylogenetic analysis of AcdS protein homologues retrieved by BLAST showed that eukaryotic AcdS sequences were distributed in three distinct clades (figure 1; electronic supplementary material, figures S1–S3). The first clade is rooted by actinobacterial AcdS sequences and corresponds exclusively to fungi (Ascomycota and a few Basidiomycota). The second clade is rooted by gammaproteobacterial AcdS sequences and only includes Ascomycota. The third clade is rooted by betaproteobacterial AcdS sequences, and includes a stramenopile subclade (Phytophthora species) and a fungal subclade. This topology was retrieved both with maximum-likelihood and Bayesian reconstructions [26] (see the electronic supplementary material, figure S1). Thus, our results strongly support a bacterial origin for acdS eukaryotic homologues, and also indicate that Actinobacteria, Betaproteobacteria and Gammaproteobacteria served as distinct acdS donors for eukaryotes. Fungal recipients belonged to several taxonomical classes, and different possibilities may account for the uneven distribution of acdS homologues in oomycetes and fungi. The first hypothesis is an ancestral acquisition of the gene followed by multiple losses in a broad range of eukaryotic lineages. The second hypothesis entails multiple HGT events, perhaps even between different types of eukaryotes.
Figure 1.

Maximum-likelihood phylogenetic tree of acdS protein sequences. The tree was rooted using d-cysteine desulfhydrase sequences as outgroup (electronic supplementary material, figure S3). Supported nodes are indicated with grey circles (bootstrap > 70).
(c). Estimated acdS gains and losses along fungal evolutionary history
To assess the extent of acdS transfer and loss events, we reconstructed the ancestral states of acdS presence/absence along fungal phylogenetic history, using 150 sequenced fungi. For actinobacterial donors, ancestral state reconstruction showed four supported acquisitions in Basidiomycota, which concerned Gymnopus luxurians, Schizophyllum commune, Gloeophyllum trabeum (Agaricomycetes class) and a Cryptococcus ancestor (Tremellomycetes class) (figure 2). Similarly, four acquisitions were identified for Ascomycota: a recent one in Oidiodendron maius (Leotiomycetes class), as well as three more ancient ones in the Eurotiomycetes class ancestor, in the Sordariomycetes class ancestor and in a Mycosphaerellaceae subclade (Dothideomycetes class). Ancestral state reconstruction for the acdS clade of betaproteobacterial origin pointed to two recent acdS acquisitions in the Basidiomycota Fomitopsis pinicola SS1 and Punctularia strigosozonata (Agaricomycetes class), and another acquisition by an unidentified ancestor of the Dothideomycetes Hysterium pulicare, Rhytidisteron rufulum and Botryosphaeria dothidea. Complete reconstructions for these two clades can be found in the electronic supplementary material, figures S4 and S5.
Figure 2.

Ancestral state character reconstruction of acdS gene gains/losses. Coloured nodes and species names indicate gene presence while black means absence. Actinobacterial, betaproteobacterial and gammaproteobacterial acdS origins are shown in, respectively, red, blue and green for these names and nodes. Uncertainty in reconstruction is indicated by a white node. Fungal classes: (1) Chytridiomycetes; (2) Monoblepharidomycetes; (3) Neocallimastigomycetes; (4) Entomophthoromycotina; (5) Kickxellomycotina; (6) Mucoromycotina; (7) Exobasidiomycetes; (8) Ustilaginomycetes; (9) Microbotryomycetes; (10) Mixiomycetes; (11) Pucciniomycetes; (12) Wallemiomycetes; (13) Tremellomycetes; (14) Dacrymycetes; (15) Agaricomycetes; (16) Taphrinomycotina; (17) Schizosaccharomycetes; (18) Saccharomycetes; (19) Pezizomycetes; (20) Leotiomycetes; (21) Sordariomycetes; (22) Lecanoromycetes; (23) Eurotiomycetes; (24) Teratosphaeriaceae; (25) Mycosphaerellaceae; (26) Dothideomycetes.
For gammaproteobacterial acdS donors, ancestral state reconstruction did not provide a clear scenario using maximum-likelihood reconstruction (see the electronic supplementary material, figure S6), but Bayesian reconstruction strongly pointed to independent acquisitions in every acdS+ fungus (see the electronic supplementary material, table S1). Yet these two reconstructions strongly support the absence of acdS in the most ancestral nodes for the Saccharomycetes, refuting the hypothesis of a single, ancestral acquisition of the gene.
For each putative ancestral recipient, no case of subsequent acdS gene loss was detected in the descent, regardless of whether the gene originated from Actinobacteria, Betaproteobacteria or Gammaproteobacteria. Thus, our results point to recurrent HGT events of acdS towards oomycetes and fungi.
(d). Functionality and selection patterns of eukaryotic acdS
Functionality of eukaryotic acdS is indicated by the conservation of the catalytic function. Indeed, the key residues K51, Y269, Y295 and E296 needed for ACC deaminase catalytic activity in the yeast C. saturnus [23] were conserved in all eukaryote sequences (both in Phytophthora and fungi), as were the amino acids adjacent to these residues (figure 3). This highlights the conservation of the bacterial acdS catalytic function across distant eukaryotic lineages that experienced independent acdS acquisitions. In addition, direct evidence for acdS transcription in eukaryotes was also obtained, as mining transcript databases allowed the identification of all or part of acdS mRNA in almost all stramenopiles and fungi studied (see the electronic supplementary material, figure S7). This indicated that differences in promoter regions between bacteria and eukaryotes were not a barrier for successful genetic transfers.
Figure 3.
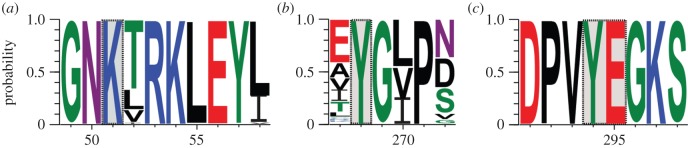
Conservation of key amino acids in acdS. Conserved amino acids are shown in (a) the Lys51 region, (b) the Tyr268 region and (c) the Tyr294-Glu295 region, which are important for catalytic activity in bacterial and eukaryotic AcdS sequences [23]. Positions indicated refer to those in the protein sequence of the Ascomycota model C. saturnus, for which key residues (boxed) were identified.
The lack of acdS deletion and the high AcdS sequence conservation suggest that this gene confers a selective advantage to microeukaryotes. In addition, comparing relative fixation rates of synonymous (silent) and non-synonymous (amino acid altering) mutations showed strong negative selection (dN/dS ratio < 1) in most species (see the electronic supplementary material, figure S8), meaning that functional mutational modifications were selected against. Despite this purifying selection, positive selection (dN/dS ratio > 1) was also found in ancestral branches, notably in the Ascomycota Aspergillus (Eurotiomycetes class), Trichoderma and Fusarium (Sordariomycetes class), and in the Dothideomycetes class. This diversifying selection, which means that functional AcdS modifications could be selected, is more likely to reflect sequence adaptation to gene biology in eukaryotes rather than a true change in protein function. This hypothesis is strengthened by the conservation of key residues implicated in the catalytic function of the protein, as well as demonstration of AcdS enzymatic activity in fungi tested [21–23].
(e). Intron acquisitions in eukaryotic acdS sequences
Unlike in prokaryotes, eukaryotic genes typically display a combination of introns and exons, and indeed we found one or several spliceosomal introns (up to eight in the Basidiomycota G. trabeum) in eukaryotic acdS sequences (figure 4). Thus, acdS acquisition by eukaryotes was followed by intron formation(s) in around half the identified transfers (31 of 65 eukaryotes). Most Sordariomycetes (corresponding to nine distinct taxonomic families) presented a conserved region of intron insertion, located 186–195 nucleotides from the acdS start codon. Introns were also found in this region in species belonging to distant taxonomic classes, such as the Lecanoromycete O. maius Zn and the Eurotiomycete Penicillium marneffei ATCC18224. However, the intron sequences themselves showed no conservation, except in closely related species (data not shown). Taken together, the data point to lineage-specific intronization, which might have gone on par with acdS evolution and domestication within these lineages.
Figure 4.

Genetic structure of eukaryotic acdS in relation to the position in the acdS phylogenetic gene tree. Coding sequences are represented by white rectangles and introns by black lines. (a) Fungal acdS of actinobacterial origin, (b) fungal acdS of betaproteobacterial origin, (c) Phytophthora acdS of betaproteobacterial origin and (d) fungal acdS of gammaproteobacterial origin.
3. Discussion
HGT is a key feature of bacterial evolution [27,28], but recent studies have also reported HGT events from prokaryotes to eukaryotes [3,7–10,12]. A significant portion of the genome of certain eukaryotic species was acquired horizontally [13–15], yet it was not clear at which order of magnitude such transfers could take place for a given gene. This work demonstrates that the bacterial gene acdS is extensively present in filamentous eukaryotes, based on the recovery of this gene in as many as 44% of the 149 sequenced genomes available. Different processes can explain the uneven distribution of genes in a given lineage, such as convergent evolution [29,30], lineage-specific gene loss [31,32] or HGT [8,33,34]. In our case, convergent evolution is unlikely as acdS nucleotide sequence identity of bacterial and eukaryotic homologues (40–78%) is particularly high, indicating a similar evolutionary origin.
Even though the gene loss hypothesis cannot be fully rejected, data clearly point to multiple and independent HGT events for acdS. In particular, eukaryotic acdS homologues do not form an independent clade outside bacterial acdS homologues (as expected if genes were vertically transmitted) but are rather interspersed within bacterial sequences. Thus, acdS phylogeny identified three major incongruences with the species tree (electronic supplementary material, figure S2), indicating HGT from three bacterial phyla to many eukaryotic lineages. Moreover, incongruences were also found inside eukaryotic clades, suggesting eukaryote-to-eukaryote transfers. Such events can take place [31,34], but here poor node supports failed to strengthen the hypothesis.
For two of the three major incongruences—namely those involving (i) branching of various fungi and stramenopiles (two distant eukaryotic lineages [35,36]) inside Betaproteobacteria and (ii) Basidiomycota and Ascomycota in relation to Actinobacteria—this could entail a unique acquisition of acdS by a eukaryotic ancestor followed by extensive gene loss during evolution and speciation of the different eukaryotic lineages. Considering vertical transfer to explain the patterns of acdS presence would imply an ancestral acquisition in an ancestor common to various eukaryotes. However, this hypothesis is rather unlikely considering the high conservation of acdS sequences in the eukaryotic lineages in each case. An alternative possibility is a series of multiple, distinct acdS transfers to different microeukaryotic clades. The two ancestral character reconstruction methods that were used clearly pointed to the latter possibility, which is very well supported statistically when considering acdS distribution in relation to fungal and stramenopile evolutionary histories.
The third incongruence involves Gammaproteobacteria and diverse Saccharomycetes/Schizosaccharomycetes. In this case, the two methods of ancestral character reconstructions gave conflicting results, thus inferences should be taken with caution. Maximum-likelihood reconstruction pointed to a unique, ancestral acquisition of acdS followed by subsequent vertical transmission and differential gene losses. However, 10 of 16 nodes of the Saccharomycetes/Schizosaccharomycetes clade were not statistically supported, limiting conclusions on the current analysis. By contrast, Bayesian reconstruction inferred the presence of the gene in a last eukaryotic common ancestor, but this was unlikely (and thus might reflect analysis bias) because only a few Saccharomycetes/Schizosaccharomycetes fungi possessed acdS. Nevertheless, a robust Bayesian reconstruction was obtained when constraining the model to infer the absence of acdS at the root of the tree. In this case, the analysis favours multiple and independent transfers with strong statistical support at each node of the Saccharomycetes/Schizosaccharomycetes clade.
In the 65 acdS+ microeukaryotes, we estimated that acdS acquisition entailed at least 15 different HGT events based on ancestral character reconstructions (figure 2). Such a magnitude for genetic transfer of a bacterial gene across a vast range of filamentous eukaryotes has never been described before [37,38] and challenges our understanding of eukaryote evolution, as most current models in evolutionary biology assume gene duplication as a key process of biochemical innovation [39]. In contrast to duplication, which gives rise to slow genetic innovation, gene acquisition might play a distinct role in enabling rapid phenotypic or ecological adaptation. Nevertheless, the actual mechanism(s) by which acdS was acquired by filamentous eukaryotes remain(s) unknown. We did not find any remnant of a mobile genetic element in the vicinity of acdS insertion sites. Furthermore, other putative bacterial genes in the vicinity of acdS were not found in eukaryotic genomes based on sequence identity search, except one encoding a putative monooxygenase downstream acdS in Eurotiomycetes, but that is also largely present in acdS-negative fungi (data not shown). Thus, it seems that acdS could have been transferred alone. Phagotrophy, the consumption of a whole cell by another one, is seen as a driving force in bacteria-to-unicellular eukaryote HGT [40], but cannot explain HGT in fungi [41].
The fact that most acdS+ microeukaryotes live in the vicinity of plants is of primary interest, because sharing a same ecological habitat may facilitate physical interaction between HGT protagonists [42,43] and is likely to promote acdS transfer. In addition, plants are the main natural source of ACC, which may represent a significant source of carbon (α-ketoglutarate) and nitrogen (ammonia) for AcdS+ plant-associated eukaryotes [44]. In return, the latter act on plants by decreasing ethylene levels. In non-pathogenic fungi like T. asperellum T203, this promotes root elongation of canola and cucumber, favouring plants' nutrient uptake and in turn their growth and yields [22,45]. In phytopathogenic oomycetes and fungi, however, the main effect of ACC degradation might be to facilitate plant infection, because ethylene signalling acts synergistically with jasmonate to induce plant defence responses [46,47].
The ACC deamination case is unusual in that the function can be ecologically important for microeukaryotes (and thus was selected), easy to perform with existing cell machinery (requiring only B6 vitamin), novel in comparison with pre-existing eukaryotic capacities (and thus was maintained as such) and involves a highly conserved gene (also suggesting that acdS transfers were recent). Consistent with this, we found that acdS was acquired by different types of filamentous eukaryotes (from both stramenopile and fungi kingdoms), which obtained the gene from contrasted bacterial phyla (both Actinobacteria and Proteobacteria). Furthermore, this took place with a conservation of the original AcdS catalytic function, a rapid domestication process (based on intronization dynamics), and effective expression of acdS in most oomycetes and fungi studied (based on mRNA database analyses). Thus, this study provides an estimate for the higher transfer rate that could be expected for this type of inter-domain HGT event.
In summary, this study shows that HGT between prokaryotes and eukaryotes can happen in high magnitude, along with the conservation of the original catabolic function and a successful adaptation of the transferred gene to very distantly related recipients.
4. Material and methods
(a). Homologous sequence retrieval and re-annotation
The AcdS protein sequence (YP_005208895) of P. fluorescens F113 was queried with BLASTp [48] against the NCBI RefSeq database [49] to retrieve prokaryotic homologues, with an E-value threshold of 1×10−20 to filter results. To retrieve eukaryotic homologues, the same sequence was simultaneously queried with BLASTp against the JGI Mycocosm [50] and NCBI RefSeq databases, with same 1×10−20 E-value threshold. ACC deaminase sequences were further selected among the retrieved homologues based on functional domain identification, using RPS-BLAST [48].
(b). Phylogenetic analysis
Protein sequences were aligned with Clustal Omega [51]. Sequences were manually filtered to discard gaps and aligned regions of low quality. For acdS sequences, the phylogenetic trees were inferred with PhyML [52] with the GTR model, 1000 bootstraps, SPR topology search [53] and the estimation of the proportion of invariant sites. Paralogues were identified by a phylogenetic approach and removed from the analysis.
(c). Fungal phylogenetic tree reconstruction
The fungal species tree was inferred from 43 concatenated protein markers [54]. For the latter, the markers were obtained for 145 sequenced fungi using HMMER3. When multiple homologues of a given marker were retrieved for a given fungal species, redundancy was resolved using a tree-based approach, and markers showing major incongruences were discarded [55]. A few sequences aligning poorly were also discarded in the final alignment to limit the possibility of false homologues, which does not compromise tree topology if sufficient data are provided [56]. Sequences were aligned as explained above (alignment of 15 813 positions) and rooted using an outgroup composed of Homo sapiens, Capsaspora owczarzaki ATCC3086, Monosiga brevicolis MX1, Nematostella vectensis and Hydra magnipapillata. The phylogenetic tree was obtained as described above, except that NNI topology search [57] was used to infer the topology and 100 bootstraps were done.
(d). Ancestral state reconstruction
Maximum-likelihood and Bayesian reconstructions were used to mitigate potential methodological biases of each approach [58,59]. The maximum-likelihood method allows quick reconstruction of ancestral state with a good sensitivity [59], whereas Bayesian methods of reconstruction have the advantage of taking into account tree uncertainty by reconstructing ancestral states over a set of phylogenetic trees [60]. Analyses were done with the previously inferred fungal tree and matrices of presence/absence of fungal acdS homologues, using Mesquite (http://mesquiteproject.org/mesquite/mesquite.html) with maximum likelihood Asymmk2 model of rate variations [61] and the reversible jump Markov chain Monte Carlo method of BayesTraits [60]. Statistical confidence was assessed with a likelihood ratio test and the Bayes factor ratio, respectively.
(e). Conservation of key amino acids in eukaryotic acdS homologues
Unfiltered alignments were used to verify the presence of amino acids required for ACC deaminase activity. Structural analysis along with site-directed mutational studies of the AcdS protein in C. saturnus identified four important amino acids (K51, Y269, Y295 and E296 [23]). Multiple alignments were numbered according to C. saturnus sequence and represented using WebLogo (http://weblogo.berkeley.edu).
(f). Intron annotation
Introns were re-annotated in nucleotide sequences using Wise v. 2.1.20 (http://www.ebi.ac.uk/Tools/psa/genewise). A hidden Markov model (HMM) profile was generated using HMMER3 and served as query to align against nucleotide sequences. Wise software was set to consider GT/AG splicing sites only.
Supplementary Material
Acknowledgements
We thank V. Daubin, N. Carraro and S. Chuzeville for useful discussions, D. Abrouk and A. Dubost (iBio) for technical support, G. Hoff and M-A. Poirier for fungal assays and the P2CHPD (Pôle de Calcul Haute Performance Dédiées) for access to computing facilities.
Funding statement
This work was supported by the Ministère Français de la Recherche.
References
- 1.Taylor JS, Raes J. 2004. Duplication and divergence: the evolution of new genes and old ideas. Annu. Rev. Genet. 38, 615–643. ( 10.1146/annurev.genet.38.072902.092831) [DOI] [PubMed] [Google Scholar]
- 2.Arsène-Ploetze F, et al. 2010. Structure, function, and evolution of the Thiomonas spp. genome. PLoS Genet. 6, e1000859 ( 10.1371/journal.pgen.1000859) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koonin E, Wolf Y. 2009. Is evolution Darwinian or/and Lamarckian? Biol. Direct 4, 42 ( 10.1186/1745-6150-4-42) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lassalle F, et al. 2011. Genomic species are ecological species as revealed by comparative genomics in Agrobacterium tumefaciens. Genome Biol. Evol. 3, 762–781. ( 10.1093/gbe/evr070) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ochman H, Lerat E, Daubin V. 2005. Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl Acad. Sci. USA 102, 6595–6599. ( 10.1073/pnas.0502035102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andres J, et al. 2013. Life in an arsenic-containing gold mine: genome and physiology of the autotrophic arsenite-oxidizing bacterium Rhizobium sp. NT-26. Genome Biol. Evol. 5, 934–953. ( 10.1093/gbe/evt061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acuña R, et al. 2012. Adaptive horizontal transfer of a bacterial gene to an invasive insect pest of coffee. Proc. Natl Acad. Sci. USA 109, 4197–4202. ( 10.1073/pnas.1121190109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keeling PJ, Palmer JD. 2008. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 9, 605–618. ( 10.1038/nrg2386) [DOI] [PubMed] [Google Scholar]
- 9.Keeling PJ. 2009. Functional and ecological impacts of horizontal gene transfer in eukaryotes. Curr. Opin. Genet. Dev. 19, 613–619. ( 10.1016/j.gde.2009.10.001) [DOI] [PubMed] [Google Scholar]
- 10.Bruto M, Prigent-Combaret C, Luis P, Hoff G, Moënne-Loccoz Y, Muller D. 2013. Horizontal acquisition of prokaryotic genes for eukaryote functioning and niche adaptation. In Evolutionary biology: exobiology and evolutionary mechanisms (ed. Pontarotti P.), pp. 165–179. Berlin, Germany: Springer. [Google Scholar]
- 11.Dunning Hotopp JC, et al. 2007. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 317, 1753–1756. ( 10.1126/science.1142490) [DOI] [PubMed] [Google Scholar]
- 12.Gladyshev EA, Meselson M, Arkhipova IR. 2008. Massive horizontal gene transfer in bdelloid rotifers. Science 320, 1210–1213. ( 10.1126/science.1156407) [DOI] [PubMed] [Google Scholar]
- 13.Boschetti C, Carr A, Crisp A, Eyres I, Wang-Koh Y, Lubzens E, Barraclough TG, Micklem G, Tunnacliffe A. 2012. Biochemical diversification through foreign gene expression in bdelloid rotifers. PLoS Genet. 8, e1003035 ( 10.1371/journal.pgen.1003035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schönknecht G, et al. 2013. Gene transfer from bacteria and archaea facilitated evolution of an extremophilic eukaryote. Science 339, 1207–1210. ( 10.1126/science.1231707) [DOI] [PubMed] [Google Scholar]
- 15.Yue J, Hu X, Sun H, Yang Y, Huang J. 2012. Widespread impact of horizontal gene transfer on plant colonization of land. Nat. Commun. 3, 1152 ( 10.1038/ncomms2148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lerat E, Daubin V, Ochman H, Moran NA. 2005. Evolutionary origins of genomic repertoires in bacteria. PLoS Biol. 3, e130 ( 10.1371/journal.pbio.0030130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. ( 10.1038/35012500) [DOI] [PubMed] [Google Scholar]
- 18.Treangen TJ, Rocha EPC. 2011. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 7, e1001284 ( 10.1371/journal.pgen.1001284) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Syvanen M. 2012. Evolutionary implications of horizontal gene transfer. Annu. Rev. Genet. 46, 341–358. ( 10.1146/annurev-genet-110711-155529) [DOI] [PubMed] [Google Scholar]
- 20.Blaha D, Prigent-Combaret C, Mirza MS, Moënne-Loccoz Y. 2006. Phylogeny of the 1-aminocyclopropane-1-carboxylic acid deaminase-encoding gene acdS in phytobeneficial and pathogenic Proteobacteria and relation with strain biogeography. FEMS Microbiol. Ecol. 56, 455–470. ( 10.1111/j.1574-6941.2006.00082.x) [DOI] [PubMed] [Google Scholar]
- 21.Jia Y-J, Ito H, Matsui H, Honma M. 2000. 1-aminocyclopropane-1-carboxylate (ACC) deaminase induced by ACC synthesized and accumulated in Penicillium citrinum intracellular spaces. Biosci. Biotechnol. Biochem. 64, 299–305. ( 10.1271/bbb.64.299) [DOI] [PubMed] [Google Scholar]
- 22.Viterbo A, Landau U, Kim S, Chernin L, Chet I. 2010. Characterization of ACC deaminase from the biocontrol and plant growth-promoting agent Trichoderma asperellum T203. FEMS Microbiol. Lett. 305, 42–48. ( 10.1111/j.1574-6968.2010.01910.x) [DOI] [PubMed] [Google Scholar]
- 23.Yao M, et al. 2000. Crystal structure of 1-aminocyclopropane-1-carboxylate deaminase from Hansenula saturnus. J. Biol. Chem. 275, 34 557–34 565. ( 10.1074/jbc.M004681200) [DOI] [PubMed] [Google Scholar]
- 24.Broekaert WF, Delauré SL, De Bolle MFC, Cammue BPA. 2006. The role of ethylene in host-pathogen interactions. Annu. Rev. Phytopathol. 44, 393–416. ( 10.1146/annurev.phyto.44.070505.143440) [DOI] [PubMed] [Google Scholar]
- 25.Li J, Ovakim DH, Charles TC, Glick BR. 2000. An ACC deaminase minus mutant of Enterobacter cloacae UW4 no longer promotes root elongation. Curr. Microbiol. 41, 101–105. ( 10.1007/s002840010101) [DOI] [PubMed] [Google Scholar]
- 26.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. ( 10.1093/bioinformatics/btg180) [DOI] [PubMed] [Google Scholar]
- 27.Dagan T, Artzy-Randrup Y, Martin W. 2008. Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc. Natl Acad. Sci. USA 105, 10 039–10 044. ( 10.1073/pnas.0800679105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koonin EV, Wolf YI. 2008. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36, 6688–6719. ( 10.1093/nar/gkn668) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersson JO. 2006. Convergent evolution: gene sharing by eukaryotic plant pathogens. Curr. Biol. 16, R804–R806. ( 10.1016/j.cub.2006.08.042) [DOI] [PubMed] [Google Scholar]
- 30.Galperin MY, Koonin EV. 2012. Divergence and convergence in enzyme evolution. J. Biol. Chem. 287, 21–28. ( 10.1074/jbc.R111.241976) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards TA, Hirt RP, Williams BAP, Embley TM. 2003. Horizontal gene transfer and the evolution of parasitic protozoa. Protist 154, 17–32. ( 10.1078/143446103764928468) [DOI] [PubMed] [Google Scholar]
- 32.Wang S, Fang W, Wang C, St. Leger RJ. 2011. Insertion of an esterase gene into a specific locust pathogen (Metarhizium acridum) enables it to infect caterpillars. PLoS Pathog. 7, e1002097 ( 10.1371/journal.ppat.1002097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards TA, Soanes DM, Jones MDM, Vasieva O, Leonard G, Paszkiewicz K, Foster PG, Hall N, Talbot NJ. 2011. Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proc. Natl Acad. Sci. USA 108, 15 258–15 263. ( 10.1073/pnas.1105100108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coelho MA, Gonçalves C, Sampaio JP, Gonçalves P. 2013. Extensive intra-kingdom horizontal gene transfer converging on a fungal fructose transporter gene. PLoS Genet. 9, e1003587 ( 10.1371/journal.pgen.1003587) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keeling PJ, Burger G, Durnford DG, Lang BF, Lee RW, Pearlman RE, Roger AJ, Gray MW. 2005. The tree of eukaryotes. Trends Ecol. Evol. (Amst.) 20, 670–676. ( 10.1016/j.tree.2005.09.005) [DOI] [PubMed] [Google Scholar]
- 36.Baldauf SL. 2008. An overview of the phylogeny and diversity of eukaryotes. J. Syst. Evol. 46, 263–273. ( 10.3724/SP.J.1002.2008.08060) [DOI] [Google Scholar]
- 37.Marcet-Houben M, Gabaldón T. 2010. Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 26, 5–8. ( 10.1016/j.tig.2009.11.007) [DOI] [PubMed] [Google Scholar]
- 38.Rogers MB. 2007. A complex and punctate distribution of three eukaryotic genes derived by lateral gene transfer. BMC Evol. Biol. 7, 89 ( 10.1186/1471-2148-7-89) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Innan H, Kondrashov F. 2010. The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108. ( 10.1038/nrg2689) [DOI] [PubMed] [Google Scholar]
- 40.Ford Doolittle W. 1998. You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 14, 307–311. ( 10.1016/S0168-9525(98)01494-2) [DOI] [PubMed] [Google Scholar]
- 41.Fitzpatrick DA. 2012. Horizontal gene transfer in fungi. FEMS Microbiol. Lett. 329, 1–8. ( 10.1111/j.1574-6968.2011.02465.x) [DOI] [PubMed] [Google Scholar]
- 42.Anderson MT, Seifert HS. 2011. Opportunity and means: horizontal gene transfer from the human host to a bacterial pathogen. mBio 2, e00005 ( 10.1128/mBio.00005-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart FJ. 2013. Where the genes flow. Nat. Geosci. 6, 688–690. ( 10.1038/ngeo1939) [DOI] [Google Scholar]
- 44.Glick BR, Penrose DM, Li J. 1998. A model for the lowering of plant ethylene concentrations by plant growth-promoting bacteria. J. Theor. Biol. 190, 63–68. ( 10.1006/jtbi.1997.0532) [DOI] [PubMed] [Google Scholar]
- 45.Brotman Y, Landau U, Cuadros-Inostroza Á, Takayuki T, Fernie AR, Chet I, Viterbo A, Willmitzer L. 2013. Trichoderma-plant root colonization: escaping early plant defense responses and activation of the antioxidant machinery for saline stress tolerance. PLoS Pathog. 9, e1003221 ( 10.1371/journal.ppat.1003221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Wees SC, Van der Ent S, Pieterse CMJ. 2008. Plant immune responses triggered by beneficial microbes. Curr. Opin. Plant Biol. 11, 443–448. ( 10.1016/j.pbi.2008.05.005) [DOI] [PubMed] [Google Scholar]
- 47.Zamioudis C, Pieterse CMJ. 2012. Modulation of host immunity by beneficial microbes. Mol. Plant Microbe Interact. 25, 139–150. ( 10.1094/MPMI-06-11-0179) [DOI] [PubMed] [Google Scholar]
- 48.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 ( 10.1186/1471-2105-10-421) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pruitt KD, Tatusova T, Brown GR, Maglott DR. 2012. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40, D130–D135. ( 10.1093/nar/gkr1079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grigoriev IV, et al. 2012. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 40, D26–D32. ( 10.1093/nar/gkr947) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sievers F, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 ( 10.1038/msb.2011.75) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. ( 10.1093/sysbio/syq010) [DOI] [PubMed] [Google Scholar]
- 53.Hordijk W, Gascuel O. 2005. Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 21, 4338–4347. ( 10.1093/bioinformatics/bti713) [DOI] [PubMed] [Google Scholar]
- 54.Ebersberger I, de Matos Simoes R, Kupczok A, Gube M, Kothe E, Voigt K, von Haeseler A. 2012. A consistent phylogenetic backbone for the fungi. Mol. Biol. Evol. 29, 1319–1334. ( 10.1093/molbev/msr285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu M, Eisen JA. 2008. A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9, R151 ( 10.1186/gb-2008-9-10-r151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wiens JJ. 2003. Missing data, incomplete taxa, and phylogenetic accuracy. Syst. Biol. 52, 528–538. ( 10.1080/10635150390218330) [DOI] [PubMed] [Google Scholar]
- 57.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. ( 10.1080/10635150390235520) [DOI] [PubMed] [Google Scholar]
- 58.Royer-Carenzi M, Pontarotti P, Didier G. 2013. Choosing the best ancestral character state reconstruction method. Math. Biosci. 242, 95–109. ( 10.1016/j.mbs.2012.12.003) [DOI] [PubMed] [Google Scholar]
- 59.Ekman S, Andersen HL, Wedin M. 2008. The limitations of ancestral state reconstruction and the evolution of the ascus in the Lecanorales (lichenized Ascomycota). Syst. Biol. 57, 141–156. ( 10.1080/10635150801910451) [DOI] [PubMed] [Google Scholar]
- 60.Pagel M, Meade A, Barker D. 2004. Bayesian estimation of ancestral character states on phylogenies. Syst. Biol. 53, 673–684. ( 10.1080/10635150490522232) [DOI] [PubMed] [Google Scholar]
- 61.Pagel M. 1999. The maximum likelihood approach to reconstructing ancestral character states of discrete characters on phylogenies. Syst. Biol. 48, 612–622. ( 10.1080/106351599260184) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.