

Abstract
Proteomics is essential for deciphering how molecules interact as a system and for understanding the functions of cellular systems in human disease; however, the unique characteristics of the human proteome, which include a high dynamic range of protein expression and extreme complexity due to a plethora of post-translational modifications (PTMs) and sequence variations, make such analyses challenging. An emerging “top-down” mass spectrometry (MS)-based proteomics approach, which provides a “bird’s eye” view of all proteoforms, has unique advantages for the assessment of PTMs and sequence variations. Recently, a number of studies have showcased the potential of top-down proteomics for unraveling of disease mechanisms and discovery of new biomarkers. Nevertheless, the top-down approach still faces significant challenges in terms of protein solubility, separation, and the detection of large intact proteins, as well as the under-developed data analysis tools. Consequently, new technological developments are urgently needed to advance the field of top-down proteomics. Herein, we intend to provide an overview of the recent applications of top-down proteomics in biomedical research. Moreover, we will outline the challenges and opportunities facing top-down proteomics strategies aimed at understanding and diagnosing human diseases.
Keywords: Proteomics, mass spectrometry, human disease, post-translational modifications, systems biology
1. Introduction
Mechanistic insights from a holistic approach at the systems level have great potential to advance our understanding of human disease and to aid in the identification of novel therapeutic targets and disease biomarkers. [1, 2] While the genome is considered to be largely static, the proteome exhibits considerable plasticity owing to alternative splicing events, protein modifications, and the amalgamation of proteins into complexes and signaling networks that are regulated both spatially and temporally. [3] Hence, in the post genomic era, proteomics is essential for deciphering how molecules interact as a system and for understanding the functions of cellular systems in healthy and disease states. [4, 5] However, the unique characteristics of the proteome, which include a high dynamic range of protein expression and extreme complexity due to a plethora of post-translational modifications (PTMs) and sequence variations, present a tremendous challenge for the field of proteomics.
PTMs modulate protein activity, stability, localization, and function, [6] playing essential roles in many critical cell signaling events in both healthy and disease states. [7] Dysregulation of a number of PTMs such as protein acetylation, glycosylation, hydroxylation, and phosphorylation, have been implicated in a spectrum of human diseases including, but not limited to, cardiovascular disease, cancer, and neurodegenerative diseases. [7, 8] Furthermore, it is generally known that sequence variations resulted from genetic mutations and alternative splicing are common causes of human diseases including cancer and heart disease. [9, 10] Consequently, a comprehensive analysis of all proteoforms (a unified term to “designate all of the different molecular forms in which the protein product of a single gene can be found, including changes due to genetic variations, alternatively spliced RNA transcripts and PTMs”[11]), is imperative for the understanding, diagnosis, and treatment of human diseases.
Mass Spectrometry (MS) is the only detection method that can unequivocally identify all protein proteoforms without a priori knowledge. [6, 12] The conventional peptide-based “bottom-up” shotgun proteomics approach is widely used but the limited sequence coverage that results from incomplete recovery of peptides following proteomic digestion reduces the amount of information that can be obtained regarding the state of the protein (e.g., the presence of sequence variations arising from point mutations, alternative splicing events, or PTMs). [13] An emerging “top-down” MS-based proteomics approach, which provides a “bird’s eye” view of all intact proteoforms, has unique advantages for the identification and localization of PTMs and sequence variations. [14–16]
In the top-down approach, intact proteins are analyzed, which results in reduced sample complexity (in terms of the number of individual species present in the sample) in comparison to the protein digests analyzed using the bottom-up approach. [14–25] Following MS analysis of all intact proteoforms in a sample, a specific proteoform of interest can be directly isolated and, subsequently, fragmented in the mass spectrometer by tandem MS (MS/MS) strategies to map both amino acid variations (arising from alternative splicing events and polymorphisms/mutations) and PTMs. The establishment of the non-ergodic MS/MS techniques, electron capture dissociation (ECD) [26] and electron transfer dissociation (ETD), [27] represents a significant advancement for top-down MS by providing reliable methods for the localization and characterization of labile PTMs such as phosphorylation and glycosylation. [18–20, 24, 28–30] Top-down MS with ECD/ETD has unique advantages for the dissection of molecular complexity via the quantification of proteoforms, unambiguous localization of PTMs and polymorphisms/mutations, discovery of unexpected PTMs and sequence variations, identification and quantification of positional isomers, and the interrogation of PTM interdependence. [18–24, 29–33]
Recently, a number of top-down proteomics studies have linked proteoform alterations to disease phenotypes, highlighting the potential for top-down proteomics in the elucidation of proteoform-associated disease mechanisms. [31–49] However, the top-down approach is still facing challenges associated with protein solubility, separation, and the detection of large intact proteins, as well as the complexity of the human proteome. Thus, new technological developments are urgently needed to advance the field of top-down proteomics. In the following sections, we provided an overview of the recent developments and applications of top-down MS in biomedical research. Moreover, we outlined the challenges and opportunities in top-down proteomics for understanding and diagnosis of human diseases.
2. Top-down MS applications in biomedical research
Given the importance of PTMs in the regulation of intracellular signaling and the link between the aberrant or altered PTM of a number of proteins and human disease, the top-down MS approach holds significant promise for the elucidation of proteoform-associated disease mechanisms by providing a powerful method for the identification, characterization and quantification of proteoforms, which 3can subsequently be correlated with disease etiology (Figure 1). The representative applications of top-down MS for the interrogation of proteoform-associated disease mechanisms are summarized in Table S1 (Supporting information) and detailed below.
Figure 1.
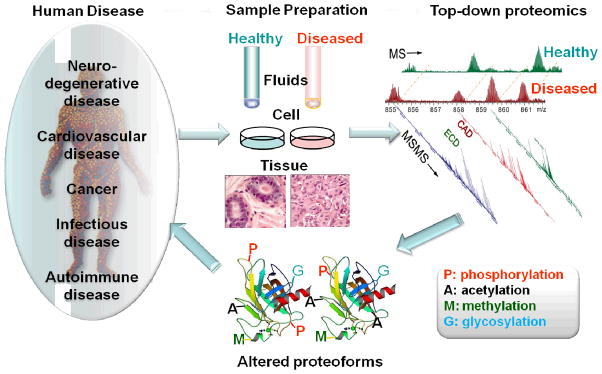
The schematic representation of the role of top-down proteomics in understanding the mechanisms of human disease.
2.1 Cardiovascular disease
Cardiovascular disease (CVD) is the leading cause of death worldwide. [50] Of the diseases classified under the umbrella of CVD, none is perhaps more devastating than heart failure (HF), which is the leading cause of death for both men and women in the US and has a 5 year 50% mortality rate. [50] While relatively little is known about the cellular and molecular mechanisms underlying HF, the altered PTM of key myofilament proteins has been implicated in the pathogenesis of HF. [34, 41, 51, 52]
Recently, we have unambiguously linked the altered PTM of cardiac troponin I (cTnI) to HF-associated contractile dysfunction in both animal models of HF and human clinical samples, using a top-down MS strategy. [34, 41] cTnI, a key myofilament protein involved in the regulation of muscular contraction, is released into the blood following cardiac injury and, thus, is currently the gold standard biomarker for chronic heart diseases. [34, 53] Phosphorylation of cTnI plays a pivotal role in the modulation of cardiac contractility by influencing myofilament calcium sensitivity. [54–56] Utilizing a top-down quantitative proteomics methodology, featuring affinity purification and high-resolution Fourier transform ion cyclotron resonance (FT-ICR)-MS, we have systematically analyzed cTnI isolated from healthy hearts and hearts with varying stages of chronic HF, and a comparison of the results from these two groups revealed an HF-associated decline in cTnI phosphorylation (Figure 2). MS/MS unambiguously localized the sites of cTnI phosphorylation in healthy and diseased samples to Ser22/23, two well-established PKA phosphorylation sites located in the cardiac-specific N-terminal extension of cTnI. Phosphorylation of these sites by PKA, activated downstream of β-adrenergic receptor stimulation, is a well-characterized facet of the “fight or flight” response that results in reduced myofilament calcium sensitivity and an increase in the cross-bridge cycling rate. [55] This study underscores the potential of PTMs as disease biomarkers and represents the first clinical application of top-down MS-based quantitative proteomics for biomarker discovery from human tissue samples. [34]
Figure 2. Identification of phosphorylation of cTnI as a potential biomarker for chronic heart failure.

The cTnI phosphorylation level was reduced in mild hypertrophy (G2), significantly reduced in severe hypotrophy (G3), and nearly abolished in chronic heart failure (G4) compared with normal human hearts (G1). Modified based on Zhang et al. 2011[34] with permission.
In addition to PKA phosphorylation at Ser22/23, phosphorylation of cTnI by protein kinase C (PKC) at Ser22/23, Ser42/44, and Thr143 has been demonstrated using an in vitro kinase assay. [55, 57] However, the role of PKC-mediated phosphorylation of cTnI in the regulation of cardiac contractility remains a topic of intense debate, in part, due to the lack of evidence of in vivo phosphorylation. Utilizing top-down ECD MS/MS, we have quantitatively determined cTnI phosphorylation changes in a spontaneously hypertensive rat (SHR) model of hypertensive heart disease and failure. MS analysis revealed increased cTnI phosphorylation in SHR rats in comparison to healthy rats and MS/MS unambiguously localized augmented phosphorylation sites to Ser22/23 and Ser42/44 in SHR, which is consistent with the upregulation of PKC –α and –δ in the SHR myocardium. [41] The identification of Ser42/44 phosphorylation by top-down MS is significant because highly specific antibodies targeting cTnI Ser42/44 phosphorylation are currently unavailable. Thus, top-down MS provided direct evidence of in vivo phosphorylation of cTnI-Ser42/44 (PKC-specific sites) in an animal model of hypertensive disease and HF, which supports the hypothesis that PKC phosphorylation of cTnI may be associated with cardiac dysfunction. [41]
Coronary artery disease represents a significant health concern because the build-up of arterial plaque in the heart can lead to myocardial infarction, which increases an individual’s risk of developing HF. [58] In a proof-of-concept study, Mazur et al. used the quantitative power of differential MS (dMS) to analyze HDL isolated from patients with high and low HDL-cholesterol levels. [31] Top-down dMS analysis, following density gradient ultracentrifugation and reverse-phase (RP) separation of plasma proteins from patients with high and low HDL-cholesterol levels, unveiled significant changes in protein abundance for 380 different m/z species. LC-MS/MS analysis using an Orbitrap-XL with ETD identified one of these m/z species as an O-glycosylated form of apolipoprotein C-III, which has been implicated in the pathogenesis of coronary artery disease. [59] Thus, as demonstrated in this study, the top-down methodology may hold promise for the identification of plasma markers of coronary artery disease.
2.2 Diabetes
In the past three decades, the global prevalence of type II diabetes (T2D) mellitus has increased dramatically and, thus, there is great interest in better understanding the mechanisms contributing to the development of this condition. [60] In addition to lifestyle changes, several therapeutic options are available for the treatment of T2D; however, treatment with certain anti-diabetes drugs has been linked to cardiovascular complications [61, 62], which necessitates the ability to distinguish subgroups of individuals who may be predisposed to cardiovascular events. To this end, Borges et al. set out to identify a panel of PTM-based biomarkers to distinguish the spectrum of CVD and T2D co-morbidities using a top-down proteomics approach. [37] They found that the presence of increased protein oxidation, primarily presenting as methionine sulfoxidation of the apolipoproteins apoAI and apoCI, was indicative of CVD while an increase in RANTES and apoCI truncation variants (produced via enzymatic cleavage) correlated with T2D. Protein glycation, a well-established marker of diabetes, was also present for albumin, VDBP, CRP, B2M, and CysC although glycation patterns did not contribute substantially to cohort separation. [37] Modified proteins were grouped according to their respective PTMs and each specific marker was analyzed in combination with other markers by principle component analysis to identify the combinations of markers allowing for the greatest separation between subgroups within the spectrum of CVD/T2D co-morbidities. [37] The end result of this work was the development of a multidimensional biomarker for T2D in the context of cardiovascular co-morbidities, which could be employed for patient screening prior to the start of a therapeutic regime to manage T2D. [37]
2.3 Infectious disease
In the field of infectious disease, a number of recent top-down MS applications have shed light on the role of bacterial protein PTMs in infection. [35, 38, 39] In a seminal report by Chamot-Rooke et al., top-down MS was employed to study the PTM of intact pilin (the major component of bacterial type IV pili) from the pathogenic bacterium N. meningitidis, the causative agent of cerebrospinal meningitis. [39] Pilin is believed to be modified by phosphocholine, phosphoethanolamine and phosphoglycerol (PG). Top-down MS analysis with a Waters Q-TOF-Premier revealed that pilin was modified with PG and localized two PG modification sites at Ser69 and Ser93 (Figure 3). In support of these findings, mutation of either Ser69 or Ser93 significantly reduced pilin glycerophosphorylation. Subsequent experiments revealed that modification of Ser93 with PG was responsible for the destabilization of type IV pili fiber bundles and enhanced bacterial detachment and migration across epithelial cells. Consequently, in this study, top-down MS analysis played a significant role in the discovery of a potential mechanism for the spread of N. meningitidis during infection.
Figure 3. Modification of the pilin subunit with PG by the PptB transferase.

Whole-protein mass spectrometry analysis of type IV pili purified from the wild-type strain (WT) (A), from a strain harboring a S69A mutation in the pilE gene (B), from a strain harboring a S93A mutation in the pilE gene (C), and from a mutant in the NMV_885 gene (ΔpptB) (D). Adapted from Chamot-Rooke et al. 2011[39] with permission.
Recently, another important study by Ansong et al. revealed a unique protein S-thiolation switch in Salmonella typhimurium in response to infection-like conditions. [35] They utilized a single-dimension ultra–high-pressure (UHP)-LC system coupled to a Velos-Orbitrap mass spectrometer to profile the intact proteome of the gram-negative bacterial pathogen S. typhimurium. [35] Top-down proteomic analysis of bacteria grown under normal and infection-like conditions resulted in the identification of 1,665 proteoforms derived from 563 different gene products, which represents the largest bacterial top-down dataset reported to date. Of particular interest was the finding that bacteria grown under infection-like conditions preferentially utilize S-cysteinylation whereas bacteria grown under basal conditions utilize S-glutathiolyation instead (Figure 4). Corroborating the finding that protein S-thiolation forms are preferentially utilized during infection, the authors identified increased expression of cysteine biosynthesis genes and reduced transcription of genes involved in glutathione biosynthesis in response to infection-like conditions. [35] Thus, top-down proteomics provided a comprehensive view of the intact proteome of the gram-negative bacterial pathogen S. typhimurium and revealed unique biological insights. [35]
Figure 4. Switch in modification from glutathionylation to cysteinylation in response to environmental conditions.

(A) Representative zero charge state spectra from top-down proteomics data showing switch in S-thiolation forms in the hypothetical protein YifE (STM14_4694). Fragmentation maps show the specific cysteine residue on which the switch occurs. (B) Estimated stoichiometry from intact protein mass spectra summed across the corresponding LC peak. Adapted from Ansong et al., 2013[35] with permission.
Burnaevskiy et al. employed a top-down MS-based methodology aimed at uncovering the mechanism of Shigella flexneri-mediated regulation of the host cell secretory pathway. [38] Transient transfection with bacterial effector proteins and infection with deletion strains revealed that the protein product of the ipaJ gene, a previously uncharacterized gene, played a major role in infection-induced Golgi destruction. [38] Top-down analysis of ARF1, a GTPase involved in the regulation of Golgi transport, revealed that the ARF1 protein was N-terminally myristolyated—a modification that allows for membrane localization of cytoplasmic proteins. Subsequent analysis of ARF1 from cells expressing IpaJ revealed cleavage of the protein consistent with cleavage of the peptide bond between Gly2 and Asp3, which led to the liberation of the GTPase domain into the cytoplasm. [38] Further study unveiled the IpaJ-mediated cleavage and liberation of myristoyl groups from a number of different proteins within infected cells. Thus, in this study, top-down analysis was able to assist in the identification of a novel mechanism for disruption of the host cell secretory pathway by the pathogenic bacteria S. flexneri.
2.4 Neurodegenerative disease
Neurodegenerative diseases are a particularly devastating class of disorders because for many neurodegenerative diseases, such as Alzheimer’s disease, treatment strategies focus only on symptom management. [63] Alterations in the sequence or PTM of a number of proteins including Parkin, [64] tau, [65] and superoxide dismutase 1 (SOD1), [66–68] have been linked to neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis (ALS). Thus, the top-down approach may be a preferred strategy for the elucidation of PTM-associated disease mechanisms underlying neurodegenerative disorders and several studies have already shown promise in this area. [36, 42, 48]
The Agar lab employed hydrogen/deuterium exchange (H/DX) MS to determine the effects of 13 familial ALS-causing polymorphisms on SOD1 structure and dynamics. [42] H/DX analysis of a series of familial ALS-causing SOD1 sequence variants revealed a single structural perturbation common to all 13 variants—destabilization of the SOD1 electrostatic loop, which has been shown to be a driving force behind protein aggregation and fibril formation in familial ALS. [69] In a more recent study published by the Agar lab, top-down MS was used to study the cross-linking of SOD1 variants as a potential therapeutic strategy for familial ALS. [36] MS analysis revealed that a single equivalent of cross-linker was responsible for mediating the chemical cross-linking of variant SOD1 monomers. [42] Additionally, fragmentation using funnel-skimmer dissociation uncovered a unique thiol-disulfide exchange reaction mechanism for SOD1 cross-linking. [36] In yet another study by Auclair et al., top-down MS analysis revealed cysteinylation and oxidation of SOD1 purified from post-mortem human nervous tissue. [70] Interestingly, cysteinylated SOD1 recovered from human tissue did not harbor additional oxidative modifications, suggesting that cysteinylation of SOD1 may protect the enzyme from oxidative damage. [70]
Interestingly, in another top-down study with relevance to neurodegenerative disease, Cabras et al. somewhat serendipitously uncovered increased levels of the protein S100A7 in the saliva proteome of patients with Down syndrome. [48] S100A7 was recently found to be elevated in the cerebrospinal fluid (CSF) and brain of patients with Alzheimer’s disease and the levels of this protein correlated with disease severity. [71] Similarly, the levels of S100A12, a ligand for the receptor for advanced glycosylation end products, was also found to increase in the saliva of Down syndrome patients in comparison to controls. [48]
2.5 Cancer
In cancer, a number of altered PTMs including phosphorylation and acetylation have been linked to the constitutive activation of cellular signaling pathways involved in the growth, proliferation, and survival of tumor cells. [72–74] Given the well-established role of PTMs in cancer, it is, perhaps, not surprising that several groups have already utilized top-down MS for the identification of disease biomarkers and to study the effects of chemotherapeutics. [40, 47, 49]
Zhang et al. developed a top-down LC/MS-based methodology for the separation and analysis of alterations in histone PTMs in primary leukemia cells from patients with refractory or relapsed acute myeloid leukemia or chronic lymphocytic leukemia in response to treatment with depsipeptide, an HDAC inhibitor. [47] Using their histone analysis platform, which employed RPLC and a Micromass Q-TOF II for the separation and detection of histone proteoforms, the authors identified increased acetylation of histones isolated from patient CD19+ B cells in response to depsipeptide treatment. Interestingly, acetylation was found to increase only on a subpopulation of histone 4 proteoforms harboring dimethylation—information that could not have been obtained by immunological detection methods. [47]
Desiderio et al. employed a top-down LC/MS platform utilizing RPLC and an LTQ Orbitrap XL to screen for potential biomarkers in the CSF of patients with posterior cranial fossa tumors. [40] Top-down MS identified two peptides, originally believed to be CSF contaminants from the blood, as LVV- and VV-hemorphin-7, two opioid peptides produced from the enzymatic cleavage of hemoglobin. Surprisingly, the absence of these peptides in the post-surgery CSF of patients was strongly indicative of residual tumor mass as a result of either subtotal resection or the presence of metastasis. Thus, these peptides may have potential as prognostic biomarkers following the removal of posterior cranial fossa tumors. [40]
In a more recent study, Hardesty et al. employed histology-directed matrix-assisted laser desorption/ionization (MALDI) imaging MS to identify protein signatures that could be used to distinguish lymph nodes harboring metastasis from healthy lymph nodes in patients with melanoma. [49] Top-down MS analysis revealed a total of 57 different signatures displaying a 2-fold or greater change in intensity between healthy and disease lymph nodes. Of these 57 signatures, 12 signals were selected based on their strong correlation with either survival or disease recurrence. Interestingly, top-down analysis also identified that three of the proteins were often present missing two of their C-terminal amino acids, which correlated with poor prognosis. [49]
2.6 Other diseases
Single amino acid changes in hemoglobin and transthyretin (TTR) can give rise to sickle cell disease and amyloidosis, respectively. Consequently, the early detection of hemoglobin and TTR variants is essential for the effective management of these diseases. To this end, Costello and co-workers developed a top-down MS platform utilizing affinity purification and direct injection of diluted whole blood for the detection of TTR and hemoglobin variants, respectively. [46] Unlike previous MS-based methods for the identification of TTR sequence variants, [43] the analytical platform employed in this study utilized a combination of nozzle-skimmer dissociation and collision activated dissociation (CAD), in addition to intact mass measurement using an LTQ-Orbitrap, to identify and precisely localize amino acid variants of both TTR and hemoglobin. [46]
More recently, Graça et al. developed a platform for the rapid analysis of hemoglobin variants from patient blood samples. [33] Although the method developed in this study employed a low resolution ion trap instrument, the fast scan speed of this instrument allowed for the detection and identification of hemoglobin variants resulting in one Dalton mass shifts on a chromatographic timescale. [33] In addition, the Cooper lab developed a top-down MS-based methodology utilizing liquid microjunction surface sampling and high-resolution MS for the analysis of hemoglobin sequence variants from neonatal dried blood spots. [32] Following intact mass measurement, hemoglobin variants were fragmented using a combination of ETD and CAD in order to precisely localize amino acid mutations in hemoglobin variants from dried blood spots that could not be diagnosed using traditional means. [32] These studies showcase the potential for top-down MS-based detection of TTR and hemoglobin structural variants in the clinic.
3. Challenges and opportunities
While the examples presented above highlight the potential of the top-down methodology for the elucidation of proteoform-associated disease mechanisms, the implementation and practice of the top-down approach still faces a number of challenges. In the following sections the specific pitfalls and shortcomings of the top-down methodology as well as potential opportunities to advance the field of top-down proteomics will be discussed.
3.1 Protein solubility
One of the most significant problems facing proteomics, not just for biomedical applications, is the issue of protein solubility. For bottom-up proteomics, protein solubility is not such a glaring problem because proteolytic digestion usually produces at least one or two soluble peptides that can be used for protein identification. However, this is not the case for top-down proteomics and protein solubility remains a significant problem; particularly for membrane proteins such as receptors and ion channels, because classical detergents such as SDS and Triton X-100, which are necessary to maintain these proteins in a soluble state, are not compatible with MS. Although approximately one-third of the proteins encoded in genome are hydrophobic membrane proteins, they are often underrepresented in proteomic studies due to their poor solubility. [75, 76]
Nonetheless, the top-down MS analysis of membrane proteins has been demonstrated in a few laboratories. The Whitelegge lab employed high concentrations of formic acid, rather than traditional surfactants, to maintain protein solubility prior to chromatographic separation, and rapid solvent transfer during HPLC (either RP or size exclusion chromatography (SEC)) followed by MS analysis to analyze integral membrane proteins. [77–79] Using this strategy, they were able to analyze a number of integral membrane proteins including bacteriorhodopsin and the cytochrome b6f complex from thylakoid membranes. [77, 79, 80] Nonetheless, a prolonged storage of proteins in high concentration of formic acid may introduce artifactual modification such as methylation. Carroll et al. extracted membrane proteins from bovine mitochondria using a high percentage of organic solvent in the presence of chaotropes and then fractionated in the same solvents by hydrophilic interaction chromatography (HILIC) before electrospray ionization (ESI) MS analysis of the integral membrane proteins. [75]
Unlike others trying to avoid the use of detergent altogether, the Robinson group developed a protocol employing these surfactants for the analysis of intact membrane complexes. [81] To combat protein signal suppression as a result of the preferential ionization of surfactant molecules, thermal activation as a result of collisions with argon molecules is utilized to liberate membrane protein complexes from detergent micelles. [81] Recently, the Kelleher group also reported the identification and characterization of membrane proteins from enriched human mitochondrial membranes by gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) coupled to LC MS/MS. [82] Despite the success in handling membrane proteins in a few experts’ labs, the solubility of membrane proteins and large intact proteins (> 70 kDa) remains a challenge for many top-down researchers, which is an area of opportunities for further development.
3.2 Challenges in protein separation
Due to the high dynamic range of protein expression and extreme complexity of the proteome, fractionation/separation of the proteome is a critical step prior to MS analysis. [83] While bottom-up proteomics boasts mature and reliable separation methods, which include gel- and LC-based separation methodologies, [84–86] effective separation strategies that are high-throughput, rapid, and compatible with top-down MS analysis are lacking.
The four-dimensional separation platform developed by the Kelleher group represents a dramatic leap forward for the separation of intact proteins. [87] This platform employs solution isoelectric focusing (separation based on isoelectric point) in the first dimension, followed by GELFrEE (separation based on size), and RPLC coupled directly to MS. Although this strategy is very robust—allowing for the identification of 3,000 intact proteoforms from HeLa cell extracts—it is also laborious. In the first dimension a number of fractions are collected offline and these fractions are again fractionated in the second dimension, resulting in the production of a large number of fractions to be analyzed by LC/MS. Furthermore, SDS is employed in the first two dimensions to improve protein solubility; however, as SDS is not compatible with MS analysis, this surfactant must be removed prior to LC/MS, lengthening the sample preparation phase.
Perhaps one of the most appealing methods for intact protein separation/purification is LC. A number of different chromatographic separation methods have already been employed for the separation of intact proteins including size exclusion chromatography, ion exchange chromatography (IEC), RP chromatography, and affinity chromatography. [88] Although far from a high throughput method, affinity chromatography represents one of the most effective means for the selective purification of a single or small subset of proteins. This method relies on biological interactions, [89] such as those between an antibody and an antigen, in order to purify a protein of interest from a complex sample such as a tissue or cell lysate. Due to the high selectivity and robustness (there have been reports of affinity columns being used hundreds of times[90]) of affinity purification, it is not surprising this strategy has been employed for the detection of a number of biologically relevant analytes from biofluids and tissues in the clinical setting. [89] Our own lab has used affinity chromatography extensively for the purification of cTnI from both animal and human myocardium followed by top-down MS analysis. [19, 21, 24, 34, 41] Affinity purification has also been used by other groups for the purification of a number of protein species. [91, 92] Nevertheless, affinity chromatography has a number of drawbacks. First, affinity chromatography is often operated offline thus the throughput of this technique is very low. Second, it commonly employs elution buffers containing salt concentrations that are not compatible with MS, thus, desalting is required prior to MS analysis. Third, the ability to purify proteins in this manner is limited by both the availability of antibodies and their variability among commercial vendors as well as the difficulty associated with developing new antibodies.
Since there is an exponential decay in the signal-to-noise ratio (S/N) as a function of increasing molecular mass, [93] it is necessary to separate high mass proteins from the low mass species in top-down proteomics. SEC is an ideal method for size-based separation of proteins because of the many advantages of SEC, which include simple operating principles, high tolerance of various solvent solutions, preservation of biological activity of proteins, and minimal sample loss as well as simple operating principles. [94, 95] Nonetheless, traditional SEC methods suffer from notoriously low resolution and detrimental sample dilution when fractions are collected over a prolonged LC analysis [95]. Recently, we reported for the first time the use of UHP-SEC for high-resolution and high-throughput separation of intact proteins for top-down proteomics. [96] We have achieved fast high-resolution separation of intact proteins (6 – 669 kDa) in less than 7 min. More importantly, such an UHP-SEC method is compatible with MS-friendly volatile solvents, making it an attractive LC strategy for top-down proteomics given that the eluted fractions can be analyzed directly by MS without an additional desalting step. The caveat of the current version of UHP-SEC is the relatively large column diameter of Waters BEH 125 and 200 columns (4.6 mm i.d. × 150 mm), which require more sample than is typically used in a proteomic study and further development of a capillary version of the BEH columns with nanoUPLC is urgently needed.
The most popular LC-based separation method is RPLC, which separates proteins based on hydrophobicity, because the mobile phase employed in this separation method is MS compatible—allowing for online separation and analysis. It should be noted that, while most LC methods can be used for the separation of proteins offline, tedious fraction collection and the increased analysis time (a number of fractions must be analyzed rather than a single sample) are undesirable; however, in order to achieve maximal proteomic depth, a certain degree of separation is necessary and, thus, two-dimensional LC methods, which can be coupled online to MS, are highly preferable for proteomic analyses. Consequently, for most two-dimensional separation platforms, especially those employing IEC methods in the first dimension, RP is often performed in the second dimension to further increase proteome fractionation (thereby reducing sample complexity), allow for online analysis, and remove salts, which have a negative impact on ESI. [97, 98] The Paša-Tolić group has optimized RPLC with the development of long (80 cm) C5 nanoLC columns. [98] Due to the convenience of this technique a number of labs have employed RPLC in either an offline or online format for the separation of intact proteins prior to MS analysis. As mentioned above IEC and SEC have also been employed for intact protein separation although these methods are often followed by RPLC in the second dimension, prior to MS analysis. [99, 100] The ability to couple LC separation directly to MS makes LC-based separation methods powerful tools for top-down proteomics.
Unfortunately, the limited lifespan of columns can necessitate the use of multiple columns during the course of a study, which can introduce separation variability as a consequence of column heterogeneity. Thus, there is great opportunity for the development of robust LC-based separation systems that can analyze large numbers of samples in a reliable manner.
3.3 Challenges of large protein MS analysis
Top-down analysis of proteins larger than ~70 kDa remains challenging due to the difficulty associated with detecting and fragmenting these molecular behemoths. [93] The use of ESI in top-down MS analysis is a double-edged sword. On one hand, ESI is generally preferred over MALDI analysis due to the fact that multiply charged precursor ions are generated allowing for more efficient fragmentation of larger proteins, particularly by electron-based dissociation methods. On the other hand, significant S/N reduction occurs for large proteins due to the spreading of the charge “pool” over a greater number of isotopic and adducted species at high mass. [93] Whereas strategies relying on depletion of the most highly abundant isotopes have demonstrated some promise, modeling suggests that, due to the decreased role of isotopic species and increased role of multiple charge states in determining S/N at high mass (>30 kDa), isotopic depletion strategies will only minimally influence S/N for high mass proteins. [93] Instead, supercharging, which leads to the preferential formation of higher charge states, appears to hold great promise for the detection and analysis of high molecular weight (MW) species. [93, 101, 102] However, the problem with commonly used supercharging reagents, such as m-nitrobenzyl alcohol, is that the presence of these reagents during LC separation can affect retention time as well as decrease chromatographic resolution. Two exciting developments stand to address this concern. First, Valeja et al. identified several different supercharging reagents that allowed for the efficient supercharging of intact proteins up to 78 kDa and did not impact chromatographic resolution when these reagents were included in the mobile phase during LC separation. [103] Second, Miladinovic et al. developed a methodology for protein supercharging following LC separation. [104] This methodology, named “in-spray supercharging” by the authors, utilizes a dual-sprayer ESI microchip, which allows for addition of the supercharging reagents to the Taylor cone following LC separation. The results obtained using in-spray supercharging were comparable to those obtained with direct injection of supercharging additives and inclusion of these reagents in the LC mobile phase and, thus, in-spray supercharging may hold promise for the detection of high MW protein species.
Another exciting advancement for the detection of large proteins is the newly developed nanomembrane detector for use in time-of-flight (TOF) mass analyzers. [105] Commonly employed TOF detectors such as electron multipliers and microchannel plates have difficulty detecting high MW proteins because these proteins move slowly in the drift tube and the electron-generating effect of a protein decreases with decreasing velocity; thus, high MW protein species will not generate sufficient secondary electrons to be detected by traditional TOF detectors. [106] However, this new detector uses the kinetic energy of the ions to produce mechanical oscillations of the nanomembrane for detection, which significantly reduces the detection bias against large proteins. The increase in the upper limit on mass detection afforded by this nanomembrane detector, combined with the essentially unlimited m/z range of TOF analyzers, represents a powerful tool for the top-down analysis of large proteins. Nonetheless, FT-ICR-MS still remains the preferred instrument for isotopic resolution of proteins with a MW greater than 70 kDa although the newest generation of Orbitrap mass spectrometers offers resolving power that is only eclipsed by FT-ICR without the need for an expensive superconducting magnet, making it a valuable alternative. [20, 107, 108] The new dynamically harmonized FT-ICR cell developed by Nikolaev and coworkers stabilizes the ICR signal over a broad mass range thus enabling broadband ultra-high-resolution detection. [109] Hence, the new generation high-field FT-ICR may provide a promising tool for ultra-high-resolution high-sensitivity analysis of large proteins for top-down proteomics.
In addition to the problems associated with the detection of large proteins, fragmentation of high MW protein species is also challenging. The ability to fragment proteins and map labile PTMs such as phosphorylation by top-down MS has been greatly dependent on the electron-based MS/MS techniques, ECD [26] (and ETD). In ECD, fragmentation via the capture of thermal electrons is believed to occur as a result of the cleavage of facile N—Cα bonds secondary to the formation of peptide cation-radicals [110] thus preserving labile PTMs. In addition, since fragmentation using ECD is localized along the peptide backbone, it often provides far more cleavages than CAD, [18] which greatly enhances the capability of top-down MS in identifying PTMs and sequence variants. [18–24] However, efficient fragmentation of large proteins with ECD remains a challenge due to the presence of extensive intramolecular interactions (e.g., electrostatic interactions, etc.) that prevent efficient dissociation of the fragmented ions. [28] Furthermore, the evolution of ion structures following ESI results in the formation of highly stable gas structures that can reduce fragmentation of protein species, particularly by electron-based fragmentation methods. [111, 112] Taking advantages of the complementary nature of ECD and CAD, we have shown that a combined ECD and CAD strategy can provide sufficient fragmentation for the sequencing of large proteins (>35 kDa). [113]
To combat the formation of highly compact gas phase structures, McLafferty and coworkers developed a novel strategy for the enhanced fragmentation of proteins in excess of 200 kDa. [114] This strategy, which is a variation on nozzle-skimmer dissociation, employs capillary heating, increased pre- and post-skimmer voltages, and electrospray additives, to reduce gas-phase structural rearrangements. Despite the fact that this technique represents a significant advancement for the top-down down field, fragmentation was localized to the N- and C-termini. The Brodbelt lab recently implemented 193 nm ultraviolet photodissociation (UVPD) in an Orbitrap mass spectrometer for the near-complete sequencing of intact proteins (up to 29 kDa) and unambiguously localized a single residue mutation and several protein modifications on Pin1 (19 kDa). [115] Nevertheless, there is opportunity for the development of novel dissociation methods to aid in the analysis of large proteins (>50 kDa) with relevance to human disease.
3.4 Protein quantification
The ability to identify changes in the expression level as well as PTMs of important proteins in signaling cascades is imperative for identifying disease mechanisms and, thus, the accurate and reliable quantification of protein abundance is a subject of increasing popularity in proteomics. [116, 117] Top-down MS can provide relative quantification of modified versus unmodified proteoforms (or proteoforms with minor sequence variations) due to the fact that the addition of modifying groups to intact proteins has minimal influence on the overall physicochemical properties of the proteins;[15] and, thus, the effect of modifying groups or minor sequence variations on the ionization efficiency and ion m/z values for intact proteins is negligible. Consequently, the MS abundances, as well as the abundances of fragment ions generated by MS/MS, have been used to determine the relative percentages of different proteoforms. [19, 20, 22, 24, 118–120]
In addition, stable isotope labeling by amino acids in cell culture (SILAC) and chemical tagging strategies, which were originally developed for use in quantitative bottom-up proteomics, have been adapted for use in top-down MS. Early work employing 14N/15N metabolic labeling for top-down MS quantification achieved 98% 15N incorporation, which allowed for the determination of 50 protein expression ratios. [121] Mann and coworkers employed SILAC labeling of an intact protein (Grb2, 28 kDa) to ascertain the feasibility of the SILAC methodology for intact protein quantification. [122] While partial incorporation (<100%) of heavy amino acids resulted in signal spreading and a severe decrease in the S/N, complete incorporation of heavy amino acids yielded accurate and reliable quantification of the target protein. [122] In an extension of this work by the Muddiman group, SILAC labeling of Aspergillus flavus and subsequent high-resolution FT-ICR-MS analysis identified 659 SILAC pairs of which 22 were confidently identified. [123] In a follow-up study, the Muddiman group employed mathematical modeling to quantitatively account for incomplete incorporation of labeled amino acids and arginine-to-proline conversion, which allowed for the identification of 11 SILAC pairs from human embryonic stem cells. [124] More recently, a quantitative approach utilizing metal-coded affinity tags (MeCAT) was used for absolute protein quantification by top-down MS. [125] This strategy, which employs inductively coupled plasma MS for the detection of lanthanide-bearing affinity tags, was able to achieve absolute quantification results that lie within the same order of magnitude as the reference method. [125] These results are particularly exciting because, unlike SILAC, which is restricted to the quantification of protein abundance changes in cell culture and mouse models, chemical tagging strategies such as MeCAT could be employed for the quantification of protein changes in human tissue samples. Although these methods represent potential quantitative options for top-down proteomics, they have not yet been widely adopted by the top-down community. However, as top-down MS continues to mature as a method for proteome analysis (beyond its traditional role as a method for the identification and analysis of PTMs on purified proteins) the ability to quantify proteins will become increasingly important and thus quantitative methods will undoubtedly play a greater role in top-down MS analyses.
3.5 Data analysis
In comparison to bottom-up proteomics, which has been significantly advanced by a host of widely-available data analysis tools, top-down MS researchers still contend with a general under-developed data analysis tools, making the analysis of complex MS and MS/MS spectra a challenging and time consuming task. There are two main types of data analysis tools available for analyzing top-down spectra: the first are tools that focus on deconvolution, which is the first step in data analysis and involves the grouping of MS peaks into isotopic envelopes; thereby allowing for the determination of the charge and monoisotopic mass. The second type of data analysis tools focus on protein identification, either by searching a database or by spectral alignment. [15, 126]
In terms of deconvolution algorithms, thorough high-resolution analysis of spectra by Horn (THRASH) is one of the most well-known algorithms for analyzing MS data. [127] The ability to subtract isotopic envelopes, determine the statistical reliability of the calculated charge states, and calculate the background noise levels allows for the comprehensive detection of isotopic envelopes present within high-resolution MS spectra. [127] However, THRASH tends to produce a high number of false positives necessitating careful validation of the results. Decon2LS, which is a software package for the automated processing and visualization of high-resolution MS spectra, uses a variation of the THRASH algorithm for deconvolution. [128] This software supports several different file formats allowing for the analysis of data from all major MS formats and provides several features for data visualization. [128] Furthermore, as an open-source software, the Decon2LS algorithms are freely available—allowing researchers to tailor the algorithms to meet their individual needs. [128] We recently developed MASH Suite, a user-friendly software for the analysis of high-resolution MS data, using the Decon2LS open source code. [129] The features of this software include a chronological workflow to guide the user through the process of uploading, analyzing, and reporting top-down MS data; the ability to adjust the theoretical isotopic distribution (created using averagine) in 1 Da increments to optimize the fit score; and a sequence table window that includes the option to add PTMs to amino acids in an uploaded sequence, which can increase the number of identified fragment ions in MS/MS spectra and aid in the localization of PTMs. [129] Additional algorithms have also been developed such as MS-Deconv, a combinatorial algorithm for spectral deconvolution created by the Pevzner lab, which compared favorably to the THRASH algorithm—recovering more true positive masses in less time. [130]
After spectral deconvolution, the next step in the analysis of top-down MS data is the identification of the proteins/peptides present in the MS or MS/MS spectra. Of the programs developed for the identification of intact proteins and protein fragments from high-resolution top-down MS spectra, ProSightPC is perhaps the most commonly used. [131] ProSightPC uses both the precursor ion mass as well as MS/MS spectral data to search a “shotgun annotated” protein database. [132] While this software can identify proteins with unexpected PTMs, the sheer number of potential proteoforms poses a problem for database-based approaches such as this. [131] The precursor ion independent top-down algorithm (PIITA) is an algorithm for protein identification that, unlike ProSightPC, is precursor-independent—meaning that protein identification is based solely on MS/MS spectral data. [133] This allows for the identification of proteins with no expectations regarding the number or identity of PTMs, thus, enabling the direct detection of PTMs and sequence variations;[133] however, this algorithm also relies on database searching to identify proteins. One recently developed software that relies on spectral alignment, rather than database searching, is MS-Align+. [134] When compared with ProSightPC, MASCOT, and OMSSA, MS-Align+ compared favorably and, thus, may hold promise for top-down data processing. [134] As the throughput of top-down proteomics continues to advance, undoubtedly there will be an increasing demand to develop better software for the analysis of top-down mass spectra—particularly software to assist in fragment assignment and scoring.
4. Conclusions and perspectives
Homeostasis depends on the precise coordination and integration of seemingly innumerable signals involved in crucial cellular processes and imbalances in or the disruption of these signals is an underlying cause of human disease. In order to understand all facets of disease etiology, it is imperative that a systems approach be employed to interrogate the far reaching consequences of alterations in one or more signaling pathways in the disease state. Consequently, large-scale “-omics” studies have great potential to assist in the unraveling of disease mechanisms.
Proteomics in particular is essential for deciphering how proteins interact as a system and for understanding the functions of cellular systems in human disease. However, due to its highly fluid nature, and the fact that the proteome is several orders of magnitude more complex than the genome, [135] in addition to other factors such as a dynamic range of protein expression and extreme molecular complexity, large-scale proteomic analysis remains challenging. Protein diversity, arising as a result of sequence variations and the addition or removal of PTMs, represents a significant challenge for bottom-up-based proteomics workflows due to the limited amount of information that can be obtained regarding the state of the protein. An emerging “top-down” mass spectrometry (MS)-based proteomics approach, which provides a “bird’s eye” view of all proteoforms present in vivo without a priori knowledge, has unique advantages for the assessment of PTMs and sequence variations and, thus, has great potential for use in biomedical as well as clinical research and probing disease mechanisms at the systems level. [136]
This review highlights a number of studies in which a top-down proteomics approach was undertaken in biomedical research. It is likely that as this methodology matures the list of biomedical studies employing top-down MS will continue to grow. Nevertheless, the top-down approach still faces significant challenges in terms of protein solubility, separation and detection of large proteins, in addition to relatively lower throughput and a lack of automation. Hopefully, as the number of labs using top-down MS increases, this will put pressure and motivation on research groups and industries focusing on methods/technology development for better instruments, separation methods, data analysis programs, etc., which will usher top-down MS to the forefront of proteomics and biomedical research.
Supplementary Material
Acknowledgments
We would like to acknowledge the financial support by NIH R01HL096971 and R01HL109810 (to YG). ZG would like to thank the NIH training grant T32GM008688.
Abbreviations and acronyms
- PTMs
post-translational modifications
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- CAD
collisionally activated dissociation
- ECD
electron capture dissociation
- FT-ICR
Fourier transform ion cyclotron resonance
- HILIC
hydrophilic interaction chromatography
- cTnI
cardiac troponin I
- SILAC
stable isotope labeling by amino acids in cell culture
- S/N
signal-to-noise ratio
- HF
heart failure
- SHR
spontaneously hypertensive rat
- dMS
differential mass spectrometry
- T2D
type II diabetes
- ALS
amyotrophic lateral sclerosis
- H/DX
hydrogen deuterium exchange
- CSF
cerebrospinal fluid
- HDAC
histone deacetylase
- MW
molecular weight
- CVD
cardiovascular disease
- SOD
superoxide dismutase
- TTR
transthyretin
- SEC
size exclusion chromatography
- RP
reverse-phase
- ETD
electron transfer dissociation
- PG
phosphoglycerol
- UHP
ultra-high-pressure
- ESI
electrospray ionization
- MALDI
matrix-assisted laser desorption/ionization
- GELFrEE
gel-eluted liquid fraction entrapment electrophoresis
- IEC
ion exchange chromatography
- TOF
time-of-flight
- THRASH
thorough high resolution analysis of spectra by Horn
- PIITA
precursor ion independent top-down algorithm
Footnotes
Conflict of interest
None.
References
- 1.Sabido E, Selevsek N, Aebersold R. Mass spectrometry-based proteomics for systems biology. Curr Opin Biotechnol. 2012;23:591–7. doi: 10.1016/j.copbio.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Drake TA, Ping PP. Proteomics approaches to the systems biology of cardiovascular diseases. Journal of Lipid Research. 2007;48:1–8. doi: 10.1194/jlr.R600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Altelaar AFMJ, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet. 2013;14:35–48. doi: 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- 4.Patterson SD, Aebersold RH. Proteomics: the first decade and beyond. Nat Genet. 2003;33 (Suppl):311–23. doi: 10.1038/ng1106. [DOI] [PubMed] [Google Scholar]
- 5.Yates JR. Mass Spectrometry as an emerging tool for systems biology. Biotechniques. 2004;36:917–9. doi: 10.2144/04366TE01. [DOI] [PubMed] [Google Scholar]
- 6.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nature Biotechnology. 2003;21:255–61. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 7.Krueger KE, Srivastava S. Posttranslational protein modifications - Current implications for cancer detection, prevention, and therapeutics. Molecular & Cellular Proteomics. 2006;5:1799–810. doi: 10.1074/mcp.R600009-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Karve TM, Cheema AK. Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J Amino Acids. 2011;2011:207691. doi: 10.4061/2011/207691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. Journal of Clinical Investigation. 2005;115:518–26. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10:186–7. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nedelkov D, Kiernan UA, Niederkofler EE, Tubbs KA, Nelson RW. Population proteomics: the concept, attributes, and potential for cancer biomarker research. Mol Cell Proteomics. 2006;5:1811–8. doi: 10.1074/mcp.R600006-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Chait BT. Mass Spectrometry: Bottom-up or Top-Down? Science. 2006;314:65–6. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- 14.Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nat Methods. 2007;4:817–21. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Ge Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ Cardiovasc Genet. 2011;4:711. doi: 10.1161/CIRCGENETICS.110.957829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tipton JD, Tran JC, Catherman AD, Ahlf DR, Durbin KR, Kelleher NL. Analysis of Intact Protein Isoforms by Mass Spectrometry. Journal of Biological Chemistry. 2011;286:25451–8. doi: 10.1074/jbc.R111.239442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. Journal of the American Chemical Society. 1999;121:806–12. [Google Scholar]
- 18.Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP, et al. Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry. Journal of the American Chemical Society. 2002;124:672–8. doi: 10.1021/ja011335z. [DOI] [PubMed] [Google Scholar]
- 19.Ayaz-Guner S, Zhang J, Li L, Walker JW, Ge Y. In vivo phosphorylation site mapping in mouse cardiac troponin I by high resolution top-down electron capture dissociation mass spectrometry: Ser22/23 are the only sites basally phosphorylated. Biochemistry. 2009;48:8161–70. doi: 10.1021/bi900739f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge Y, Rybakova IN, Xu Q, Moss RL. Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc Natl Acad Sci U S A. 2009;106:12658–63. doi: 10.1073/pnas.0813369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sancho Solis R, Ge Y, Walker JW. Single amino acid sequence polymorphisms in rat cardiac troponin revealed by top-down tandem mass spectrometry. J Muscle Res Cell Motil. 2008;29:203–12. doi: 10.1007/s10974-009-9168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zabrouskov V, Ge Y, Schwartz J, Walker JW. Unraveling molecular complexity of phosphorylated human cardiac troponin I by top down electron capture dissociation/electron transfer dissociation mass spectrometry. Mol Cell Proteomics. 2008;7:1838–49. doi: 10.1074/mcp.M700524-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Zabrouskov V, Han X, Welker E, Zhai H, Lin C, van Wijk KJ, et al. Stepwise deamidation of ribonuclease A at five sites determined by top down mass spectrometry. Biochemistry. 2006;45:987–92. doi: 10.1021/bi0517584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Dong X, Hacker TA, Ge Y. Deciphering modifications in swine cardiac troponin I by top-down high-resolution tandem mass spectrometry. J Am Soc Mass Spectrom. 2010;21:940–8. doi: 10.1016/j.jasms.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armirotti A, Damonte G. Achievements and perspectives of top-down proteomics. Proteomics. 2010;10:3566–76. doi: 10.1002/pmic.201000245. [DOI] [PubMed] [Google Scholar]
- 26.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, et al. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem. 2000;72:563–73. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 27.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–33. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper HJ, Hakansson K, Marshall AG. The role of electron capture dissociation in biomolecular analysis. Mass Spectrom Rev. 2005;24:201–22. doi: 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- 29.Eliuk SM, Maltby D, Panning B, Burlingame AL. High resolution electron transfer dissociation studies of unfractionated intact histones from murine embryonic stem cells using on-line capillary LC separation: determination of abundant histone isoforms and post-translational modifications. Mol Cell Proteomics. 2010;9:824–37. doi: 10.1074/mcp.M900569-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian Z, Tolic N, Zhao R, Moore RJ, Hengel SM, Robinson EW, et al. Enhanced top-down characterization of histone post-translational modifications. Genome Biol. 2012;13:R86. doi: 10.1186/gb-2012-13-10-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazur MT, Cardasis HL, Spellman DS, Liaw A, Yates NA, Hendrickson RC. Quantitative analysis of intact apolipoproteins in human HDL by top-down differential mass spectrometry. Proc Natl Acad Sci U S A. 2010;107:7728–33. doi: 10.1073/pnas.0910776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards RL, Griffiths P, Bunch J, Cooper HJ. Top-down proteomics and direct surface sampling of neonatal dried blood spots: diagnosis of unknown hemoglobin variants. J Am Soc Mass Spectrom. 2012;23:1921–30. doi: 10.1007/s13361-012-0477-9. [DOI] [PubMed] [Google Scholar]
- 33.Coelho Graca D, Lescuyer P, Clerici L, Tsybin YO, Hartmer R, Meyer M, et al. Electron transfer dissociation mass spectrometry of hemoglobin on clinical samples. J Am Soc Mass Spectrom. 2012;23:1750–6. doi: 10.1007/s13361-012-0446-3. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Guy MJ, Norman HS, Chen YC, Xu Q, Dong X, et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res. 2011;10:4054–65. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, et al. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc Natl Acad Sci U S A. 2013;110:10153–8. doi: 10.1073/pnas.1221210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auclair JR, Boggio KJ, Petsko GA, Ringe D, Agar JN. Strategies for stabilizing superoxide dismutase (SOD1), the protein destabilized in the most common form of familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2010;107:21394–9. doi: 10.1073/pnas.1015463107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borges CR, Oran PE, Buddi S, Jarvis JW, Schaab MR, Rehder DS, et al. Building multidimensional biomarker views of type 2 diabetes on the basis of protein microheterogeneity. Clin Chem. 2011;57:719–28. doi: 10.1373/clinchem.2010.156976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burnaevskiy N, Fox TG, Plymire DA, Ertelt JM, Weigele BA, Selyunin AS, et al. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature. 2013;496:106–9. doi: 10.1038/nature12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chamot-Rooke J, Mikaty G, Malosse C, Soyer M, Dumont A, Gault J, et al. Posttranslational modification of pili upon cell contact triggers N. meningitidis dissemination. Science. 2011;331:778–82. doi: 10.1126/science.1200729. [DOI] [PubMed] [Google Scholar]
- 40.Desiderio C, D’Angelo L, Rossetti DV, Iavarone F, Giardina B, Castagnola M, et al. Cerebrospinal fluid top-down proteomics evidenced the potential biomarker role of LVV- and VV-hemorphin-7 in posterior cranial fossa pediatric brain tumors. Proteomics. 2012;12:2158–66. doi: 10.1002/pmic.201100499. [DOI] [PubMed] [Google Scholar]
- 41.Dong X, Sumandea CA, Chen YC, Garcia-Cazarin ML, Zhang J, Balke CW, et al. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J Biol Chem. 2012;287:848–57. doi: 10.1074/jbc.M111.293258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molnar KS, Karabacak NM, Johnson JL, Wang Q, Tiwari A, Hayward LJ, et al. A common property of amyotrophic lateral sclerosis-associated variants: destabilization of the copper/zinc superoxide dismutase electrostatic loop. J Biol Chem. 2009;284:30965–73. doi: 10.1074/jbc.M109.023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nepomuceno AI, Mason CJ, Muddiman DC, Bergen HR, 3rd, Zeldenrust SR. Detection of genetic variants of transthyretin by liquid chromatography-dual electrospray ionization fourier-transform ion-cyclotron-resonance mass spectrometry. Clin Chem. 2004;50:1535–43. doi: 10.1373/clinchem.2004.033274. [DOI] [PubMed] [Google Scholar]
- 44.Pflieger D, Przybylski C, Gonnet F, Le Caer JP, Lunardi T, Arlaud GJ, et al. Analysis of human C1q by combined bottom-up and top-down mass spectrometry: detailed mapping of post-translational modifications and insights into the C1r/C1s binding sites. Mol Cell Proteomics. 2010;9:593–610. doi: 10.1074/mcp.M900350-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rauser S, Marquardt C, Balluff B, Deininger SO, Albers C, Belau E, et al. Classification of HER2 receptor status in breast cancer tissues by MALDI imaging mass spectrometry. J Proteome Res. 2010;9:1854–63. doi: 10.1021/pr901008d. [DOI] [PubMed] [Google Scholar]
- 46.Theberge R, Infusini G, Tong W, McComb ME, Costello CE. Top-Down Analysis of Small Plasma Proteins Using an LTQ-Orbitrap. Potential for Mass Spectrometry-Based Clinical Assays for Transthyretin and Hemoglobin. Int J Mass Spectrom. 2011;300:130–42. doi: 10.1016/j.ijms.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Freitas MA, Wickham J, Parthun MR, Klisovic MI, Marcucci G, et al. Differential expression of histone post-translational modifications in acute myeloid and chronic lymphocytic leukemia determined by high-pressure liquid chromatography and mass spectrometry. J Am Soc Mass Spectrom. 2004;15:77–86. doi: 10.1016/j.jasms.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 48.Cabras T, Pisano E, Montaldo C, Giuca MR, Iavarone F, Zampino G, et al. Significant modifications of the salivary proteome potentially associated with complications of Down syndrome revealed by top-down proteomics. Mol Cell Proteomics. 2013;12:1844–52. doi: 10.1074/mcp.M112.026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hardesty WM, Kelley MC, Mi D, Low RL, Caprioli RM. Protein signatures for survival and recurrence in metastatic melanoma. J Proteomics. 2011;74:1002–14. doi: 10.1016/j.jprot.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–59. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 52.van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, et al. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 53.Babuin L, Jaffe AS. Troponin: the biomarker of choice for the detection of cardiac injury. Canadian Medical Association Journal. 2005;173:1191–202. doi: 10.1503/cmaj.050141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solaro RJ, Rosevear P, Kobayashi T. The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochemical and Biophysical Research Communications. 2008;369:82–7. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovascular Research. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 56.Sumandea MP, Burkart EM, Kobayashi T, De Tombe PP, Solaro RJ. Molecular and integrated biology of thin filament protein phosphorylation in heart muscle. Ann N Y Acad Sci. 2004;1015:39–52. doi: 10.1196/annals.1302.004. [DOI] [PubMed] [Google Scholar]
- 57.Westfall MV, Borton AR. Role of troponin I phosphorylation in protein kinase C-mediated enhanced contractile performance of rat myocytes. Journal of Biological Chemistry. 2003;278:33694–700. doi: 10.1074/jbc.M305404200. [DOI] [PubMed] [Google Scholar]
- 58.Kannel WB. Incidence and epidemiology of heart failure. Heart Fail Rev. 2000;5:167–73. doi: 10.1023/A:1009884820941. [DOI] [PubMed] [Google Scholar]
- 59.Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus--present and future perspectives. Nat Rev Endocrinol. 2012;8:228–36. doi: 10.1038/nrendo.2011.183. [DOI] [PubMed] [Google Scholar]
- 61.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 62.Nissen SE, Wolski K. Rosiglitazone revisited: an updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch Intern Med. 2010;170:1191–201. doi: 10.1001/archinternmed.2010.207. [DOI] [PubMed] [Google Scholar]
- 63.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7:137–52. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–21. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- 65.Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, et al. Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A. 1994;91:7787–91. doi: 10.1073/pnas.91.16.7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kabashi E, Valdmanis PN, Dion P, Rouleau GA. Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann Neurol. 2007;62:553–9. doi: 10.1002/ana.21319. [DOI] [PubMed] [Google Scholar]
- 67.Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A. Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem. 2004;279:15499–504. doi: 10.1074/jbc.M313295200. [DOI] [PubMed] [Google Scholar]
- 68.Shibata N, Hirano A, Kobayashi M, Sasaki S, Kato T, Matsumoto S, et al. Cu/Zn superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci Lett. 1994;179:149–52. doi: 10.1016/0304-3940(94)90956-3. [DOI] [PubMed] [Google Scholar]
- 69.Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–7. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 70.Auclair JR, Johnson JL, Liu Q, Salisbury JP, Rotunno MS, Petsko GA, et al. Post-translational modification by cysteine protects Cu/Zn-superoxide dismutase from oxidative damage. Biochemistry. 2013;52:6137–44. doi: 10.1021/bi4006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin W, Ho L, Wang J, Peskind E, Pasinetti GM. S100A7, a novel Alzheimer’s disease biomarker with non-amyloidogenic alpha-secretase activity acts via selective promotion of ADAM-10. PLoS One. 2009;4:e4183. doi: 10.1371/journal.pone.0004183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 73.Julien SG, Dube N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 74.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 75.Carroll J, Fearnley IM, Walker JE. Definition of the mitochondrial proteome by measurement of molecular masses of membrane proteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16170–5. doi: 10.1073/pnas.0607719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Catherman AD, Li M, Tran JC, Durbin KR, Compton PD, Early BP, et al. Top Down Proteomics of Human Membrane Proteins from Enriched Mitochondrial Fractions. Analytical Chemistry. 2013;85:1880–8. doi: 10.1021/ac3031527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Whitelegge J, Halgand F, Souda P, Zabrouskov V. Top-down mass spectrometry of integral membrane proteins. Expert Rev Proteomics. 2006;3:585–96. doi: 10.1586/14789450.3.6.585. [DOI] [PubMed] [Google Scholar]
- 78.Whitelegge JP. HPLC and mass spectrometry of intrinsic membrane proteins. Methods Mol Biol. 2004;251:323–40. doi: 10.1385/1-59259-742-4:323. [DOI] [PubMed] [Google Scholar]
- 79.Whitelegge JP, le Coutre J, Lee JC, Engel CK, Prive GG, Faull KF, et al. Toward the bilayer proteome, electrospray ionization-mass spectrometry of large, intact transmembrane proteins. Proc Natl Acad Sci U S A. 1999;96:10695–8. doi: 10.1073/pnas.96.19.10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Whitelegge JP, Zhang H, Aguilera R, Taylor RM, Cramer WA. Full subunit coverage liquid chromatography electrospray ionization mass spectrometry (LCMS+) of an oligomeric membrane protein: cytochrome b(6)f complex from spinach and the cyanobacterium Mastigocladus laminosus. Mol Cell Proteomics. 2002;1:816–27. doi: 10.1074/mcp.m200045-mcp200. [DOI] [PubMed] [Google Scholar]
- 81.Laganowsky A, Reading E, Hopper JT, Robinson CV. Mass spectrometry of intact membrane protein complexes. Nat Protoc. 2013;8:639–51. doi: 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Catherman AD, Li M, Tran JC, Durbin KR, Compton PD, Early BP, et al. Top down proteomics of human membrane proteins from enriched mitochondrial fractions. Anal Chem. 2013;85:1880–8. doi: 10.1021/ac3031527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Doucette AA, Tran JC, Wall MJ, Fitzsimmons S. Intact proteome fractionation strategies compatible with mass spectrometry. Expert Rev Proteomics. 2011;8:787–800. doi: 10.1586/epr.11.67. [DOI] [PubMed] [Google Scholar]
- 84.Kellie JF, Tran JC, Lee JE, Ahlf DR, Thomas HM, Ntai I, et al. The emerging process of Top Down mass spectrometry for protein analysis: biomarkers, protein-therapeutics, and achieving high throughput. Mol Biosyst. 2010;6:1532–9. doi: 10.1039/c000896f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 86.Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73:5683–90. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 87.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–8. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Neverova I, Van Eyk JE. Role of chromatographic techniques in proteomic analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;815:51–63. doi: 10.1016/j.jchromb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 89.Hage DS. Affinity chromatography: a review of clinical applications. Clin Chem. 1999;45:593–615. [PubMed] [Google Scholar]
- 90.de Frutos M, Regnier FE. Tandem chromatographic-immunological analyses. Anal Chem. 1993;65:17A–25A. doi: 10.1021/ac00049a001. [DOI] [PubMed] [Google Scholar]
- 91.Nelson RW, Krone JR, Bieber AL, Williams P. Mass spectrometric immunoassay. Anal Chem. 1995;67:1153–8. doi: 10.1021/ac00103a003. [DOI] [PubMed] [Google Scholar]
- 92.Niederkofler EE, Tubbs KA, Kiernan UA, Nedelkov D, Nelson RW. Novel mass spectrometric immunoassays for the rapid structural characterization of plasma apolipoproteins. J Lipid Res. 2003;44:630–9. doi: 10.1194/jlr.D200034-JLR200. [DOI] [PubMed] [Google Scholar]
- 93.Compton PD, Zamdborg L, Thomas PM, Kelleher NL. On the scalability and requirements of whole protein mass spectrometry. Anal Chem. 2011;83:6868–74. doi: 10.1021/ac2010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alpert AJ. Size Exclusion High-Performance Liquid Chromatography of Small Solutes. In: Wu C-S, editor. Column Handbook for Size Exclusion Chromatography. San Diego: Academic Press; 1999. pp. 249–66. [Google Scholar]
- 95.Doucette AA, Tran JC, Wall MJ, Fitzsimmons S. Intact proteome fractionation strategies compatible with mass spectrometry. Expert Review of Proteomics. 2011;8:787–800. doi: 10.1586/epr.11.67. [DOI] [PubMed] [Google Scholar]
- 96.Chen X, Ge Y. Ultrahigh pressure fast size exclusion chromatography for top-down proteomics. Proteomics. 2013;13:2563–6. doi: 10.1002/pmic.201200594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sharma S, Simpson DC, Tolic N, Jaitly N, Mayampurath AM, Smith RD, et al. Proteomic profiling of intact proteins using WAX-RPLC 2-D separations and FTICR mass spectrometry. J Proteome Res. 2007;6:602–10. doi: 10.1021/pr060354a. [DOI] [PubMed] [Google Scholar]
- 98.Simpson DC, Ahn S, Pasa-Tolic L, Bogdanov B, Mottaz HM, Vilkov AN, et al. Using size exclusion chromatography-RPLC and RPLC-CIEF as two-dimensional separation strategies for protein profiling. Electrophoresis. 2006;27:2722–33. doi: 10.1002/elps.200600037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Neverova I, Van Eyk JE. Role of chromatographic techniques in proteomic analysis. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2005;815:51–63. doi: 10.1016/j.jchromb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 100.Gundry RL, Fu Q, Jelinek CA, Van Eyk JE, Cotter RJ. Investigation of an albumin-enriched fraction of human serum and its albuminome. Proteomics Clinical Applications. 2007;1:73–88. doi: 10.1002/prca.200600276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iavarone AT, Williams ER. Mechanism of charging and supercharging molecules in electrospray ionization. J Am Chem Soc. 2003;125:2319–27. doi: 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lomeli SH, Yin S, Ogorzalek Loo RR, Loo JA. Increasing charge while preserving noncovalent protein complexes for ESI-MS. J Am Soc Mass Spectrom. 2009;20:593–6. doi: 10.1016/j.jasms.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Valeja SG, Tipton JD, Emmett MR, Marshall AG. New reagents for enhanced liquid chromatographic separation and charging of intact protein ions for electrospray ionization mass spectrometry. Anal Chem. 2010;82:7515–9. doi: 10.1021/ac1016858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miladinovic SM, Fornelli L, Lu Y, Piech KM, Girault HH, Tsybin YO. In-spray supercharging of peptides and proteins in electrospray ionization mass spectrometry. Anal Chem. 2012;84:4647–51. doi: 10.1021/ac300845n. [DOI] [PubMed] [Google Scholar]
- 105.Park J, Qin H, Scalf M, Hilger RT, Westphall MS, Smith LM, et al. A mechanical nanomembrane detector for time-of-flight mass spectrometry. Nano Lett. 2011;11:3681–4. doi: 10.1021/nl201645u. [DOI] [PubMed] [Google Scholar]
- 106.Chen X, Westphall MS, Smith LM. Mass spectrometric analysis of DNA mixtures: instrumental effects responsible for decreased sensitivity with increasing mass. Anal Chem. 2003;75:5944–52. doi: 10.1021/ac030127h. [DOI] [PubMed] [Google Scholar]
- 107.Kelleher NL, Senko MW, Siegel MM, McLafferty FW. Unit resolution mass spectra of 112 kDa molecules with 3 Da accuracy. Journal of the American Society for Mass Spectrometry. 1997;8:380–3. [Google Scholar]
- 108.Valeja SG, Kaiser NK, Xian F, Hendrickson CL, Rouse JC, Marshall AG. Unit Mass Baseline Resolution for an Intact 148 kDa Therapeutic Monoclonal Antibody by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Analytical Chemistry. 2011;83:8391–5. doi: 10.1021/ac202429c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kostyukevich YI, Vladimirov GN, Nikolaev EN. Dynamically harmonized FT-ICR cell with specially shaped electrodes for compensation of inhomogeneity of the magnetic field. Computer simulations of the electric field and ion motion dynamics. J Am Soc Mass Spectrom. 2012;23:2198–207. doi: 10.1007/s13361-012-0480-1. [DOI] [PubMed] [Google Scholar]
- 110.Turecek F. N[bond]C(alpha) bond dissociation energies and kinetics in amide and peptide radicals. Is the dissociation a non-ergodic process? J Am Chem Soc. 2003;125:5954–63. doi: 10.1021/ja021323t. [DOI] [PubMed] [Google Scholar]
- 111.Breuker K, McLafferty FW. Stepwise evolution of protein native structure with electrospray into the gas phase, 10(-12) to 10(2) s. Proc Natl Acad Sci U S A. 2008;105:18145–52. doi: 10.1073/pnas.0807005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Breuker K, Oh H, Horn DM, Cerda BA, McLafferty FW. Detailed unfolding and folding of gaseous ubiquitin ions characterized by electron capture dissociation. J Am Chem Soc. 2002;124:6407–20. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- 113.Peng Y, Chen X, Zhang H, Xu Q, Hacker TA, Ge Y. Top-down Targeted Proteomics for Deep Sequencing of Tropomyosin Isoforms. Journal of Proteome Research. 2013;12:187–98. doi: 10.1021/pr301054n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Han X, Jin M, Breuker K, McLafferty FW. Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons. Science. 2006;314:109–12. doi: 10.1126/science.1128868. [DOI] [PubMed] [Google Scholar]
- 115.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, et al. Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J Am Chem Soc. 2013;135:12646–51. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389:1017–31. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 117.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1:252–62. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 118.Pesavento JJ, Mizzen CA, Kelleher NL. Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Anal Chem. 2006;78:4271–80. doi: 10.1021/ac0600050. [DOI] [PubMed] [Google Scholar]
- 119.Jiang L, Smith JN, Anderson SL, Ma P, Mizzen CA, Kelleher NL. Global assessment of combinatorial post-translational modification of core histones in yeast using contemporary mass spectrometry. LYS4 trimethylation correlates with degree of acetylation on the same H3 tail. J Biol Chem. 2007;282:27923–34. doi: 10.1074/jbc.M704194200. [DOI] [PubMed] [Google Scholar]
- 120.Pesavento JJ, Bullock CR, LeDuc RD, Mizzen CA, Kelleher NL. Combinatorial modification of human histone H4 quantitated by two-dimensional liquid chromatography coupled with top down mass spectrometry. J Biol Chem. 2008;283:14927–37. doi: 10.1074/jbc.M709796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Du Y, Parks BA, Sohn S, Kwast KE, Kelleher NL. Top-down approaches for measuring expression ratios of intact yeast proteins using Fourier transform mass spectrometry. Anal Chem. 2006;78:686–94. doi: 10.1021/ac050993p. [DOI] [PubMed] [Google Scholar]
- 122.Waanders LF, Hanke S, Mann M. Top-down quantitation and characterization of SILAC-labeled proteins. J Am Soc Mass Spectrom. 2007;18:2058–64. doi: 10.1016/j.jasms.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 123.Collier TS, Hawkridge AM, Georgianna DR, Payne GA, Muddiman DC. Top-down identification and quantification of stable isotope labeled proteins from Aspergillus flavus using online nano-flow reversed-phase liquid chromatography coupled to a LTQ-FTICR mass spectrometer. Anal Chem. 2008;80:4994–5001. doi: 10.1021/ac800254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Collier TS, Sarkar P, Rao B, Muddiman DC. Quantitative top-down proteomics of SILAC labeled human embryonic stem cells. J Am Soc Mass Spectrom. 2010;21:879–89. doi: 10.1016/j.jasms.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 125.Bergmann U, Ahrends R, Neumann B, Scheler C, Linscheid MW. Application of metal-coded affinity tags (MeCAT): absolute protein quantification with top-down and bottom-up workflows by metal-coded tagging. Anal Chem. 2012;84:5268–75. doi: 10.1021/ac203460b. [DOI] [PubMed] [Google Scholar]
- 126.Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA. Identification of post-translational modifications by blind search of mass spectra. Nat Biotechnol. 2005;23:1562–7. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- 127.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J Am Soc Mass Spectrom. 2000;11:320–32. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 128.Jaitly N, Mayampurath A, Littlefield K, Adkins JN, Anderson GA, Smith RD. Decon2LS: An open-source software package for automated processing and visualization of high resolution mass spectrometry data. BMC Bioinformatics. 2009;10:87. doi: 10.1186/1471-2105-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guner H, Close PL, Cai W, Zhang H, Peng Y, Gregorich ZR, et al. MASH Suite: A User-Friendly and Versatile Software Interface for High-Resolution Mass Spectrometry Data Interpretation and Visualization. J Am Soc Mass Spectrom. 2014;25:464–70. doi: 10.1007/s13361-013-0789-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu X, Inbar Y, Dorrestein PC, Wynne C, Edwards N, Souda P, et al. Deconvolution and database search of complex tandem mass spectra of intact proteins: a combinatorial approach. Mol Cell Proteomics. 2010;9:2772–82. doi: 10.1074/mcp.M110.002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perkel JM. Tearing the top off ‘Top-Down’ Proteomics. Biotechniques. 2012;53:75–8. doi: 10.2144/000113900. [DOI] [PubMed] [Google Scholar]
- 132.Zamdborg L, LeDuc RD, Glowacz KJ, Kim YB, Viswanathan V, Spaulding IT, et al. ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic Acids Res. 2007;35:W701–6. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tsai YS, Scherl A, Shaw JL, MacKay CL, Shaffer SA, Langridge-Smith PR, et al. Precursor ion independent algorithm for top-down shotgun proteomics. J Am Soc Mass Spectrom. 2009;20:2154–66. doi: 10.1016/j.jasms.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 134.Liu X, Sirotkin Y, Shen Y, Anderson G, Tsai YS, Ting YS, et al. Protein identification using top-down. Mol Cell Proteomics. 2012;11:M111 008524. doi: 10.1074/mcp.M111.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Brower V. Proteomics: biology in the post-genomic era. Companies all over the world rush to lead the way in the new post-genomics race. EMBO Rep. 2001;2:558–60. doi: 10.1093/embo-reports/kve144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Savaryn JP, Catherman AD, Thomas PM, Abecassis MM, Kelleher NL. The emergence of top-down proteomics in clinical research. Genome Med. 2013;5:53. doi: 10.1186/gm457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.