

Abstract
AIM: To investigate interleukin-18 (IL-18) in patients with chronic panreatitis (CP).
METHODS: We studied 29 patients with CP and 30 healthy controls. Peripheral blood mononuclear cells (PBMC) were isolated and incubated with 50 mmol/L ethanol, lipopolysaccharide (LPS) (doses 25 g/L, 250 g/L, 2500 g/L) and both agents for 24 h. Levels of IL-18 in the supernatants, and levels of IL-18, IL-12, interferon (IFN)-γ and soluble CD14 in the serum were analysed by ELISA technique. Expression of IL-18 in PBMC was investigated by reverse-transcription (RT)-PCR. IL-18 protein levels in CP tissue and in normal pancreas were studied by ELISA technique. IL-18 levels in PBMC and pancreatic tissue were determined by Westernblot. Immunohistochemistry for pancreatic IL-18 expression was performed.
RESULTS: In patients, IL-18 serum levels were significantly enhanced by 76% (mean: 289.9 ± 167.7 ng/L) compared with controls (mean: 165.2 ± 43.6 ng/L; P < 0.0005). IL-12 levels were enhanced by 25% in patients (18.3 ± 7.3 ng/L) compared with controls (14.7 ± 6.8 ng/L, P = 0.0576) although not reaching the statistical significance. IFN-γ and soluble CD14 levels were not increased. In vitro, LPS stimulated significantly and dose-dependently IL-18 secretion from PBMC. Incubation with ethanol reduced LPS-stimulated IL-18 secretion by about 50%. The mRNA expression of IL-18 in PBMC and the response of PBMC to ethanol and LPS was similar in CP patients and controls. In PBMC, no significant differences in IL-18 protein levels were detected between patients and controls. IL-18 protein levels were increased in CP tissues compared to normal pancreatic tissues. IL-18 was expressed by pancreatic acinar cells and by infiltrating inflammatory cells within the pancreas.
CONCLUSION: IL-18 originates from the chronically inflammed pancreas and appears to be involved in the fibrotic destruction of the organ.
Keywords: Chronic pancreatitis, Cytokines, Interleukin-18, Pancreatic fibrosis
INTRODUCTION
Chronic pancreatitis represents an inflammatory disease characterized by repeated attacks of acute pancreatitis, severe abdominal pain, progressive destruction of the pancreatic tissue with fibrous replacement of the parenchyma leading to both exocrine and endocrine insufficiency[1]. In industrialized countries, excessive alcohol consumption is associated with the development of chronic pancreatitis in the majority of patients[2,3]. The early stages of the human disease remain almost inaccessible to investigation. Recent genetic findings suggest that premature digestive enzyme activation with subsequent pancreatic autodigestion represents a dominant factor in the initiation of acute pancreatitis[4,5]. These genetic studies also support a progressive link between repeated episodes of acute pancreatitis and the development of chronic pancreatitis[5]. However, the exact immune mechanisms underlying the progression of chronic pancreatitis remain unclear.
The identification and characterization of pancreatic stellate cells (PSC) provided deep insights into the development of pancreatic fibrosis[6,7]. Activated PSC synthesize and secrete increased amounts of extracellular matrix proteins resulting in the fibrotic destruction of the pancreas[6,7]. Recent in vitro studies have demonstrated that alcohol and its metabolite acetaldehyde[8], oxidative stress[8], growth factors such as platelet derived growth factor[6], and the cytokine transforming growth factor (TGF)-β1[6] have the capacity to activate PSC during pancreatic injury. More recently, it was shown that PSC also responds to additional proinflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, and antiinflammatory cytokines such as IL-10[9].
During chronic pancreatitis, lymphocytes and mononuclear cells infiltrate the pancreas and contribute to the local progression of the disease through T-lymphocyte mediated cytotoxicity and production of cytokines[10-14]. However, data regarding the role of cytokines in chronic pancreatitis remain limited. IL-18 represents a proinflammatory cytokine that plays an important role in the Th-1 response due to its ability to induce interferon (IFN)-γ production in T-cells and natural killer cells[15,16]. IL-18 has been investigated in a variety of inflammatory and autoimmune human diseases[17]. In previous studies, our group and others have demonstrated an upregulation of serum IL-18 levels in patients with acute pancreatitis[18-20]. In vitro studies have shown that endotoxin induces IL-18 gene expression and secretion in human peripheral blood mononuclear cells (PBMC)[21]. In a previous investigation, the serum levels and gene expression of IL-18 in PBMC of patients with alcoholic liver cirrhosis were significantly enhanced compared to healthy controls, and IL-18 levels correlated with plasma endotoxin levels[22].
These data raise the possibility that IL-18 also participates in the immune mechanisms that result in the fibrotic destruction of the pancreas during chronic pancreatitis. Thus, the aim of the present study was to investigate this cytokine in patients with chronic pancreatitis. We determined the serum levels of IL-18, IFN, IL-12 and soluble CD14 in patients with chronic pancreatitis and healthy controls. We performed several in vitro studies with PBMC. We determined the protein expression of IL-18 in chronic pancreatitis tissue and in normal pancreas, and conducted immunohistochemical investigations in human pancreatic tissues.
MATERIALS AND METHODS
Patients
A total of 29 patients (22 males, 7 females; mean age 52 ± 11 years) with alcoholic and non-alcoholic chronic pancreatitis and 30 healthy controls with no history of alcohol abuse (10 males, 20 females; mean age 39 ± 11 year) were prospectively enrolled into the study at the University Hospital of Heidelberg at Mannheim, Mannheim, Germany. The study was approved by the Ethics Committee of the Faculty of Clinical Medicine Mannheim, University of Heidelberg, Germany. Written informed consent was obtained from each participant.
Data on the history of both alcohol consumption and the clinical course of pancreatic disease were assessed by patient self-report and review of the medical records. In each participant, a detailed history of alcohol intake was established using different screening methods, including the Lübeck alcohol dependence and abuse screening test (LAST)[23], the alcohol use disorders identification test (AUDIT)[24], the lifetime drinking history (LDH)[25], and a patient interview questioning ICD-10 criteria of chronic alcohol dependence[26]. Alcoholic disease etiology was established with the presence of at least one of the following criteria: (1) patient self-report of alcohol abuse as cause of the disease, (2) patient self-report with a history of excessive alcohol intake of at least 80 g per day in males and 60 g per day in females for some years, or (3) smaller amounts of daily alcohol intake in combination with answers gained from the above mentioned screening methods for alcohol consumption that allowed the diagnosis of chronic alcohol abuse.
The diagnosis of chronic pancreatitis required the typical clinical features of chronic pancreatitis with or without recurrent episodes of acute pancreatitis, and was based on the determination of pancreatic exocrine and endocrine function, pancreatic imaging or histological tissue examination. Pancreatic imaging was performed either by endoscopic retrograde pancreatography (ERP), computed tomography (CT), magnetic resonance imaging (MRI), or endosonography. In each patient, abdominal ultrasound was performed. According to the definitions of an international workshop on chronic pancreatitis[27], patients were further classified as suffering “definite” or “probable” chronic pancreatitis. Briefly, the classification of “definite” chronic pancreatitis required a typical clinical history of chronic pancreatitis and one or more of the following criteria: (1) calcifications in the pancreas, (2) moderate to marked ductal lesions, (3) marked exocrine insufficiency, and (4) typical histology of an adequate surgical specimen[27]. For the diagnosis of “probable” chronic pancreatitis, one or more of the following criteria were present in addition to the typical clinical features: (1) mild ductal alterations, (2) recurrent or persistent pseudocysts, (3) pathological secretin test, and (4) endocrine insufficiency[27].
In all patients, an episode of acute pancreatitis at the time of recruitment or within the last two months before inclusion into the study was excluded. Additional exclusion criteria were infections with fever or leukocytosis, liver cirrhosis and surgical or endoscopic interventions within 2 mo before examination.
Routine laboratory parameters
Routine laboratory parameters were determined in serum samples taken at the same time as samples for cytokine and endotoxin measurement. These parameters included serum concentrations of amylase, lipase, C-reactive protein (CRP), creatinine, and total white blood cell count. Endocrine pancreatic insufficiency was determined by the presence of diabetes mellitus requiring antidiabetic treatment or records of an abnormal oral glucose tolerance test. Exocrine pancreatic insufficiency was defined by diarrhea, steatorrhea or maldigestion that was markedly reduced by enzyme supplementation. In some patients, pancreatic exocrine function was determined by using one or more of the following commercially available tests according to the manufacturer’s recommendations: measurement of fecal chymotrypsin by a colorimetric method (Chymo, Boehringer, Germany), determination of fecal fat excretion by infrared reflection method (Esetek Analyser Fenir 8820, TSZ Stimotron AG, Wettenberg-Launsbach, Germany), measurement with the Pankreolauryl test N (Temmler Pharma GmbH, Marburg, Germany) or application of the pancreozymin-secretin-test (Sekretolin Diagnostikum, Hoechst AG, Germany or Takus, Pharmacia GmbH, Erlangen, Germany).
PBMC isolation and incubation with ethanol and endotoxin
Peripheral venous blood was collected from patients and healthy controls into sterile, pyrogen-free disposable syringes with endotoxin-free heparin (10 ku/L). As reported previously[22], PBMC was separated by standard density gradient centrifugation (Ficoll-Paque method) and adjusted to 3 × 109 cells/L in RPMI 1640 supplemented with 100 mL/L heat inactivated fetal bovine serum. PBMC were incubated with or without lipopolysaccharide (LPS) (doses 25 g/L, 250 g/L, 2500 g/L), in the absence or presence of 50 mol/L ethanol for 24 h. Cells were spun down, and PBMC supernatants were stored at -20°C until measurement of cytokine levels. For RNA extraction, cells were stored at -80°C in 4 mol/L GTC extraction buffer.
Immunoassay for IL-18, IFN, IL-12, soluble CD14
Concentrations of IL-18 in the serum, supernatants of PBMC and pancreatic tissues were determined by a specific sandwich enzyme linked immunoassay (ELISA; Fujisaki Institute, Hayashibara Biochemical Laboratories, Inc., Okayama, Japan) with minor modifications as described previously[22]. Serum concentrations of IFN were determined by a specific ELISA using two monoclonal antibodies as described previously[28]. Serum concentrations of IL-12 and soluble CD14 were also determined by ELISA technique as described previously[28].
RNA isolation and IL-18 RT-PCR analysis in PBMC
RNA extraction from PBMC was performed using acid phenol-chloroform extraction[22]. The RNA concentration was quantified spectrophotometrically. Complementary DNA of PBMC was obtained by reverse-transcription (RT) using 1 μg RNA and oligo d(T) primers. PCR for IL-18 and the housekeeping enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was performed as described previously[22]. Densitometric assessment of the PCR products was performed using EASY Plus 3.2 software (Herolab, Wiesloch, Germany). Semiquantitative PCR results were obtained by grading a ratio between the densitometry results of IL-18 and GAPDH.
Endotoxin assay
Plasma endotoxin levels were determined using an automated kinetic turbodimetric limulus amoebocyte lysate microtiter test as described previously[22].
Pancreatic tissue samples
Pancreatic tissue samples were obtained from 8 patients with chronic pancreatitis and from 2 individuals without pancreatic disease. Two of these patients with chronic pancreatitis were obtained from the present investigation and had later to be operated due to the development of a benign pancreatic mass and intractable pancreatic pain. The remaining 6 patients with chronic pancreatitis and the 2 individuals without pancreatic disease were obtained from clinical routine interventions. In 4 of these patients with chronic pancreatitis, surgery was necessary due to the development of pancreatic cancer.
Human pancreatic tissue and cell lysates of PBMC were homogenized in 5 mmol/L Hepes, pH 7.0, 280 mmol/L mannitol, 10 mmol/L KCl, 1 mmol/L MgCl2, 1 mmol/L benzamidine, 20 mg/L trypsin inhibitor, 1 μmol/L leupeptin, and 0.2 mmol/L PMSF and boiled in electrophoresis sample buffer. Proteins were electrophoretically separated on SDS-125 g/L polyacrylamide gels. Electrotransfer to nitrocellulose membranes was done as described previously[29]. After staining with 2 g/L Ponceau S to check the efficiency of the transfer, free binding sites of the membrane were blocked with 10 g/L bovine serum albumin in Tris-buffered saline (10 mmol/L Tris-HCl, pH 8.0, and 150 mmol/L NaCl) for 60 min, followed by 75 min incubation with anti-IL-18 antibody (Natutec, Frankfurt, Germany) diluted in Tris-buffered saline plus 2 mL/L Tween 20. Bound antibodies were visualized with secondary antibodies conjugated to horse radish peroxidase using an enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech, Freiburg, Germany) and autoradiography film (Fujifilm Super HR-E 30, Fuji photo film, Düsseldorf, Germany).
Immunohistochemistry
Frozen tissue specimen were cut in 10 μm cryostat sections, transferred on glass slides and air-dried overnight. Immunohistochemistry was performed according to the streptavidin-biotin method. Sections were washed in tris-buffered saline (TBS) and incubated with 30 g/L bovine serum albumine (BSA) for 10 min at room temperature to block non-specific antibody reactions. The sections were incubated overnight at 4°C with the primary antibody at a 1:5000 dilution (anti-human IL-18 polyclonal antibody, NatuTec, Frankfurt am Main, Germany). The slides were then rinsed repeatedly with TBS and were incubated with a biotin-streptavidin-conjugated secondary antibody (goat anti-rabbit immunoglobulin-specific antibody, Jackson Immuno Research, West Grove, USA) for 30 min at room temperature. The slides were washed again with TBS and were treated with a streptavidin-alkaline-phosphatase complex. Liquid diaminobenzidine was added as chromogen, and counterstaining was performed with hematoxylin. Controls were performed by using mouse serum (Sigma, Saint Louis, USA) as primary antibody and by administration of recombinant human IL-18 (MoBiTec, Göttingen, Germany) in excess to the primary antibody before incubation. Light microscopical investigations were performed using a Zeiss Axioskop microscope.
Statistical analysis
We used the t test, if necessary with the Welch correction for unequal variances, for analysis of IL-18, IFN, IL-12 and soluble CD14 levels between patients and controls. These data are expressed as mean ± SD. P < 0.05 was considered significant. We used paired t tests for the comparison of IL-18 secretion from PBMC. The Bonferroni-Holm correction was applied to adjust for calculating three tests at a time to compare the IL-18 secretion from PBMC after different stimulations.
RESULTS
Clinical characteristics
Alcoholic chronic pancreatitis was diagnosed in 23 patients, and non-alcoholic chronic pancreatitis was found in 6 patients (Table 1). According to the recommendations of a workshop on chronic pancreatitis, 21 patients were classified with “definite” chronic pancreatitis, and 8 patients were classified with “probable” chronic pancreatitis (Table 1). An episode of acute pancreatitis was excluded in all patients by physical examination, abdominal ultrasound and determination of routine laboratory parameters (Table 2).
Table 1.
Clinical characteristics of patients with chronic pancreatitis and healthy controls
| Clinical characteristics | Patients | Controls |
| n | 29 | 30 |
| Age (yr) | 52 ± 11 | 39 ± 11 |
| Sex (Male/Female) | 22/7 | 10/20 |
| Alcoholic chronic pancreatitis | 23/29 | NA1 |
| Non-alcoholic chronic pancreatitis | 6/29 | NA1 |
| Definite chronic pancreatitis | 21/29 | NA1 |
| Probable chronic pancreatitis | 8/29 | NA1 |
| Exocrine insufficiency | 16/29 | NA1 |
| Endocrine insufficiency | 11/29 | NA1 |
Not applicable.
Table 2.
Routine laboratory parameters in patients with chronic pancreatitis and healthy controls (mean ± SD)
| Parameter | Normal range | Patients | Controls |
| White cell blood count | 3.6-11.0 × 109/L | 7.0 ± 1.9 | 6.0 ± 1.2 |
| C-reactive protein | < 5 mg/L | 6.4 ± 7.7 | 3.2 ± 0.9 |
| Creatinine | 6-11 mg/L | 0.9 ± 0.3 | 0.9 ± 0.2 |
| Amylase | 8.4-31.7 U/L | 33.0 ± 22.6 | 33.0 ± 17.4 |
| Lipase | < 190 U/L | 182.0 ± 98.0 | 161.0 ± 64.0 |
Serum levels of IL-18, IL-12 and IFN in CP patients
The fasted serum IL-18 levels were significantly enhanced by 76% in 29 patients with chronic pancreatitis (mean ± SD: 289.9 ± 167.7 ng/L) compared to healthy controls (n = 30; 165.2 ± 43.6 ng/L; Welch’s T test, P < 0.0005). The IL-12 levels were enhanced by 25% in patients (n = 27 due to lack of sample in 2 patients; mean ± SD: 18.3 ± 7.3 ng/L) compared to controls (n = 30, 14.7 ± 6.8 ng/L). We observed a trend towards a statistically significant difference between patients and control subjects (T test, P = 0.0576). Serum IFN levels were not increased in patients (n = 25 due to lack of sample in 4 patients; 28 ± 15.4 ng/L) compared to controls (n = 24 due to lack of sample in 6 controls; 34.4 ± 18.0 ng/L; T test, P = 0.18).
Serum soluble CD14 and plasma endotoxin levels in CP patients
Serum levels of soluble CD14 were similar in patients (n = 16, mean ± SD: 2723.3 ± 649.2 ng/L) and healthy controls (n = 26, mean ± SD: 2630.6 ± 480.3 ng/L; Welch’s T-test: P = 0.63). Endotoxemia was not detectable in patients (n = 29) and controls (n = 30).
Expression of IL-18 mRNA in PBMC of CP patients
We investigated the IL-18 mRNA expression in PBMC of patients with chronic pancreatitis by RT-PCR to determine if the enhanced serum IL-18 levels in chronic pancreatitis represent a result of an increased gene expression in PBMC. No significant difference was found between the mRNA expression of IL-18 in patients with chronic pancreatitis (n = 5 patients with highest IL-18 serum levels) in comparison to healthy controls (n = 5 control individuals with lowest IL-18 serum levels) (Figure 1). The semiquantitative analysis of the ratio between the densitometric results from the PCR-products of IL-18 and GAPDH did not reveal a significant difference. The ratio for patients with chronic pancreatitis and healthy controls was 0.438 ± 0.074 and 0.410 ± 0.082, respectively.
Figure 1.
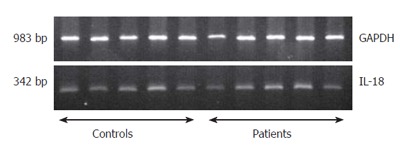
Analysis of RT-PCR amplification of IL-18 in PBMC of CP patients.
IL-18 protein levels in pancreatic tissues
IL-18 protein levels in pancreatic tissues were determined by ELISA technique in two patients with chronic pancreatitis, and in two individuals without pancreatic disease. The protein levels of IL-18 in chronic pancreatitis tissue (n = 2, 50.5 ng/L and 86.1 ng/L) were enhanced compared to pancreatic tissue from individuals without pancreatic disease (n = 2, 33.6 ng/L and 25.1 ng/L). In these patients and control subjects, IL-18 protein levels in the pancreas were also determined by Westernblot (Figure 2A). The IL-18 protein levels were again enhanced in patients with chronic pancreatitis compared to the control individuals (Figure 2A).
Figure 2.
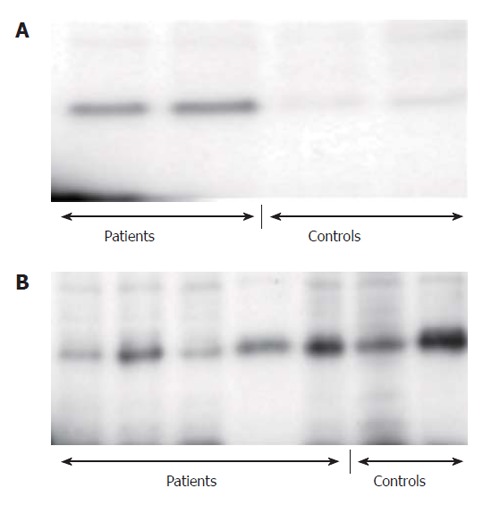
IL-18 in cell lysates of PBMC and in pancreatic tissue of CP patients. A: In the pancreatic tissue; B: In cell lysates of PBMC.
IL-18 protein levels in cell lysates of PBMC
IL-18 protein levels were determined in cell lysates of PBMC by Westernblot investigations (Figure 2B). We studied PBMC cell lysates from 5 patients with chronic pancreatitis and from 2 control individuals to determine if the elevated levels of IL-18 in patients with chronic pancreatitis result from an increased expression of IL-18 in PBMC. We confirmed IL-18 protein expression in the cell lysates of both patients and controls. However, we did not detect significant differences in IL-18 protein expression between cell lysates from patients and controls (Figure 2B).
IL-18 levels in the supernatants from PBMC after incubation with ethanol and LPS
We investigated the IL-18 levels in the supernatants of PBMC from all patients (n = 29) and all controls (n = 30). We stimulated the PBMC with ethanol and LPS to detect a possible influence of these agents on IL-18 secretion from PBMC and to reveal possible differences between PBMC from patients and control subjects. These in vitro studies demonstrated that the basal secretion of IL-18 from PBMC was similar in patients and control individuals.
The stimulation of PBMC with LPS (25 g/L, 250 g/L and 2500 g/L) and the stimulation with LPS (25 g/L, 250 g/L and 2500 g/L) together with ethanol (50 mmol/L) resulted in a dose related and statistically significant enhancement of IL-18 secretion from PBMC of patients and controls (T tests; P < 0.0007) (Figure 3A and B). However, we did not detect significant differences in the secretion patterns of IL-18 between PBMC from patients and control individuals.
Figure 3.
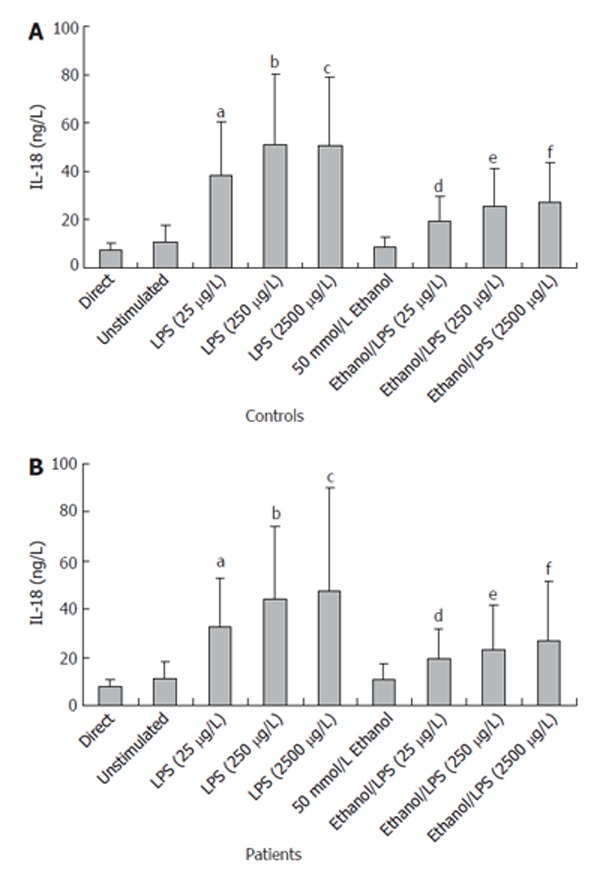
In vitro IL-18 secretion from PBMC of CP patients. aP < 0.0007 25 μg/L LPS vs unstimulated; bP < 0.0007 250 μg/L LPS vs unstimulated; cP < 0.0007 2500 μg/L LPS vs unstimulated; dP < 0.0001 Ethanol/25 μg/L LPS vs 25 μg/L LPS; eP < 0.0001 Ethanol/250 μg/L LPS vs 250 μg/L LPS; fP < 0.0001 Ethanol/2500 μg/L LPS vs 2500 μg/L LPS.
The incubation with ethanol alone for 24 h did not affect basal IL-18 secretion, but ethanol significantly reduced LPS-stimulated IL-18 secretion by about 50% compared to LPS stimulation alone (T tests; P < 0.0001) (Figure 3A and B).
The application of the Bonferroni-Holm correction did not change the significance of previously obtained P values.
Immunohistochemistry
We performed immunohistochemical investigations for IL-18 in pancreatic tissues from 6 patients with chronic pancreatitis. In 4 of these patients with chronic pancreatitis, invasive ductal adenocarcinoma of the pancreas had developed and was the reason for pancreatic surgery. In these patients, we investigated the chronic pancreatitis tissue which was free of pancreatic carcinoma tissues. The immunohistochemical analysis of the chronic pancreatitis tissue revealed that clusters of infiltrating mononuclear cells were stained positive for IL-18 by using anti-IL-18 antibody (Figure 4A). In chronic pancreatitis tissue, clusters of pancreatic acinar cells were also stained positive for the expression of IL-18 (Figure 4B). Thus, IL-18 appears to be expressed in both infiltrating inflammatory cells and pancreatic acinar cells during chronic pancreatitis. In addition, we studied sections from pancreatic carcinoma tissue, and detected positive staining for anti-IL-18 antibody in pancreatic carcinoma cells as well. In general, staining for IL-18 was more pronounced in the samples with pancreatic carcinoma than in the samples from patients with chronic pancreatitis without pancreatic carcinoma. Control investigations with mouse serum and with the IL-18-antigene-antibody-mixture revealed no positive staining for IL-18 and confirmed the accuracy of the method (Figure 4C).
Figure 4.
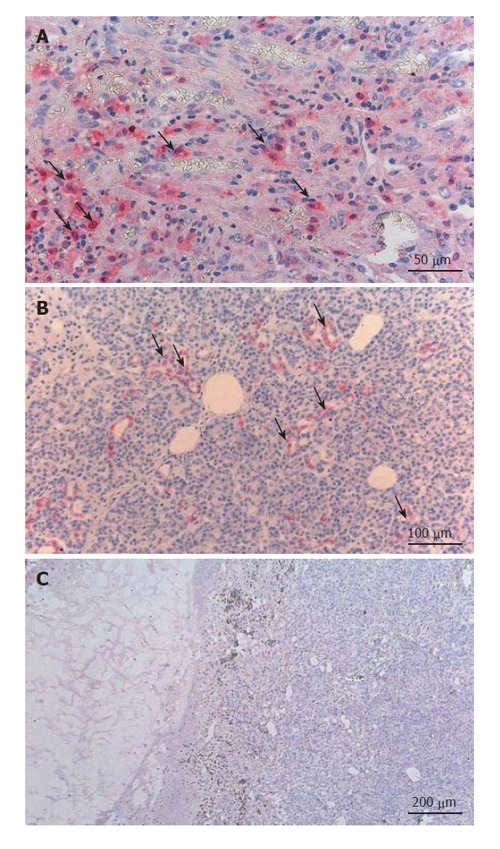
IL-18 expression in pancreatic tissue of CP patients. A: Clusters of infiltrating mononuclear cells stained positive; B: Clusters of pancreatic acinar cells also stained positive for the expression of IL-18; C: No positive staining for IL-18 in the negative control (mouse serum with IL-18 antigen-antibody mixture).
DISCUSSION
The major findings of the present investigation are: (1) the fasted serum levels of IL-18 are significantly enhanced by 76% in patients with chronic pancreatitis compared to healthy control individuals; (2) the mRNA expression of IL-18, the protein levels of IL-18, and the in vitro secretion of IL-18 after stimulation with ethanol and endotoxin is similar in PBMC from patients with chronic pancreatitis and from healthy control subjects; (3) in vitro, ethanol significantly reduces endotoxin-stimulated IL-18 secretion by about 50%; (4) the IL-18 protein levels in chronic pancreatitis tissue are increased; and (5) IL-18 is expressed in pancreatic acinar cells and infiltrating inflammatory cells within the pancreas.
These results suggest that PBMC are not the main source of the enhanced serum levels of IL-18 in patients with chronic pancreatitis. It rather appears that IL-18 originates from the chronically inflammed pancreas. Indeed, there is increasing evidence that immunological mechanisms play an important role in the development and progression of chronic pancreatitis[9-13,30,31]. Cytokines, growth factors and other immunological mediators are produced by resident cells and recruited cells within the chronically inflammed pancreas and contribute to pancreatic fibrosis[14]. In addition, pancreatic acinar cells also produce, release and respond to cytokines[32]. Thus, we assume that IL-18 participates in the destruction of the pancreas during chronic pancreatitis.
Alcohol consumption represents an important risk factor for the development of chronic pancreatitis. Chronic alcohol consumption leads to an increased gut permeability with subsequent endotoxemia, and endotoxin has been reported as a mediator of alcohol induced liver damage[33]. Studies in ethanol-fed rats also suggest a role of endotoxin in the development of pancreatic injury[34]. Endotoxin strongly induced IL-18 gene expression in PBMC, and the response to endotoxin was mainly regulated by the expression of CD14[21]. In our study, we did not detect endotoxin in the plasma of patients with chronic pancreatitis, and the soluble CD14 levels were similar in patients and control individuals. However, it has to be stressed that the present study was not designed to investigate the full impact of endotoxin on the development of pancreatic damage. Enhanced endotoxin plasma levels may only be detectable for short periods of time during acute ethanol consumption in humans, whereas the patients in our study had not consumed alcohol on the day of the investigation. In addition, not all patients with alcoholic chronic pancreatitis were still abusing alcohol at the time of recruitment into the study. Therefore, it appears that the enhanced blood levels of IL-18 do not result from endotoxin-mediated mechanisms.
Interestingly, our in vitro studies showed that the incubation of PBMC with ethanol decreased the endotoxin-stimulated secretion of IL-18 by PBMC thereby suggesting that ethanol consumption modulates the IL-18 expression in the pancreas. However, the role of IL-18 in alcoholic pancreatitis remains speculative since only limited data are available regarding the probably pleiotropic immunological function of IL-18 in chronic pancreatitis.
IL-18 participates both in Th1 and Th2 immune responses[16]. IL-18 induces the production of cytokines such as TNF-α and IL-1 and enhances the production of further chemokines such as IL-8 and MCP-1[35] that are increased in chronic pancreatitis tissue[10,31,36]. Of note, IL-8 plays a major role in the recruitment of infiltrating neutrophils to the site of inflammation[37]. The simultaneous presence of IL-12 and IL-18 results in a marked production of nitric oxide and reactive oxygen intermediates in macrophages and neutrophils[38] which may also facilitate premature intrapancreatic trypsin activation and pancreatic autodigestion[39]. The predominantly antiinflammatory cytokine IL-10 inhibits the synthesis of proinflammatory cytokines such as TNF-α, IL-1, IL-6 and IL-8 and may play a dominant role in protecting the pancreas during pancreatic inflammation[40,41]. There was only minimal induction of the antiinflammatory cytokines IL-1 receptor and IL-10 through IL-18[35], and IL-10 failed to inhibit IL-18 production in response to inflammatory stimuli[42]. Therefore, IL-18 may escape the influence of IL-10 during pancreatic inflammation.
Finally, among its pleiotropic effects, IL-18 strongly induces IFN-γ production[15,16]. Increased levels of IFN were reported in chronic pancreatitis tissue from humans and animals[13,43,44]. Interestingly, the fibrosis in chronic pancreatitis seems to be driven by TGF-β1[45], and IFN-γ and TGF-β1 are linked by an antagonistic relationship[46,47]. INF-γ possesses several antifibrotic characteristics, and IFN-γ has already been used therapeutically in animals and humans with fibrotic diseases[47-49]. The recruitment and activation of infiltrating cells may depend on the local production of inflammatory mediators such as TGF-β1 and IFN-γ[11,31]. Recent in vitro studies have demonstrated that TGF-β1 strongly suppressed the production of IFN-γ that had been induced by costimulation with IL-18 and phytohaemagglutinin, a strong stimulator of IFN-γ synthesis, or by costimulation with IL-18 and IL-12[50]. However, the role of IFN-γ in chronic pancreatitis is not completely clarified, and further studies are required to reveal the role of IL-18 during pancreatic fibrosis.
In conclusion, PSC are activated during pancreatic injury, and these cells produce and release increased amounts of extracellular matrix proteins thereby leading to the fibrotic destruction of the pancreas[6,7]. Pancreatic stellate cells are activated and regulated on exposure to various proinflammatory cytokines[9]. Thus, future studies should address the role of IL-18 in the context of PSC activation and regulation.
ACKNOWLEDGMENTS
We thank Karin Kaiser for her excellent technical assis-tance.
Footnotes
S- Editor Wang GP L- Editor Ma JY E- Editor Ma WH
References
- 1.Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med. 1995;332:1482–1490. doi: 10.1056/NEJM199506013322206. [DOI] [PubMed] [Google Scholar]
- 2.Niebergall-Roth E, Harder H, Singer MV. A review: acute and chronic effects of ethanol and alcoholic beverages on the pancreatic exocrine secretion in vivo and in vitro. Alcohol Clin Exp Res. 1998;22:1570–1583. doi: 10.1111/j.1530-0277.1998.tb03951.x. [DOI] [PubMed] [Google Scholar]
- 3.Apte MV, Wilson JS. Alcohol-induced pancreatic injury. Best Pract Res Clin Gastroenterol. 2003;17:593–612. doi: 10.1016/s1521-6918(03)00050-7. [DOI] [PubMed] [Google Scholar]
- 4.Pfützer RH, Whitcomb DC. SPINK1 mutations are associated with multiple phenotypes. Pancreatology. 2001;1:457–460. doi: 10.1159/000055847. [DOI] [PubMed] [Google Scholar]
- 5.Schneider A, Whitcomb DC. Hereditary pancreatitis: a model for inflammatory diseases of the pancreas. Best Pract Res Clin Gastroenterol. 2002;16:347–363. doi: 10.1053/bega.2002.0311. [DOI] [PubMed] [Google Scholar]
- 6.Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grunert A, Adler G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115:421–432. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- 7.Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Apte MV, Phillips PA, Fahmy RG, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Naidoo D, Wilson JS. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology. 2000;118:780–794. doi: 10.1016/s0016-5085(00)70148-x. [DOI] [PubMed] [Google Scholar]
- 9.Mews P, Phillips P, Fahmy R, Korsten M, Pirola R, Wilson J, Apte M. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut. 2002;50:535–541. doi: 10.1136/gut.50.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunger RE, Mueller C, Z’graggen K, Friess H, Buchler MW. Cytotoxic cells are activated in cellular infiltrates of alcoholic chronic pancreatitis. Gastroenterology. 1997;112:1656–1663. doi: 10.1016/s0016-5085(97)70048-9. [DOI] [PubMed] [Google Scholar]
- 11.Ebert MP, Ademmer K, Müller-Ostermeyer F, Friess H, Büchler MW, Schubert W, Malfertheiner P. CD8+CD103+ T cells analogous to intestinal intraepithelial lymphocytes infiltrate the pancreas in chronic pancreatitis. Am J Gastroenterol. 1998;93:2141–2147. doi: 10.1111/j.1572-0241.1998.00610.x. [DOI] [PubMed] [Google Scholar]
- 12.Emmrich J, Weber I, Nausch M, Sparmann G, Koch K, Seyfarth M, Lohr M, Liebe S. Immunohistochemical characterization of the pancreatic cellular infiltrate in normal pancreas, chronic pancreatitis and pancreatic carcinoma. Digestion. 1998;59:192–198. doi: 10.1159/000007488. [DOI] [PubMed] [Google Scholar]
- 13.Hasel C, Rau B, Perner S, Strater J, Moller P. Differential and mutually exclusive expression of CD95 and CD95 ligand in epithelia of normal pancreas and chronic pancreatitis. Lab Invest. 2001;81:317–326. doi: 10.1038/labinvest.3780240. [DOI] [PubMed] [Google Scholar]
- 14.Vyas SK. Growth factors and cytokines in chronic pancreatitis. In: Johnson CD, Imrie CW, editors. Pancreatic disease: towards the year 2000. London: Springer Verlag; 1999. pp. 155–165. [Google Scholar]
- 15.Dinarello CA. IL-18: A TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J Allergy Clin Immunol. 1999;103:11–24. doi: 10.1016/s0091-6749(99)70518-x. [DOI] [PubMed] [Google Scholar]
- 16.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. 2001;19:423–474. doi: 10.1146/annurev.immunol.19.1.423. [DOI] [PubMed] [Google Scholar]
- 17.Gracie JA, Robertson SE, McInnes IB. Interleukin-18. J Leukoc Biol. 2003;73:213–224. doi: 10.1189/jlb.0602313. [DOI] [PubMed] [Google Scholar]
- 18.Hanck C, Bertsch T, Rossol S, Kurimoto M, Singer MV, Richter A. Enhanced serum levels of IL-18 in patients with severe acute pancreatitis. Digestion. 1999;60:379 (abstract). [Google Scholar]
- 19.Rau B, Baumgart K, Paszkowski AS, Mayer JM, Beger HG. Clinical relevance of caspase-1 activated cytokines in acute pancreatitis: high correlation of serum interleukin-18 with pancreatic necrosis and systemic complications. Crit Care Med. 2001;29:1556–1562. doi: 10.1097/00003246-200108000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Wereszczynska-Siemiatkowska U, Mroczko B, Siemiatkowski A. Serum profiles of interleukin-18 in different severity forms of human acute pancreatitis. Scand J Gastroenterol. 2002;37:1097–1102. doi: 10.1080/003655202320378310. [DOI] [PubMed] [Google Scholar]
- 21.Manigold T, Böcker U, Traber P, Dong-Si T, Kurimoto M, Hanck C, Singer MV, Rossol S. Lipopolysaccharide/endotoxin induces IL-18 via CD14 in human peripheral blood mononuclear cells in vitro. Cytokine. 2000;12:1788–1792. doi: 10.1006/cyto.2000.0783. [DOI] [PubMed] [Google Scholar]
- 22.Hanck C, Manigold T, Bocker U, Kurimoto M, Kolbel CB, Singer MV, Rossol S. Gene expression of interleukin 18 in unstimulated peripheral blood mononuclear cells of patients with alcoholic cirrhosis. Gut. 2001;49:106–111. doi: 10.1136/gut.49.1.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rumpf HJ, Hapke U, Hill A, John U. Development of a screening questionnaire for the general hospital and general practices. Alcohol Clin Exp Res. 1997;21:894–898. [PubMed] [Google Scholar]
- 24.Saunders JB, Aasland OG, Babor TF, de la Fuente JR, Grant M. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO Collaborative Project on Early Detection of Persons with Harmful Alcohol Consumption--II. Addiction. 1993;88:791–804. doi: 10.1111/j.1360-0443.1993.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 25.Skinner HA, Sheu WJ. Reliability of alcohol use indices. The Lifetime Drinking History and the MAST. J Stud Alcohol. 1982;43:1157–1170. doi: 10.15288/jsa.1982.43.1157. [DOI] [PubMed] [Google Scholar]
- 26.Wittchen HU, Zaudig M, Fydrich T. SKID - Strukturiertes Klinisches Interview für DSM-IV. Achse I und II. Hogrefe, Göttingen. 1997. [Google Scholar]
- 27.Ammann RW. A clinically based classification system for alcoholic chronic pancreatitis: summary of an international workshop on chronic pancreatitis. Pancreas. 1997;14:215–221. doi: 10.1097/00006676-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Rossol S, Marinos G, Carucci P, Singer MV, Williams R, Naoumov NV. Interleukin-12 induction of Th1 cytokines is important for viral clearance in chronic hepatitis B. J Clin Invest. 1997;99:3025–3033. doi: 10.1172/JCI119498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feick P, Gilhaus S, Schulz I. Pervanadate stimulates amylase release and protein tyrosine phosphorylation of paxillin and p125(FAK) in differentiated AR4-2J pancreatic acinar cells. J Biol Chem. 1998;273:16366–16373. doi: 10.1074/jbc.273.26.16366. [DOI] [PubMed] [Google Scholar]
- 30.Hanck C, Rossol S, Hartmann A, Singer MV. Cytokine gene expression in peripheral blood mononuclear cells reflects a systemic immune response in alcoholic chronic pancreatitis. Int J Pancreatol. 1999;26:137–145. doi: 10.1385/IJGC:26:3:137. [DOI] [PubMed] [Google Scholar]
- 31.Saurer L, Reber P, Schaffner T, Buchler MW, Buri C, Kappeler A, Walz A, Friess H, Mueller C. Differential expression of chemokines in normal pancreas and in chronic pancreatitis. Gastroenterology. 2000;118:356–367. doi: 10.1016/s0016-5085(00)70218-6. [DOI] [PubMed] [Google Scholar]
- 32.Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurman RG, Bradford BU, Knecht KT, Iimuro Y, Arteel GE, Yin M, Connor HD, Wall C, Raleigh JA, von Frankenberg M, et al. Endotoxin, kupffer cells and alcoholic liver injury. In: Blum HE, Bode C, Bode JC, Sartor RB, editors. Gut and the liver. Dordrecht, Boston, London: Kluwer Academic Publishers; 1998. pp. 222–240. [Google Scholar]
- 34.Kono H, Nakagami M, Rusyn I, Connor HD, Stefanovic B, Brenner DA, Mason RP, Arteel GE, Thurman RG. Development of an animal model of chronic alcohol-induced pancreatitis in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1178–G1186. doi: 10.1152/ajpgi.2001.280.6.G1178. [DOI] [PubMed] [Google Scholar]
- 35.Puren AJ, Fantuzzi G, Gu Y, Su MS, Dinarello CA. Interleukin-18 (IFNgamma-inducing factor) induces IL-8 and IL-1beta via TNFalpha production from non-CD14+ human blood mononuclear cells. J Clin Invest. 1998;101:711–721. doi: 10.1172/JCI1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Sebastiano P, di Mola FF, Di Febbo C, Baccante G, Porreca E, Innocenti P, Friess H, Büchler MW. Expression of interleukin 8 (IL-8) and substance P in human chronic pancreatitis. Gut. 2000;47:423–428. doi: 10.1136/gut.47.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mueller C, Saurer L. Cellular immune reactions in chronic pancreatitis. In: Büchler MW, Friess H, Uhl W, Malfertheiner P, editors. Chronic pancreatitis. Novel concepts in biology and therapy. Berlin, Wien: Blackwell Wissenschafts-Verlag GmbH; 2002. pp. 184–195. [Google Scholar]
- 38.Kashiwamura S, Ueda H, Okamura H. Roles of interleukin-18 in tissue destruction and compensatory reactions. J Immunother. 2002;25 Suppl 1:S4–11. doi: 10.1097/00002371-200203001-00002. [DOI] [PubMed] [Google Scholar]
- 39.Gukovskaya AS, Vaquero E, Zaninovic V, Gorelick FS, Lusis AJ, Brennan ML, Holland S, Pandol SJ. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology. 2002;122:974–984. doi: 10.1053/gast.2002.32409. [DOI] [PubMed] [Google Scholar]
- 40.Pezzilli R, Billi P, Miniero R, Barakat B. Serum interleukin-10 in human acute pancreatitis. Dig Dis Sci. 1997;42:1469–1472. doi: 10.1023/a:1018814710291. [DOI] [PubMed] [Google Scholar]
- 41.Demols A, Van Laethem JL, Quertinmont E, Degraef C, Delhaye M, Geerts A, Deviere J. Endogenous interleukin-10 modulates fibrosis and regeneration in experimental chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1105–G1112. doi: 10.1152/ajpgi.00431.2001. [DOI] [PubMed] [Google Scholar]
- 42.Zediak VP, Hunter CA. IL-10 fails to inhibit the production of IL-18 in response to inflammatory stimuli. Cytokine. 2003;21:84–90. doi: 10.1016/s1043-4666(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 43.Sparmann G, Behrend S, Merkord J, Kleine HD, Graser E, Ritter T, Liebe S, Emmrich J. Cytokine mRNA levels and lymphocyte infiltration in pancreatic tissue during experimental chronic pancreatitis induced by dibutyltin dichloride. Dig Dis Sci. 2001;46:1647–1656. doi: 10.1023/a:1010689117772. [DOI] [PubMed] [Google Scholar]
- 44.Xie MJ, Motoo Y, Su SB, Sawabu N. Expression of tumor necrosis factor-alpha, interleukin-6, and interferon-gamma in spontaneous chronic pancreatitis in the WBN/Kob rat. Pancreas. 2001;22:400–408. doi: 10.1097/00006676-200105000-00011. [DOI] [PubMed] [Google Scholar]
- 45.van Laethem JL, Deviere J, Resibois A, Rickaert F, Vertongen P, Ohtani H, Cremer M, Miyazono K, Robberecht P. Localization of transforming growth factor beta 1 and its latent binding protein in human chronic pancreatitis. Gastroenterology. 1995;108:1873–1881. doi: 10.1016/0016-5085(95)90152-3. [DOI] [PubMed] [Google Scholar]
- 46.Ulloa L, Doody J, Massagué J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 47.Sime PJ, O'Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 48.Oldroyd SD, Thomas GL, Gabbiani G, El Nahas AM. Interferon-gamma inhibits experimental renal fibrosis. Kidney Int. 1999;56:2116–2127. doi: 10.1046/j.1523-1755.1999.00775.x. [DOI] [PubMed] [Google Scholar]
- 49.Menke A, Vogelmann R, Bachem M, Adler G. Mechanisms of fibrosis and potential antifibrotic agents. In: Lankisch PG, DiMagno EP, editors. Pancreatic disease. Berlin, Heidelberg, New York: Springer Verlag; 1999. pp. 132–139. [Google Scholar]
- 50.Kämpfer H, Paulukat J, Mühl H, Wetzler C, Pfeilschifter J, Frank S. Lack of interferon-gamma production despite the presence of interleukin-18 during cutaneous wound healing. Mol Med. 2000;6:1016–1027. [PMC free article] [PubMed] [Google Scholar]