

Abstract
AIM: To study the presence of sustained low diffusing capacity (DLCO) after liver transplantation (LT) in patients with hepatopulmonary syndrome (HPS).
METHODS: Six patients with mild-to-severe HPS and 24 without HPS who underwent LT were prospectively followed before and after LT at mid-term (median, 15 mo). HPS patients were also assessed at long-tem (median, 86 mo).
RESULTS: Before LT, HPS patients showed lower PaO2 (71 ± 8 mmHg), higher AaPO2 (43 ± 10 mmHg) and lower DLCO (54% ± 9% predicted), due to a combination of moderate-to-severe ventilation-perfusion (VA/Q) imbalance, mild shunt and diffusion limitation, than non-HPS patients (94 ± 4 mmHg and 19 ± 3 mmHg, and 85% ± 3% predicted, respectively) (P < 0.05 each). Seven non-HPS patients had also reduced DLCO (70% ± 4% predicted).
At mid- and long-term after LT, compared to pre-LT, HPS patients normalized PaO2 (91 ± 3 mmHg and 87 ± 5 mmHg), AaPO2 (14 ± 3 mmHg and 23 ± 5 mmHg) and all VA/Q descriptors (P < 0.05 each) without changes in DLCO (53% ± 8% and 56% ± 7% predicted, respectively). Post-LT DLCO in non-HPS patients with pre-LT low DLCO was unchanged (75% ± 6% predicted).
CONCLUSION: While complete VA/Q resolution in HPS indicates a reversible functional disturbance, sustained low DLCO after LT also present in some non-HPS patients, points to persistence of sub-clinical liver-induced pulmonary vascular changes.
Keywords: Carbon monoxide diffusing capacity, Multiple inert gas elimination technique, Pulmonary gas exchange, Pulmonary vascular disorders, Ventilation-perfusion relationships
INTRODUCTION
Single-breath diffusing capacity (DLCO) for carbon monoxide (transfer factor) impairment is the single most commonly abnormal lung function test in patients with end-stage hepatic disease[1]. Although the mechanism of this isolated abnormality in advanced hepatic patients without chronic respiratory co-morbidities remains largely unknown, its high prevalence suggests sub-clinical liver-induced changes in the pulmonary vascular bed. Pathophysiologically, alveolar ventilation to pulmonary blood flow (VA/Q) imbalance secondary to narrowing and early closure of airways to dependent lung zones as a consequence of interstitial edema and/or ascites in the context of fluid retention has been suggested as the most plausible mechanism[1,2].
Alternatively, in patients with hepatopulmonary syndrome (HPS), an arterial oxygenation defect caused by intrapulmonary vascular dilatations (IPVD) in patients with liver disease[3,4], VA/Q mismatching along with intrapulmonary shunt (i.e., zero VA/Q units) and diffusion limitation for oxygen essentially reflecting a diffusion-perfusion defect[5] encompass the frequent finding of low DLCO[4]. It is highly likely that in HPS DLCO is reduced because the distance between the alveoli and the red cells in the central stream of the dilated pulmonary micro-vessels is too great for complete equilibration of CO with hemoglobin[4]. Diffusion limitation for oxygen may be aggravated in part by a high cardiac output (QT) resulting in a shorter transit time of the red blood cell, hence contributing to the development of the diffusion-perfusion abnormality[4].
We have shown that, in patients with HPS, several gas exchange markers, namely PaO2, alveolar-to-arterial PO2 difference (AaPO2), intrapulmonary shunt and increased low VA/Q regions, correlate with DLCO[6]. Similarly, greater predicted (calculated according to the multiple inert gas elimination technique [MIGET][7]) than measured PaO2, an indirect estimate of diffusion limitation for oxygen, correlates with DLCO in hypoxemic HPS patients[6]. Low transfer factor is also a reliable predictor for the diagnosis of HPS[6].
Liver transplantation (LT) is the only therapeutic approach leading conclusively to complete resolution of advanced hepatic disease, including gas exchange abnormalities in HPS[4,8-10]. Although sustained low DLCO in hepatic patients with and without HPS has been reported after LT[8,9,11,12], this finding remains controversial. Persistence of low DLCO after LT would point to an underlying pulmonary vascular derangement.
We therefore decided to investigate comprehensively gas exchange status before and after LT in hepatic patients with and without HPS, to shed further light into the pathophysiology of low DLCO in advanced hepatic disease states.
MATERIALS AND METHODS
Subjects
From our original cohort of 80 candidates for LT (14 patients with and 66 without HPS)[6], a subset of 42 recipients of LT (53%) (9 with and 33 without HPS) between 1995 and 1997 was prospectively followed. An additional patient with HPS transplanted in 2000 was also included. From these 43 patients, 4 HPS (3 died before follow-up, 1 declined consent) and 9 non-HPS patients (6 died before follow-up, 3 refused to participate) were lost, the final population including 30 patients (6 with and 24 without HPS) (Table 1). Initial immunosuppressive therapy consisted of systemic corticosteroids, azathioprine and cyclosporine. Azathioprine was administered only during the first postoperative month. The dosage of corticosteroids was progressively tapered until discontinuation at the 18-24th mo. Cyclosporine was maintained through the period. The doses of these immunosuppressive drugs were also appropriately modified according to development of serious adverse effects or organ rejection. The study was approved by the Ethics Committee of our centre and all patients gave their written informed consent.
Table 1.
Physical and clinical characteristics of patients with and without hepatopulmonary syndrome (HPS)
| Characteristics | HPS | Non-HPS |
| Patients (n) | 6 | 24 |
| Gender (F/M) | 2/4 | 9/15 |
| Age (yr) | 43 ± 6 | 54 ± 2 |
| Smoking Habits (n) | ||
| Smokers Ex-smokers | 1 2 | 5 5 |
| Etiology (n) | ||
| Hepatitis C | 3 | 12 |
| Alcohol | 1 | 4 |
| Idiopathic | 0 | 4 |
| Other | 2 | 4 |
| Child-Pugh, n (%) | ||
| A | 1 (17) | 3 (13) |
| B | 4 (66) | 13 (54) |
| C | 1 (17) | 8 (33) |
| Gastrointestinal bleeding, n (%) | 4 (66) | 10 (42) |
| Ascites, n (%) | 3 (50) | 18 (75) |
| Cutaneous spider naevi, n (%) | 6 (100) | 17 (71) |
| Concomitant respiratory Symptoms, n (%) | ||
| Digital clubbing | 4 (66) | 2 (8)a |
| Dyspnea | 6 (100) | 3 (12)a |
| Cyanosis | 2 (33) | 0 (0)a |
P < 0.05 vs non-HPS.
The diagnosis of HPS was based on the presence of an oxygenation defect [namely, increased AaPO2 ≥ 15 mmHg while breathing air in upright position, irrespective of the presence of hypoxemia (PaO2 < 80 mmHg)] and indicative evidence of IPVD [using contrast-enhanced echocardiography (CEE)] in a patient with underlying chronic hepatic disease, according to the European Respiratory Society (ERS) recommendations[4].
Study design
Lung function tests were assessed in all patients before LT (median, 4 mo; range, 2 wk-18 mo; HPS patients, 2 wk-12 mo) and between 12 and 18 mo (median, 15 mo; mid-term ≤ 1.5 yr) following LT. Patients with HPS were also evaluated at long-term (≥ 3 yr after LT) (median, 86 mo; range, 45-117 mo) (one patient refused arterial puncture at this time point only). CEE measured as previously described[6], was performed before LT in all patients and also after LT in HPS patients (at mid-term only). In the latter individuals, VA/Q distributions were also estimated before and at mid-term after LT.
Measurements
Forced spirometry, static lung volumes, DLCO after correction for anemia[13], minute ventilation (VE), arterial respiratory blood gases (PaO2, PaCO2, AaPO2) and pH, and oxygen consumption (VO2) were measured or calculated as previously described[6]. In the 6 patients with HPS, VA/Q distributions were calculated using MIGET as previously described[6]. A pulmonary artery catheter was inserted (Swan-Ganz catheter, Baxter Healthcare, Irvine, CA, USA) in 4 patients and MIGET was computed using mixed venous respiratory and inert gases and cardiac output (QT) determined by thermodilution. In the remaining 2 HPS patients, QT was measured by dye dilution (DC-410; Waters Instruments, Rochester, MN, USA) such that mixed venous inert gases and PO2 were estimated by mass balance in the customary manner[7]. The agreement between these two MIGET approaches has been previously validated in our laboratory[14]. The dispersion of the distributions of pulmonary blood flow (Log SDQ) and alveolar ventilation (Log SDV) on a logarithmic scale (upper normal limit, 0.60 and 0.65, respectively)[15], key descriptors of the amount of low and high VA/Q units respectively, and the difference among measured retentions and excretions of the inert gases corrected for the elimination of acetone (DISP R-E*) (normal values < 3.0), an overall (mathematical) index of VA/Q heterogeneity[16], all dimensionless, were the most accurately used indices of VA/Q imbalance. Intrapulmonary shunt and low VA/Q regions were defined as the fraction of blood flow diverted to lung units with low VA/Q ratios < 0.005 and between 0.005 and 0.1, respectively. Dead space was defined as the fraction of alveolar ventilation diverted to lung units with VA/Q ratios >100.
Statistical analysis
Descriptive data are expressed as mean ± SE. Paired and unpaired Student’s t tests were used to compare data before and after LT and Pearson’s correlations test was used when appropriate. P ≤ 0.05 was considered statistically significant at all effects.
RESULTS
Before LT
Lung function tests before and after LT in patients with and without HPS are set out in Table 2. Respiratory symptoms and signs were more prevalent in HPS patients. Although mean spirometric and static lung volumes were within normal limits without differences between the two subgroups, individually one non-smoker with HPS (No.3) had a mild restrictive pattern (due to pleural thickening) and 3 other HPS patients (No.4-6) (one smoker and two ex-smokers) had mild to moderate airflow limitation (Table 3). Compared to patients without HPS (Table 2), DLCO and DLCO/alveolar volume (KCO) were mildly to severely decreased (< 80% predicted) in each HPS patient (P < 0.05 each). In patients without HPS, DLCO was mildly to moderately reduced (70% ± 4% predicted; range, 54%-79% predicted) in 7 (Figure 1), all but 2 (PaO2, 58 and 74 mmHg each) normoxemic (PaO2, 92 ± 8 mmHg; FEV1, 93% ± 5%; FEV1/FVC ratio, 78% ± 2%). In contrast, DLCO was normal (91% ± 3% predicted; range, 80%-131% predicted; PaO2, 95 ± 5 mmHg) in the 17 remainders without other differences between these two subgroups. Both VE and VO2 in all patients and QT, measured in HPS patients only, were increased. Chest high-resolution CT scans in HPS patients excluded the coexistence of emphysematous and/or diffuse interstitial changes.
Table 2.
Lung function test and blood gas analysis before and after liver transplantation (LT) (mean ± SE)
|
HPS1 |
Non-HPS1 |
||||
| Parameter | Pre-LT | Mid-term Post-LT | Long-term Post-LT | Pre-LT | Mid-term Post-LT |
| FEV12 (% pred) | 89 ± 7 | 92 ± 8 | 87 ± 9 | 90 ± 4 | 89 ± 4 |
| FVC 3 (% pred) | 97 ± 6 | 100 ± 7 | 93 ± 11 | 89 ± 3 | 88 ± 3 |
| FEV12/FVC3 4 (%) | 72 ± 4 | 77 ± 5 | 74 ± 5 | 76 ± 2 | 76 ± 2 |
| TLC4 (% pred) | 100 ± 7 | 100 ± 9 | 99 ± 9 | 96 ± 2 | 93 ± 18 |
| DLCO5 (mL/min/mmHg) | 16.8 ± 3.4c | 16.6 ± 3.5 | 16.9 ± 3.3 | 23.1 ± 2.1 | 20.7 ± 3.5 |
| DLCO 5 (% pred) | 54 ± 9c | 53 ± 8 | 56 ± 7 | 85 ± 3 | 79 ± 3 |
| KCO6 (%pred) | 59 ± 10c | 54 ± 6 | 60 ± 7 | 91 ± 3 | 86 ± 4 |
| VE 7 (l/min) | 10.3 ± 1.2 | 7.4 ± 0.4a | na13 | 10.3 ± 1.0 | 8.4 ± 0.6 |
| VO2 8 (mL/min) | 263.7±22.8 | 247.0 ± 26.2a | na13 | 270.3 ± 17.6 | 254.3 ± 13.2 |
| Hemoglobin (g/L) | 111 ± 22 | 121 ± 20 | 134 ± 21 | 104 ± 18 | 114 ± 16 |
| PHa9 | 7.45 ± 0.02 | 7.39 ± 0.01a | 7.42 ± 0.01 | 7.47 ± 0.01 | 7.41 ± 0.01a |
| PaO210 (mmHg) | 71 ± 8c | 91 ± 3a | 87 ± 5a | 94 ± 4 | 94 ± 2 |
| PaCO211 (mmHg) | 28 ± 2 | 36 ± 1a | 34 ± 0a | 31 ± 1 | 35 ± 1a |
| AaPO2 12 (mmHg) | 43 ± 10c | 14 ± 3a | 23 ± 5a | 19 ± 3 | 14 ± 2 |
Hepatopulmonary syndrome;
Forced expiratory volume at 1 s;
Forced vital capacity;
Total lung capacity;
Single-breath carbon monoxide diffusing capacity;
carbon monoxide diffusing capacity normalized per liter alveolar volume;
Minute ventilation;
Oxygen uptake;
Arterial pH;
Partial pressure of arterial oxygen;
Partial pressure of arterial carbon dioxide;
Alveolar to arterial oxygen partial pressure gradient;
Not available.
P < 0.05 vs pre-LT;
P < 0.05 vs non-HPS.
Table 3.
Individual lung function test before and after liver transplantation (LT) in hepatopulmonary syndrome patients (% pred)
| Pre-LT | Mid-term post-LT | Long-Term Post-LT | ||||||||||||||||
| Patients No. | 1 | 2 | 3 | 4 2 | 5 1 | 6 2 | 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 3 | 4 | 5 | 6 |
| FEV1 | 106 | 110 | 77 | 90 | 71 | 80 | 106 | 120 | 74 | 83 | 96 | 81 | 102 | 123 | 76 | 71 | 66 | 87 |
| FVC | 99 | 116 | 77 | 106 | 87 | 97 | 99 | 126 | 64 | 100 | 83 | 93 | 99 | 131 | 70 | 106 | 66 | 87 |
| FEV1/FVC | 84 | 76 | 81 | 65 | 62 | 61 | 81 | 78 | 94 | 63 | 80 | 65 | 80 | 76 | 89 | 52 | 75 | 74 |
| TLC | 93 | 118 | 70 | 117 | 96 | 104 | 92 | 125 | 69 | 122 | 88 | 102 | 92 | 138 | 68 | 114 | 85 | 96 |
| DLCO | 34 | 70 | 24 | 43 | 77 | 78 | 47 | 77 | 29 | 39 | 71 | 58 | 46 | 85 | 34 | 46 | 57 | 69 |
Smoker;
Ex-smoker. For other abbreviations see Table 2.
Figure 1.
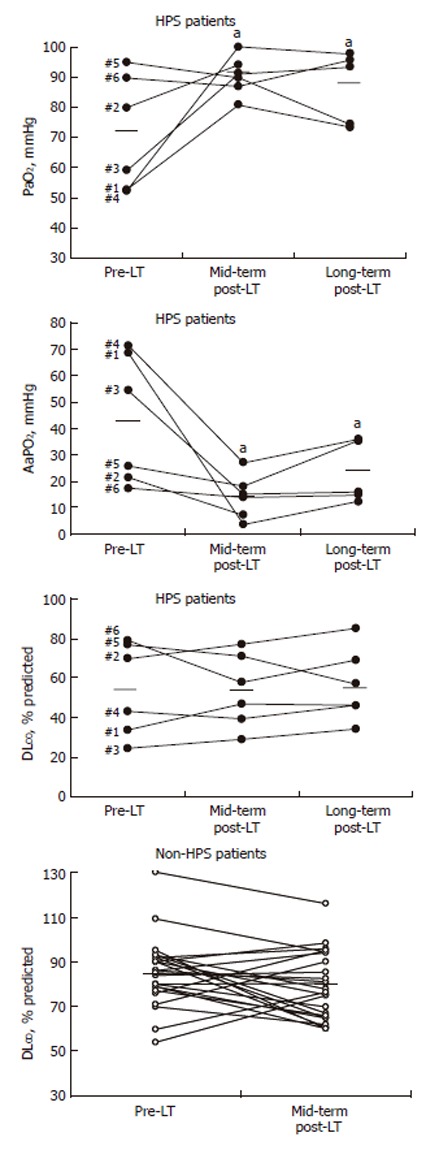
Individual values of arterial PO2, AaPO2 and DLCO in patients with and without HPS before and after LT. Bold bars denote mean values. aP < 0.05 between pre- and post-LT measurements.
Three HPS patients (No.1, No.3 and No.4) had severe disease (PaO2 < 60 - ≥ 50 mmHg), one (No.2) moderate (PaO2 < 80 - ≥ 60 mmHg) and the two remainders (No.5 and No.6) a mild stage (PaO2 ≥ 80 mmHg; AaPO2, 26 and 17 mmHg each)[4] (Figure 1). Patients with HPS had lower PaO2 and higher AaPO2 than non-HPS patients (P < 0.05 each), while PaCO2 was equally decreased in the two groups. Distributions of VA/Q ratios were abnormal in each HPS patient, as shown by mild to severe increases in Log SDQ and DISP R-E*, mild to moderate increases in intrapulmonary shunt and mildly increased low VA/Q regions (Table 4). The mean distribution of pulmonary blood flow (mean Q) was mildly reduced (normal, ~1.0). By contrast, Log SDV was normal and the mean distribution of alveolar ventilation (mean V) was moderately increased. Dead space was severely reduced. In 3 hypoxemic HPS patients, predicted (according to MIGET) PaO2 (68 ± 2 mmHg) was greater than actual measured PaO2 (55 ± 2 mmHg). There was a negative correlation between the latter PO2 difference and low DLCO in all HPS patients (r = -0.85; P < 0.05), suggesting a close relationship between diffusion impairment for oxygen and DLCO.
Table 4.
Ventilation-perfusion distributions in patients with hepatopulmonary syndrome before and after liver transplantation (LT) (mean ± SE)
| Pre-LT | Post-LT | |
| Shunt1, % QT2 | 7.8 ± 3.3 | 0.6 ± 0.1a |
| Low VA/Q mode3, % of QT | 4.1 ± 2.1 | 0.0 ± 0.0a |
| Mean Q4 | 0.85 ± 0.13 | 0.84 ± 0.04 |
| Log SDQ5 | 0.95 ± 0.17 | 0.49 ± 0.08a |
| Mean V6 | 1.50 ± 0.25 | 1.11 ± 0.15 |
| Log SDV7 | 0.64 ± 0.04 | 0.47 ± 0.07a |
| DISP R-E*8 | 11 ± 2 | 4 ± 1a |
| Dead space, % | 18.8 ± 3.9 | 19.3 ± 4.6 |
| Pred-meas PaO29, mmHg | 6 ± 4 | 7 ± 3 |
Percentage of blood flow to unventilated units (VA/Q < 0.005) (normal, 0% of QT);
Cardiac output;
Perfusion to units with VA/Q ratios between 0.005 and 0.1 (normal, 0% of QT);
Mean VA/Q of the perfusion distribution;
Dispersion of blood flow distribution (normal < 0.60);
Mean VA/Q of the ventilation distribution;
Dispersion of ventilation distribution (normal < 0.65);
The difference among measured retentions and excretions of the inert gases corrected for the excretion of acetone (normal < 3.0 );
Predicted PaO2 (by MIGET)-actual PaO2 (measured).
P < 0.05 vs pre-LT.
After LT
Liver function tests were within normal limits in all patients and diseases such as de novo autoimmune hepatitis, non-alcoholic steato-hepatitis, chronic rejection, and severe hepatitis C recurrence were excluded. At mid-term, all patients improved clinically while mean FEV1 and lung volumes remained unchanged.
In patients with HPS, CEE-evidence for IPVD normalized, while both breathlessness and exercise tolerance substantially ameliorated or disappeared. Three HPS patients showed progressive resolution of finger clubbing. As opposed to the absence of any improvement in both DLCO and KCO, the three arterial blood gas markers substantially ameliorated reaching normal limits (P < 0.05 each) close to those of non-HPS patients before LT. This was essentially due to resolution of all VA/Q descriptors, namely intrapulmonary shunt, regions of low VA/Q units, Log SDQ and DISP R-E* (P < 0.05 each) (Table 4). Dead space and mean V remained unchanged. Predicted (98 ± 2 mmHg) and actual (91 ± 3 mmHg) PaO2 in pre-LT hypoxemic HPS normalized and there was no correlation between the latter PO2 difference and low DLCO.
In patients without HPS, mean PaO2, AaPO2 and DLCO remained unvaried (Table 2) including the DLCO values of the 7 patients with pre-LT low DLCO (75% ± 6% predicted; range, 60%-95% predicted; PaO2, 91 ± 3 mmHg), in whom 2 had normalized DLCO. Mean DLCO in the remaining 17 non-HPS patients slightly decreased (to 81% ± 4% predicted; range, 65%-116% predicted; PaO2, 94 ± 2 mmHg) (P < 0.05), whose physiological significance remained uncertain.
Mean PaCO2 increased (P < 0.05 in HPS patients only) and arterial pH decreased (P < 0.05) in both HPS and non-HPS patients, to reach normal limits without differences between the two populations. Increased PaCO2 kept pace with decreased VE, although only significantly in HPS patients (P < 0.05).
Pulmonary vascular resistance (PVR) increased (from 92 ± 69 dyn.s.cm-5 to 123 ± 78 dyn.s.cm-5), QT decreased (from 10.6 ± 4 to 6.6 ± 2 L/min) (P < 0.05 each) and pulmonary artery pressure was unchanged (from 18 ± 8 mmHg to 17 ± 6 mmHg) in the 4 patients with HPS in whom hemodynamics were carried out. Mean VO2 decreased in HPS patients only (P < 0.05).
At long-term, 3 HPS patients had abnormal spirometry (Table 3): patient No.3 had a similar moderate restrictive pattern shown before LT, patient No.4 (ex-smoker) asymptomatic moderate airflow limitation possibly due to coexisting chronic obstructive pulmonary disease (COPD), and patient No.5 (smoker) mild-to-moderate undefined ventilatory pattern. Patient No.6 had normal spirometry. Unlike the persisting normalcy of arterial blood gases, mean DLCO and KCO in HPS remained unchanged at the same pre-LT levels (Figure 1).
DISCUSSION
The most salient finding of the present study is that all patients with HPS and a small subset of patients without HPS showed sustained reduced DLCO as the most remarkable abnormal lung function test following successful LT. More importantly, in HPS patients these findings were observed in conjunction with normalization of all respiratory (PaO2, PaCO2 and AaPO2) and inert gas exchange indices, namely regions with low VA/Q units, increased dispersion of pulmonary blood flow (Log SDQ) and increased overall index of VA/Q heterogeneity (DISP R-E*), intrapulmonary shunt and diffusion-perfusion defect (as shown by a greater predicted MIGET than actual measured PaO2). Furthermore, most of the clinical, functional and hemodynamic abnormalities in HPS patients ablated or decreased and CEE-based IPVD disappeared. The absence of noticeable DLCO changes after LT in non-HPS patients who received the same immunosuppressive therapy compared with patients with HPS, rules out at first glance any complementary iatrogenic interstitial lung effect. All in all, our findings point to persistence of sub-clinical structural changes at the pulmonary vascular bed.
Our results complement and extend a few prospective, although inconclusive and contentious, studies on the outcome of gas exchange disturbances after LT in patients with advanced hepatic disease[8,9,11,12]. Eriksson et al[8] observed in 6 patients with end-stage liver disease, but without proven-diagnosis of HPS, complete resolution of respiratory and inert gas exchange after LT, but they have not reported post-DLCO. Krowka et al[9] found that PaCO2 and steady-state diffusing capacity are improved after LT without any reference to HPS. In contrast, Laberge et al[11] reported that HPS is resolved in two children with HPS who normalized DLCO and do particularly well after LT. It is postulated that, in juvenile HPS, the response of the pulmonary vasculature to LT appears to be more favorable than in adults[17]. Finally, Battaglia et al[12] have demonstrated sustained low DLCO after LT although improvement or resolution of hypoxemia and/or widened AaPO2 is related to pulmonary capillary thickening[18]. Notwithstanding, both the poor resolution of the gas exchange tools used and the lack of a solid definition of HPS in these studies have limited partially more evidence-based conclusions.
We are confronted therefore with the pathological basis of this intriguing sustained reduced DLCO after LT in patients with and without HPS. We hypothesize that the persistence of low DLCO after LT in patients with HPS may be consistent with structural pulmonary vascular changes already present before LT, such as increased thickness of the walls of small veins originally shown in a post-mortem case of liver cirrhosis complicated by cyanosis strongly suggestive of HPS[19]. Ultrastructurally, this thickening is essentially due to a layer of collagen fibers interspersed with fine filamentous material whilst the basement membrane and many capillary walls are thickened with collagen, which provokes an approximately two-fold increase in the minimum blood-gas distance and contributes to the reduction in DLCO[19]. This structural vascular abnormality in HPS can be sub-clinical and still consistent with post-LT VA/Q normalization and disappearance of intrapulmonary shunt and diffusion-perfusion defect. Persisting low DLCO after LT in most of the subset of patients without HPS may be due either to a similar, albeit plausibly less severe, derangement than the one alluded to[19] or alternatively to a different unknown abnormality of the pulmonary vascular bed as yet. Very recently, we have shown that pulmonary exchange defects in HPS patients remain unchanged after administration of NG-nitro-L-arginine methyl ester (L-NAME), an inhibitor of nitric oxide (NO) synthase, hence suggesting that HPS-induced gas exchange disturbances may be related to pulmonary vascular remodeling rather than to an ongoing vasodilator effect of enhanced NO production[20].
Decline of DLCO occurs after heart transplantation (HT) and persists for up to three years[21], a finding that appears to contribute to exercise limitation in HT recipients[22]. Post-HT DLCO decline has been related to increased intra-capillary resistance to CO transfer secondary to a combination of anemia and reduced pulmonary capillary blood volume (Vc)[23]. Post-HT normalization of pulmonary hemodynamics along with persisting structural pulmonary vascular changes due to pre-HT severe chronic heart failure may lie behind reduced Vc after HT[23]. Notwithstanding severe pulmonary arterial hypertension in HT candidates is never present in patients with HPS. Accordingly, it may be unlikely that pulmonary hemodynamic changes after LT in our study might play a major role in post-LT persisting low DLCO. Moreover, although three out of six HPS patients had an active (1) or past smoking (2) history, only one (patient No.4) had associated asymptomatic moderate airflow limitation that could explain, at least in part, persisting low DLCO after LT. We have shown, however, that HPS-induced gas exchange disturbances and hemodynamic findings predominate in the face of coexisting chronic respiratory disorders, such as COPD[24]. We did not assess the components of DLCO because our primary aim was to ascertain in HPS patients whether or not pre-LT could reduce DLCO persisted within the context of gas exchange status following LT as compared to patients without HPS.
There are two strengths in our study, namely its prospective nature and lengthy design using clear-cut diagnostic criteria for patients with[4] and without HPS, and the combined use of routine lung function tests along with one of the most robust tools to unravel the intrapulmonary determinants of arterial deoxygenation in HPS. We acknowledge, however, three limitations. The number of HPS patients studied is relatively small; the absence of pathological basis represents a weakness; and diffusion impairment cannot be estimated with MIGET when arterial hypoxemia is not present.
Persisting low DLCO after LT in end-stage hepatic patients with and without coexisting HPS has not been previously reported in such a comprehensive manner. Whatever the ultimate mechanism for this unique and intriguing post-LT DLCO impairment is, our findings point to the presence of pulmonary vascular involvement in advanced liver disease.
Footnotes
Supported by Red Respira-ISCIII-RTIC-03/11 and Generalitat de Catalunya, No. 2005SGR-00822
S- Editor Pan BR L- Editor Wang XL E- Editor Liu WF
References
- 1.Hourani JM, Bellamy PE, Tashkin DP, Batra P, Simmons MS. Pulmonary dysfunction in advanced liver disease: frequent occurrence of an abnormal diffusing capacity. Am J Med. 1991;90:693–700. [PubMed] [Google Scholar]
- 2.Rodríguez-Roisin R, Agustí AG, Roca J. The hepatopulmonary syndrome: new name, old complexities. Thorax. 1992;47:897–902. doi: 10.1136/thx.47.11.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berthelot P, Walker JG, Sherlock S, Reid L. Arterial changes in the lungs in cirrhosis of the liver--lung spider nevi. N Engl J Med. 1966;274:291–298. doi: 10.1056/NEJM196602102740601. [DOI] [PubMed] [Google Scholar]
- 4.Rodríguez-Roisin R, Krowka MJ, Hervé P, Fallon MB. Pulmonary-Hepatic vascular Disorders (PHD) Eur Respir J. 2004;24:861–880. doi: 10.1183/09031936.04.00010904. [DOI] [PubMed] [Google Scholar]
- 5.Genovesi MG, Tierney DF, Taplin GV, Eisenberg H. An intravenous radionuclide method to evaluate hypoxemia caused by abnormal alveolar vessels. Limitation of conventional techniques. Am Rev Respir Dis. 1976;114:59–65. doi: 10.1164/arrd.1976.114.1.59. [DOI] [PubMed] [Google Scholar]
- 6.Martínez GP, Barberà JA, Visa J, Rimola A, Paré JC, Roca J, Navasa M, Rodés J, Rodriguez-Roisin R. Hepatopulmonary syndrome in candidates for liver transplantation. J Hepatol. 2001;34:651–657. doi: 10.1016/s0168-8278(00)00108-2. [DOI] [PubMed] [Google Scholar]
- 7.Roca J, Wagner PD. Contribution of multiple inert gas elimination technique to pulmonary medicine. 1. Principles and information content of the multiple inert gas elimination technique. Thorax. 1994;49:815–824. doi: 10.1136/thx.49.8.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eriksson LS, Söderman C, Ericzon BG, Eleborg L, Wahren J, Hedenstierna G. Normalization of ventilation/perfusion relationships after liver transplantation in patients with decompensated cirrhosis: evidence for a hepatopulmonary syndrome. Hepatology. 1990;12:1350–1357. doi: 10.1002/hep.1840120616. [DOI] [PubMed] [Google Scholar]
- 9.Krowka MJ, Dickson ER, Wiesner RH, Krom RA, Atkinson B, Cortese DA. A prospective study of pulmonary function and gas exchange following liver transplantation. Chest. 1992;102:1161–1166. doi: 10.1378/chest.102.4.1161. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Roisin R, Krowka MJ. Is severe arterial hypoxaemia due to hepatic disease an indication for liver transplantation A new therapeutic approach. Eur Respir J. 1994;7:839–842. [PubMed] [Google Scholar]
- 11.Laberge JM, Brandt ML, Lebecque P, Moulin D, Veykemans F, Paradis K, Pelletier L, Lacroix J, Blanchard H, de Ville de Goyet J. Reversal of cirrhosis-related pulmonary shunting in two children by orthotopic liver transplantation. Transplantation. 1992;53:1135–1138. [PubMed] [Google Scholar]
- 12.Battaglia SE, Pretto JJ, Irving LB, Jones RM, Angus PW. Resolution of gas exchange abnormalities and intrapulmonary shunting following liver transplantation. Hepatology. 1997;25:1228–1232. doi: 10.1002/hep.510250527. [DOI] [PubMed] [Google Scholar]
- 13.Cotes JE, Chinn DJ, Quanjer PH, Roca J, Yernault JC. [Standardization of the measurement of transfer factor (diffusing capacity). Work Group on Standardization of Respiratory Function Tests. European Community for Coal and Steel. Official position of the European Respiratory Society] Rev Mal Respir. 1994;11 Suppl 3:41–52. [PubMed] [Google Scholar]
- 14.Rodriguez-Roisin R, Ballester E, Roca J, Torres A, Wagner PD. Mechanisms of hypoxemia in patients with status asthmaticus requiring mechanical ventilation. Am Rev Respir Dis. 1989;139:732–739. doi: 10.1164/ajrccm/139.3.732. [DOI] [PubMed] [Google Scholar]
- 15.Cardús J, Burgos F, Diaz O, Roca J, Barberà JA, Marrades RM, Rodriguez-Roisin R, Wagner PD. Increase in pulmonary ventilation-perfusion inequality with age in healthy individuals. Am J Respir Crit Care Med. 1997;156:648–653. doi: 10.1164/ajrccm.156.2.9606016. [DOI] [PubMed] [Google Scholar]
- 16.Gale GE, Torre-Bueno JR, Moon RE, Saltzman HA, Wagner PD. Ventilation-perfusion inequality in normal humans during exercise at sea level and simulated altitude. J Appl Physiol (1985) 1985;58:978–988. doi: 10.1152/jappl.1985.58.3.978. [DOI] [PubMed] [Google Scholar]
- 17.Kikuchi H, Ohkohchi N, Orii T, Satomi S. Living-related liver transplantation in patients with pulmonary vascular disease. Transplant Proc. 2000;32:2177–2178. doi: 10.1016/s0041-1345(00)01623-7. [DOI] [PubMed] [Google Scholar]
- 18.Matsubara O, Nakamura T, Uehara T, Kasuga T. Histometrical investigation of the pulmonary artery in severe hepatic disease. J Pathol. 1984;143:31–37. doi: 10.1002/path.1711430106. [DOI] [PubMed] [Google Scholar]
- 19.Stanley NN, Williams AJ, Dewar CA, Blendis LM, Reid L. Hypoxia and hydrothoraces in a case of liver cirrhosis: correlation of physiological, radiographic, scintigraphic, and pathological findings. Thorax. 1977;32:457–471. doi: 10.1136/thx.32.4.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gómez FP, Barberà JA, Roca J, Burgos F, Gistau C, Rodríguez-Roisin R. Effects of nebulized N(G)-nitro-L-arginine methyl ester in patients with hepatopulmonary syndrome. Hepatology. 2006;43:1084–1091. doi: 10.1002/hep.21141. [DOI] [PubMed] [Google Scholar]
- 21.al-Rawas OA, Carter R, Stevenson RD, Naik SK, Wheatley DJ. The time course of pulmonary transfer factor changes following heart transplantation. Eur J Cardiothorac Surg. 1997;12:471–478; discussion 478-479. doi: 10.1016/s1010-7940(97)00127-9. [DOI] [PubMed] [Google Scholar]
- 22.Al-Rawas OA, Carter R, Stevenson RD, Naik SK, Wheatley DJ. Exercise intolerance following heart transplantation: the role of pulmonary diffusing capacity impairment. Chest. 2000;118:1661–1670. doi: 10.1378/chest.118.6.1661. [DOI] [PubMed] [Google Scholar]
- 23.Al-Rawas OA, Carter R, Stevenson RD, Naik SK, Wheatley DJ. Mechanisms of pulmonary transfer factor decline following heart transplantation. Eur J Cardiothorac Surg. 2000;17:355–361. doi: 10.1016/s1010-7940(00)00359-6. [DOI] [PubMed] [Google Scholar]
- 24.Martinez G, Barberà JA, Navasa M, Roca J, Visa J, Rodriguez-Roisin R. Hepatopulmonary syndrome associated with cardiorespiratory disease. J Hepatol. 1999;30:882–889. doi: 10.1016/s0168-8278(99)80143-3. [DOI] [PubMed] [Google Scholar]