

Abstract
Objectives
Emerging drug resistance to antiviral therapies is an increasing challenge for the treatment of influenza virus infections. One new antiviral compound, BTA938, a dimeric derivative of the viral neuraminidase inhibitor zanamivir, contains a 14-carbon linker bridging two zanamivir moieties. In these studies, we evaluated antiviral efficacy in cell cultures infected with influenza virus and in mouse models of lethal influenza using H1N1pdm09, H3N2 and oseltamivir-resistant (H275Y) viruses.
Methods
In vitro activity was evaluated against 22 strains of influenza virus. Additionally, in vivo studies compared the efficacy of BTA938 or zanamivir after intranasal treatment. We also tested the hypothesis of a dual mode of action for BTA938 using scanning electron microscopy (SEM).
Results
BTA938 inhibited the viruses at nanomolar concentrations in vitro with a median 50% effective concentration value of 0.5 nM. In mouse models, the dimer provided ∼10-fold greater protection than zanamivir. The data also showed that a single low dose (3 mg/kg) protected 100% of mice from an otherwise lethal oseltamivir-resistant (H275Y) influenza virus infection. Remarkably, a single prophylactic treatment (10 mg/kg) administered 7 days before the challenge protected 70% of mice and when administered 1 or 3 days before the challenge it protected 90% of mice. Additionally, SEM provides evidence that the increased antiviral potency may be mediated by an enhanced aggregation of virus on the cell surface.
Conclusions
In vitro and in vivo experiments showed the high antiviral activity of BTA938 for the treatment of influenza virus infections. Moreover, we demonstrated that a single dose of BTA938 is sufficient for prophylactic and therapeutic protection in mouse models.
Keywords: neuraminidase inhibitors, prophylactic therapy, oseltamivir-resistant influenza virus
Introduction
Influenza viruses cause highly contagious respiratory disease in susceptible humans. Most seasonal influenza infections of healthy adults are uncomplicated and the symptoms typically clear within 1–2 weeks without a need for hospitalization.1 However, young children, the elderly, pregnant women, those who are morbidly obese and people with underlying chronic or immunocompromising conditions are recognized as being at risk of developing severe or complicated illness following influenza virus infections.2 Viral or bacterial pneumonia represents one of these complications with a potentially fatal outcome.3 The control of human influenza virus infections continues to be a major public health challenge. Despite widespread vaccination, influenza remains a significant burden that can give rise to potential crisis situations even in communities with advanced healthcare. It is estimated that, in the USA alone, influenza causes an average of 23 607 deaths per year.4 The WHO estimates that, worldwide, annual influenza epidemics cause 3–5 million cases of severe illness and 250 000–500 000 deaths.5,6 Complicating the picture are continuously emerging influenza virus subtypes and variants that are new to man and therefore have the potential to cause pandemics in an immunologically naive population, as exemplified by the most recent influenza pandemic in 2009.7
In addition to vaccines, antiviral drugs are employed to reduce morbidity and to prevent complications from seasonal as well as from pandemic influenza virus infections. In the USA, four anti-influenza agents have been approved. These are the viral M2 ion channel blockers amantadine and rimantadine (adamantane group) and the viral neuraminidase (NA) inhibitors oseltamivir and zanamivir.8 Due to widespread resistance to the adamantanes among influenza A(H1N1)pdm09 and influenza A(H3N2) viruses currently circulating globally, the Advisory Committee on Immunization Practices does not recommend their usage.9 Instead, oseltamivir and zanamivir are the primary antiviral agents recommended for the prevention and treatment of influenza.9 However, few and sporadic cases of oseltamivir resistance have been reported for A(H1N1)pdm09. Fortunately, the pandemic virus isolate with the H275Y mutation in the NA that confers oseltamivir resistance is still zanamivir-sensitive.10–12 During the 2009–10 influenza season, oseltamivir resistance was mainly seen in patients who had received oseltamivir prophylaxis or treatment,13–17 but during the 2010–11 season, the proportion of oseltamivir-resistant A(H1N1)pdm09-virus-infected patients without prior oseltamivir exposure had increased dramatically from 11% to 73.5%.17,18 This trend, suggestive of a low level of community transmission of the mutated virus, is of concern, although, thankfully, these viruses have been reported to be sensitive to zanamivir and laninamivir. Notably, resistance to zanamivir and laninamivir has been very rare to date. Nonetheless, a recent report of a widespread community cluster of H275Y oseltamivir-resistant A(H1N1)pdm09 influenza in Australia,19 with improved virus fitness, emphasizes the importance of ongoing vigilance and continued investigation of new or improved antiviral medications with higher potency and reduced dosing frequency.
The apparent inability of the viral NA to readily mutate to avoid the action of zanamivir indicates the suitability of this drug as a starting point for manipulation to improve the longevity of action. This is critical, as both oseltamivir and zanamivir currently require twice-daily administration to be effective. Dimeric derivatives of zanamivir have recently been synthesized and have been shown to be potent and long-acting inhibitors of influenza NA from different type A and B viruses.20,21 Of the wide range of linker lengths and linker groups tested to connect two zanamivir monomers via a 7-carbamate group, a zanamivir dimer with an unsubstituted 14-carbon alkyl linker showed the most promise.20 This compound was designated BTA938 and corresponds to compound #9 and #3, respectively, in the studies performed by Macdonald and colleagues.20,21 Although a closely related dimeric derivative with a slightly shorter linker showed greater inhibition of virus replication, BTA938 had superior in vivo efficacy.20 In addition, it was demonstrated that BTA938 had a much higher lung retention time in rats when compared with zanamivir and other dimeric zanamivir derivatives.20,21 The increased level of compound retention in the lung suggests that BTA938 might be a suitable candidate for once-weekly prophylactic/therapeutic application instead of the currently twice-daily zanamivir dosing regimen for several consecutive days.21,22
BTA938 was synthesized with the assumption that multivalent binding might potentiate the drug effect of zanamivir and it appears that dimeric zanamivir derivatives do have higher antiviral activity. However, the underlying mechanism of action is not clear. Based on data from transmission electron microscopy indicating that purified influenza viruses formed clusters after treatment with BTA938, Macdonald et al. speculated that the increased antiviral potency was a consequence of enhanced virus particle aggregation through intervirion NA binding,20 thereby further impeding successful virion release and spread.
Here, we report expanded in vitro and in vivo studies of the 14-carbon-linked zanamivir dimer BTA938, with an emphasis on the possibility of once-weekly prophylactic/therapeutic application. Previous in vivo experiments were limited to an assessment of the reduction in pulmonary viral loads following the non-lethal influenza challenge of mice treated 7 days previously with the compound. We now describe the therapeutic efficacy in our model of lethal influenza infections using influenza A/California/04/2009 (H1N1)pdm09 virus, A/Victoria/3/75 (H3N2) virus and oseltamivir-resistant influenza A/Mississippi/03/2001 (H1N1) H275Y virus. We also provide data on prophylactic efficacy after challenge with influenza A(H1N1)pdm09 virus. Our results highlight the potential of this particular zanamivir derivative for both the treatment and prophylaxis of infections caused by highly virulent influenza viruses. In addition, we present ultrastructural data supporting the dual-action mode hypothesis of this compound: as an NA inhibitor and as an enhancer of intervirion aggregation.
Materials and methods
Antiviral compounds
BTA938 and laninamivir were provided by Biota Holdings Ltd (Melbourne, Australia). Zanamivir was obtained from Haorui Pharma-Chem Inc. (Edison, NJ, USA) and Biota Holdings Ltd. Oseltamivir carboxylate was sourced from Epichem (Murdoch, WA, Australia) and peramivir from SYNthesis Med Chem (Melbourne, Australia). Ribavirin was acquired from the former ICN Pharmaceuticals (Costa Mesa, CA, USA).
Cell culture and media
Madin-Darby canine kidney (MDCK) cells used in cytopathic effect (CPE) assays were purchased from the ATCC (Manassas, VA, USA) and grown in antibiotic-free minimum essential medium (MEM) with Earle's balanced salts and l-glutamine (MEM/EBSS; HyClone, Fisher Scientific, USA) containing 5% fetal bovine serum (HyClone, Fisher Scientific) at 37°C under a 5% CO2 atmosphere. The test medium consisted of MEM/EBSS supplemented with 10 U/mL trypsin, 1 μg/mL EDTA and 50 μg/mL gentamicin.
The MDCK cells used in the plaque diameter reduction assay (PDRA) were sourced from the European Collection of Cell Cultures (Wiltshire, UK). Cells were grown in antibiotic-free MEM supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Gibco, Australia) and 1% (w/v) l-glutamine (Invitrogen-Gibco, Australia). The overlay medium used for plaque formation comprised Dulbecco's modified Eagle's medium: Nutrient Mix F12 (DMEM-F12; Gibco, Australia), supplemented with 0.6 μg/mL TPCK-trypsin, 1×insulin–transferrin–selenium (ITS), 0.2% (w/v) BSA and 0.5% (w/v) SeaKem GTG agarose (Lonza, Australia).
Influenza virus strains
A panel of 22 influenza A and B viruses were used for in vitro growth inhibition assays. These included A/Sydney/601/2009 (H1), A/Brisbane/59/2007 (H1N1), A/New Caledonia/20/99 (H1N1), A/Puerto Rico/8/34 (H1N1) (PR8), A/Solomon Islands/03/2006 (H1N1), A/WSN/33 (H1N1), A/Auckland/3/2009 (H1N1)pdm09, A/California/07/2009 (H1N1)pdm09, A/Brisbane/10/07 (H3N2), A/New York/55/2004 (H3N2), A/Panama/2007/99 (H3N2), A/Sydney/5/97 (H3N2), A/Victoria/3/75 (H3N2), A/Victoria/502/2009 (H3N2), A/Wisconsin/67/2005 (H3N2), A/Duck/MN/1525/81 (H5N1), A/Vietnam/1203/2004 (H5N1)H, B/Brisbane/60/2008, B/Florida/4/2006, B/Harbin/7/94, B/Hong Kong/5/72 and B/Malaysia/2506/2006. Influenza A/Vietnam/1203/2004 (H5N1)H virus is a hybrid containing the haemagglutinin (HA) and NA gene segments from influenza A/Vietnam/1203/2004 in a genetic background of influenza A/Ann Arbor/6/60 (H1N1) and can therefore be safely handled in BSL-2 laboratories.
Influenza virus seed stocks were provided by the WHO Collaborating Centre for Reference and Research on Influenza (North Melbourne, Australia). PR8 virus was used in the electron microscopic imaging studies and was provided by Lorena Brown, University of Melbourne. Working stocks of influenza viruses were generated by a further three passages in MDCK cells cultured in MEM supplemented with 1% (w/v) l-glutamine (Invitrogen-Gibco, Australia), 1.2 μg/mL TPCK-trypsin (Worthington, USA), 1×ITS (BioWhittaker MD, USA) and 0.2% (w/v) BSA in PBS (BSA/PBS; Sigma-Aldrich, Australia). Virus titres were determined using either a quantal assay in MDCK cells and the 50% cell culture infectious dose (CCID50) calculated as described by Reed and Muench23 or by plaque formation in MDCK cells24 and expressed as pfu/mL.
The efficacy of the NA inhibitors was tested against selected virus strains in the mouse model. Mouse-passaged influenza A/California/04/2009 (H1N1)pdm09, strain designation 175190, was received from Dr Elena Govorkova, Department of Infectious Diseases, St Jude Children's Hospital, Memphis, TN, USA.25 A virus stock was prepared by growth in MDCK cells. Influenza A/Victoria/3/75 (H3N2) virus was obtained from ATCC (Manassas, VA, USA) and oseltamivir-resistant influenza A/Mississippi/3/2001 (H1N1) virus was obtained from the Neuraminidase Inhibitor Surveillance Network (Melbourne, Australia). These viruses were passaged seven times in mice to enhance their virulence, and then stocks made in MDCK cells and the lethal dose determined. The NA of the mouse-adapted A/Mississippi/3/2001 (H1N1) virus was sequenced to confirm the retention of the H275Y mutation conferring the oseltamivir-resistant phenotype.
Viral CPE inhibition assay
A stock solution of BTA938 (10 mg/mL) was prepared in DMSO and diluted to a working solution (200 μg/mL) in MEM/EBSS supplemented with gentamicin (50 μg/mL; Sigma, St Louis, MO, USA). For the same in vitro tests, ribavirin was dissolved in MEM/EESS with gentamicin at a working concentration of 640 μg/mL.
Cells were cultured in 96-well flat-bottom microtitre plates in growth medium until they reached confluence. Cell growth medium was replaced by test medium. Eight serial dilutions (half log10) of BTA938 (0.032–100 μg/mL) were added in triplicate, 5–10 min prior to the addition of 50–100 CCID50 of virus. Ribavirin was used as an internal control for optimal virus load. After 3 or 6 days of incubation at 37°C under a 5% CO2 atmosphere, the cells were visually scored for virally induced CPE and then stained with 0.011% (final concentration) neutral red (NR). After 2 h at 37°C, cells were washed with sterile PBS and the stain was eluted with extraction buffer (50% ethanol/50% Sorensen's citrate buffer). The colour intensity was determined by the method of Finter26 using a computerized microplate reader (SPECTRAmax® Plus384; Molecular Devices Corporation, Sunnyvale, CA, USA) with absorbance measurements at 540 nm. 50% effective concentration (EC50) values, defined as the effective concentration of a compound required to reduce the viral CPE by 50%, were calculated based on visual (microscopic) scoring of the cell monolayers and on the reduction in NR dye uptake. Assays were independently run a minimum of three times.
The cytotoxicity of BTA938 was assessed in parallel with the antiviral determinations, in wells on the same microtitre plates to which no virus was added. The cytotoxic effects of the compound were expressed as the 50% cytotoxic concentration (CC50), defined as the concentration required to reduce cell growth by 50%. Based on the EC50 and CC50 values, a selectivity index (SI) was computed as the ratio of CC50 to EC50.
Plaque diameter reduction assay (PDRA)
Stock solutions of BTA938 (2.5 mM) and zanamivir, oseltamivir and laninamivir (10 mM) were made by dissolving the compounds in water and were further diluted in virus growth medium consisting of MEM, supplemented with 1% (w/v) l-glutamine, 1.2 μg/mL TPCK-trypsin, 1× ITS and 0.2% (w/v) BSA for use in the PDRA assay. MDCK cells (6 × 105/2 mL) were seeded into six-well plates (Corning Labware, Corning, NY, USA) and, following incubation for 3 days at 37°C in a humidified 5% CO2 atmosphere, the monolayer of cells was ∼90% confluent. The supernatant was then removed from each well and the wells were washed once in PBS previously warmed to 37°C. Serial 5-fold dilutions of each compound in virus growth medium were added to wells. The cell monolayers were inoculated with virus at a concentration previously determined to yield ∼20–40 plaques per well. The cell monolayers were then incubated at 37°C in a humidified 5% CO2 atmosphere for 1 h. The supernatant containing virus and compound was then removed and the plaque overlay medium (generally 1%) added, containing identical concentrations of the compound(s). The assay plates were incubated at 37°C in a humidified 5% CO2 atmosphere to allow plaques to develop (between 2 and 4 days).
After removal of the agar overlay, the plaques were visualized by the addition of 0.1% (w/v) Crystal Violet (Sigma, Australia), 0.85 M sodium chloride (Sigma, Australia) and 10% (v/v) formaldehyde (Ajax Finechem, Australia). Following a 24 h incubation at room temperature, the staining solution and overlay were removed and the diameters of the plaques in each well were evaluated after capturing images using Adobe Photoshop CS3 extended version 10.0.1.
The diameters of the plaques present at each dilution of the compound were expressed as a percentage of the diameter of plaques present in the negative control. These percentages were used to graphically determine the effective concentration of compound that produces a 50% reduction in influenza-induced plaque diameter (EC50). The EC50 values were calculated by non-linear regression using IDBS XLFit4 Excel Add-in Version 4.2.0 (ID Business Solutions Inc., Alameda, CA, USA).
Animals and experimental design
Female 18–20 g BALB/c mice were obtained from Charles River Laboratories International Inc. (Wilmington, MA, USA). The mice were maintained on Teklad Rodent Diet (Harlan Laboratories Inc., Indianapolis, IN, USA) and tap water at the Laboratory Animal Research Center of Utah State University. The animals were quarantined for 3 to 6 days prior to use.
In all animal experiments, zanamivir and the zanamivir dimer BTA938 were administered by the intranasal route to anaesthetized mice in a 50 μL volume. BTA938 and zanamivir were prepared in PBS for administration.
Doses for all treatments and placebo were given 12 h apart except for experiments with single occasion administration. The placebo group received intranasally administered PBS in a dosing regimen similar to that used for the test treatments. Mice were anaesthetized by the intraperitoneal injection of 0.1 mL of ketamine/xylazine (50 mg/kg/5 mg/kg) and then challenged by the intranasal route with approximately three 50% lethal doses of virus in 50 μL. This dose corresponded to 400 CCID50/mL for A/California/04/2009 (H1N1)pdm09, 100 CCID50/mL for A/Victoria/3/75 (H3N2) and 40 CCID50/mL for A/Mississippi/03/2001 (H1N1) H275Y viruses. The challenge dose for each virus was carefully determined by titration in mice following treatment with intranasal saline, because intranasal challenge with influenza virus is exacerbated by the intranasal administration of liquids.27 The mice were weighed prior to treatment and then every other day thereafter to assess the effects of treatment on ameliorating the weight loss due to virus infection. Mice not receiving any treatment served as normal controls for weight comparisons. All groups were observed for weight loss and mortality through day 21. Mice losing ≥70% of their initial body weight were euthanized.
Statistical analysis of animal studies
Kaplan–Meier survival curves were generated and compared by the log-rank (Mantel–Cox) test followed by pairwise comparison using the Gehan–Breslow–Wilcoxon test in GraphPad Prism 5.0f (GraphPad Software Inc., La Jolla, CA, USA). Mean body weights were analysed by one-way analysis of variance followed by Tukey's multiple comparison tests using Prism 5.0f.
Ethics statement for animal studies
These studies were completed in accordance with the approval of the Institutional Animal Care and Use Committee of Utah State University dated 20 September 2010 (renewed 30 August 2013). The work was done in the AAALAC-accredited Laboratory Animal Research Center of Utah State University. The US Government (NIH) approval was renewed on 7 April 2010 (Animal Welfare Assurance no. A3801-01) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Revision; 2010).
Scanning electron microscopy (SEM)
MDCK cells were infected with PR8 virus at a multiplicity of infection of 10 for 1 h at 37°C/5% CO2. After infection, the cells were washed twice with medium and resuspended in medium containing 50 μg/mL zanamivir or BTA938. Negative controls were prepared with infected or uninfected cells suspended in an equal volume of medium alone. Cells were then placed onto 13 mm gold-coated coverslips (NUNC) in 24-well plates and incubated for 10 h at 37°C/5% CO2. Coverslips (13 mm2) were gold coated in an Edwards S150B sputter coater (Edwards, West Sussex, England) for 5 min to give a coating of ∼30–40 nm thickness. The coverslips were removed and dehydrated in increasing concentrations (10%, 30%, 50%, 70% and 90%) of ethanol in water followed by two changes of 100% ethanol for 10 min at each step. The coverslips were dried in a Bal-Tec CPD 030 critical point dryer (Balzers, Liechtenstein, Germany) and mounted onto 25 mm aluminum stubs with double-sided carbon tabs. The coverslips were then coated with 10 nm of chromium using a Xenosput sputter coater (Dynavac, Wantirna South, Australia). The cells on the coverslips were imaged with a Philips XL30 field-emission scanning electron microscope (Philips, Eindhoven, Netherlands) at a voltage of 2.0 kV and a spot size of 2.
Results
BTA938 is potent against a broad range of influenza virus strains in vitro
The viral NA inhibitor BTA938, a zanamivir dimer, was evaluated independently in two laboratories for activity against 22 strains of influenza virus as shown in Tables 1 and 2. Visual and colorimetric determination of the inhibition of CPEs plus plaque diameter reduction data both demonstrated the exceptionally high antiviral activity of BTA938 against the influenza virus strains examined, as well as the low cytopathology as reflected by the high SI. The EC50 values were in the low nanomolar range, with BTA938 exhibiting higher potency than zanamivir, oseltamivir and laninamivir for the majority of viruses tested. However, why the EC50 values for BTA938 were higher than those for oseltamivir against the A/Brisbane/59/2007 (H1N1) and A/Victoria/502/2009 (H3N2) virus strains is not known. In addition, differences in the results from visual and NR assays may reflect the fact that NR assays have the ability to detect toxicity that cannot be observed visually. Uninfected MDCK cells did not exhibit any signs of cytotoxicity at the drug concentrations tested (data not shown).
Table 1.
Antiviral activity of BTA938 against influenza viruses by CPE inhibition
| Virus | EC50 (μM)a visual | EC50 (μM)a NR | SIb visual | SIb NR |
|---|---|---|---|---|
| H1N1 | ||||
| A/California/04/2009 | 0.36 ± 0.18 | 0.93 ± 0.58 | 2200 | 1667 |
| A/California/07/2009 | 0.25 ± 0.49 | 0.59 ± 0.77 | 2497 | 1887 |
| A/NWS/33 | <0.03 ± 0.00 | 0.04 ± 0.03 | >3100 | 2680 |
| A/Solomon Islands/03/2006 | 0.06 ± 0.05 | 0.46 ± 0.50 | 2343 | 427 |
| A/Mississippi/03/2001(H275Y) | 1.40 ± 0.49 | 2.10 ± 1.40 | >417 | >278 |
| H3N2 | ||||
| A/Panama/10/2007 | <0.03 ± 0.00 | <0.03 ± 0.00 | >3100 | >3100 |
| A/Victoria/3/75 | <0.03 ± 0.00 | 0.20 ± 0.33 | >3100 | 2360 |
| A/Wisconsin/67/2005 | 0.19 ± 0.23 | 0.26 ± 0.37 | 1958 | 1936 |
| H5N1 | ||||
| A/Duck/MN/1525/81 | <0.03 ± 0.00 | <0.03 ± 0.00 | >3100 | >3100 |
| A/Vietnam/1203/2004H | <0.03 ± 0.00 | <0.03 ± 0.00 | >3100 | >3100 |
| B | ||||
| Malaysia/2506/2004 | <0.03 ± 0.00 | <0.03 ± 0.00 | >3100 | >3100 |
Results are the means from five independent assays except for A/Panama/10/2007 and A/Victoria/3/75, which were tested independently four times, and A/Solomon Islands/03/2006, A/California/04/2009 and A/Mississippi/03/2001(H275Y), which were tested three times.
aDefined as the effective concentration of a compound required to reduce viral CPEs by 50%.
bDetermined by dividing values for CC50 (>95 μM for all replicates) by values for EC50. SI values <4.0, compound not active; SI values 4.0–9.9, compound slightly active; SI values 10–49.9, compound moderately active; and SI values >50, compound highly active.
Table 2.
Antiviral activity of BTA938, zanamivir, oseltamivir and laninamivir against influenza viruses by plaque diameter reduction
| Virus strain | Mean EC50 (nM) |
|||
|---|---|---|---|---|
| BTA938 | zanamivir | oseltamivir | laninamivir | |
| H1N1 | ||||
| A/Sydney/601/2009 | 0.2 ± 0.1 | 893.6 ± 398.3* | >2500 | 412.2 ± 65.5* |
| A/Auckland/3/2009 | 1.3 ± 0.9 | 446.9 ± 716.3 | 194.5 ± 205.4* | 143.2 ± 138.5* |
| A/Brisbane/59/07 | 50.1 ± 14.4 | 252.1 ± 172.8 | <0.001* | 87.5 ± 66 |
| A/Solomon Islands/3/2006 | 0.001 ± 0.01 | 52.2 ± 45.4 | 110.8 ± 47.9* | 12.0 ± 3.8* |
| A/New Caledonia/20/99 | 0.5 ± 0.4 | 22.5 ± 13.8* | 4.4 ± 4.3 | 65.5 ± 52.9* |
| A/WSN/33 | 0.4 ± 0.5 | 269.7 ± 77.5* | 203.8 ± 122.7* | 236.4 ± 196.9 |
| H3N2 | ||||
| A/Victoria/502/2009 | 0.3 ± 0.2 | 0.20 ± 0.1 | 0.1 ± 0.1 | 0.07 ± 0.05 |
| A/Brisbane/10/07 | 1.1 ± 0.4 | 52.4 ± 30.4 | 2.6 ± 1.6 | 18.8 ± 13.3 |
| A/New York/55/2004 | 1.3 ± 0.9 | 261.4 ± 142.9* | 34.2 ± 12.8* | 19.9 ± 7.7* |
| A/Panama/2007/99 | 0.6 ± 0.6 | 21.5 ± 6.9* | 2.6 ± 0.8* | 20.9 ± 16.3 |
| A/Sydney/5/97 | 0.2 ± 0.3 | 10.5 ± 0.2* | 2.0 ± 1.7 | 38.7 ± 25.0* |
| A/Victoria/3/75 | 0.3 ± 0.2 | 49.0 ± 33.7 | 5.4 ± 2.4* | 98.8 ± 115.6 |
| B | ||||
| B/Brisbane/60/2008 | 0.4 ± 0.4 | 436.8 ± 216.7* | 698.8 ± 481.1 | 319.3 ± 174.1* |
| B/Florida/4/2006 | 2.2 ± 3.0 | 271.5 ± 59.6* | 456.8 ± 385.2* | 95.1 ± 73.6* |
| B/Yamanashi/166/98 | 0.7 ± 0.3 | 13.8 ± 17.2 | 5.8 ± 3.8 | 13.6 ± 7.8* |
| B/Harbin/7/94 | 0.5 ± 0.3 | 47.0 ± 32.3 | 102.2 ± 23.1* | 78.6 ± 48.6 |
| B/Hong Kong/5/72 | 0.7 ± 0.5 | 101 ± 53.7* | 123.4 ± 45.6* | 37.0 ± 29.5 |
Results are the means from three independent assays.
*BTA938 shows a significant difference (P ≤ 0.05) from mean EC50 values for other NA inhibitors.
BTA938 shows greater therapeutic potency than zanamivir against H1N1 and H3N2 virus infections
Initially, the in vivo efficacy of BTA938 for the treatment of influenza was tested against A/California/04/2009 (H1N1)pdm09 virus infections using an established BALB/c mouse challenge model. Groups of 10 BALB/c mice (15 placebo) were treated by the intranasal route twice daily for 5 days with varying doses of BTA938 (1, 0.3, 0.1 and 0.03 mg/kg/day) or zanamivir (10, 3, 1 and 0.3 mg/kg/day), beginning 4 h post-challenge with virus. Five mice did not receive any treatment and served as the weight control group. Animals were monitored for weight loss and mortality for 21 days.
Results indicated a significant difference in the mean day of death, compared with placebo, for the groups receiving 0.1, 0.3 and 1.0 mg/kg doses of BTA938 and the 3 and 10 mg/kg doses of zanamivir (Figure 1a and c). However, significant differences in mean body weight were not observed (Figure 1b and d). The 1 mg/kg dose of BTA938 provided 80% protection from death following virus challenge (Figure 1a). By comparison, the 10 mg/kg dose of zanamivir provided 40% protection from challenge (Figure 1c). Consequently, the zanamivir dimer was ∼10-fold more potent by dose, or 5-fold by molar ratio, than the monomer in protecting against death.
Figure 1.

Effect of twice-daily treatment with BTA938 and zanamivir for an influenza A/California/04/2009 (H1N1)pdm09 virus infection. Mice were treated twice daily with the indicated doses of BTA938 or zanamivir for 5 days, beginning 4 h post-virus challenge. Survival (a and c) and body weight (b and d) were monitored for 21 days post-challenge. A significant difference in survival, compared with placebo, was observed for the groups receiving 0.1, 0.3 and 1.0 mg/kg doses of BTA938 and 3 and 10 mg/kg doses of zanamivir (a and c). However, significant differences in mean body weights were not observed (b and d). **P < 0.01. ***P < 0.001.
We also evaluated the treatment effect of the zanamivir dimer against an H3N2 subtype influenza virus (A/Victoria/3/1975). The same treatment regimen as in the previous experiment was applied with the exception that the 0.03 mg/kg dose of dimer was replaced by 3 mg/kg, based on the findings that the 0.03 mg/kg dose did not provide significant protection against the H1N1pdm09 virus. Groups of 10 mice (15 for placebo) received treatment and five mice served as weight controls (no treatment group).
In this study, a significant difference in the mean day of death, compared with placebo, was observed for all treatment groups (Figure 2a and c). A significant difference in mean body weight was also observed for all treatments groups (Figure 2b and d). All of the mice were protected from challenge infection by the 0.1, 0.3, 1 and 3 mg/kg doses of BTA938 and the 1, 3 and 10 mg/kg doses of zanamivir (Figure 2). These data confirm the greater potency of BTA938 compared with zanamivir against a challenge infection with influenza A/Victoria/3/1975 (H3N2) virus.
Figure 2.

BTA938 and zanamivir treatment for an influenza A/Victoria/3/75 (H3N2) virus infection. Mice were treated twice daily with the indicated doses of BTA938 or zanamivir for 5 days, beginning 4 h post-virus challenge. Survival (a and c) and body weight (b and d) were monitored for 21 days post-challenge. A significant difference in survival, compared with placebo, was observed for all the treatment groups (a and c). A significant difference in mean body weight was also observed for all the treatment groups (b and d). ***P < 0.001.
Single-dose BTA938 protects mice from pandemic H1N1 virus infection
Any treatment regimen involving repeated doses or other complexities is at risk of incomplete patient compliance. A reduction in dosing frequency would therefore be advantageous on many levels. We thus assessed the dose–response of a single therapeutic treatment of BTA938 administered 4 h post-virus challenge with influenza A/California/04/2009 (H1N1)pdm09 virus. Four drug concentrations were evaluated (0.3, 1, 3 and 10 mg/kg/day). Again, groups of 10 mice (15 for placebo) were treated and 5 mice served as weight controls.
A significant difference from placebo was observed in the mean day of death for all four treatment groups (Figure 3a), although higher mortality was observed in the group treated with the 3 mg dose than the 1 mg dose. The immediate reason for these observations in unclear. However, we often observe a higher variability in survival following challenge for groups of mice treated with antiviral drugs at suboptimal doses. In addition, the difference in the mean day of death for the groups treated with the 1 or 3 mg doses of BTA938 was not significant. Following treatment with a single 10 mg/kg dose of BTA938, mice lost significantly less weight than mice receiving the placebo (Figure 3b). This study demonstrated the efficacy of a single 10 mg/kg dose of BTA938 administered 4 h post-virus challenge in preventing virus-induced weight loss and mortality in mice.
Figure 3.
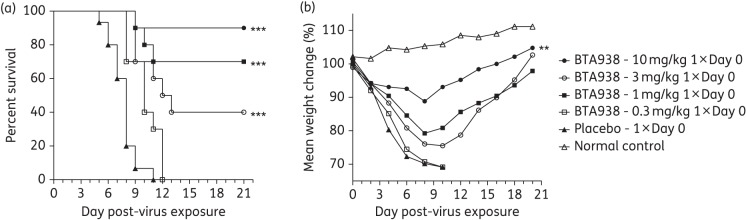
Effect of single-dose therapeutic treatment using BTA938 for an influenza A/California/04/2009 (H1N1)pdm09 virus infection. Survival and body weight were monitored following a single treatment with the indicated doses of BTA938, beginning 4 h post-virus challenge. A significant difference in survival, compared with placebo, was observed for the 1, 3 and 10 mg/kg treatment groups (a). A significant difference in mean body weight, compared with placebo, was only observed for the 10 mg/kg treatment group (b). **P < 0.01. ***P < 0.001.
Single-dose BTA938 is equivalent to multiple doses of zanamivir for an oseltamivir-resistant (H275Y) influenza virus infection
Oseltamivir resistance has been reported for pandemic influenza A(H1N1)2009 viruses with the single amino acid substitution of histidine to tyrosine at position 275. Fortunately, the H275Y mutant is susceptible to zanamivir and laninamivir. However, with the exception of Japan, zanamivir is currently the only approved alternative therapeutic treatment option available in most countries. We speculated that the zanamivir derivative BTA938 could also be usefully applied in the treatment of oseltamivir-resistant influenza virus infections even when administered as a single dose. We tested our hypothesis by challenging mice with oseltamivir-resistant influenza A/Mississippi/03/2001 (H1N1) virus and treating them 4 h later with a single dose of BTA938. Three different concentrations (3, 10 and 30 mg/kg) were evaluated and compared with groups of 10 mice receiving zanamivir administered intranasally twice daily for 5 days at the same dosage levels. Groups of 15 mice received a placebo and 3 mice received no treatment.
Kaplan–Meier survival curves showed that all doses of BTA938 and zanamivir completely protected mice from the challenge infection (Figure 4a). This was mirrored by the results obtained from body weight measurements (Figure 4b). These data clearly demonstrate that a single dose of BTA938 is equivalent to comparable doses of zanamivir administered twice per day for 5 days against an H275Y oseltamivir-resistant virus infection in mice.
Figure 4.
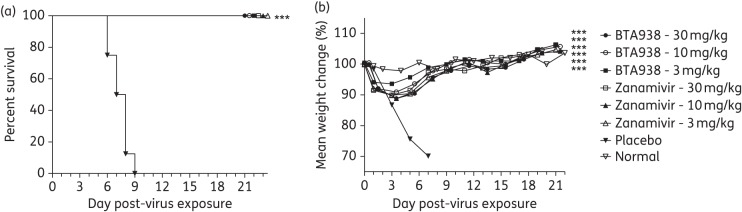
Effect of single-dose therapeutic treatment using BTA938 for an oseltamivir-resistant influenza A/Mississippi/03/2001 (H1N1) H275Y virus infection. Survival and body weight were monitored following a single treatment with BTA938 at the indicated doses, or twice daily for 5 days with zanamivir, beginning 4 h post-virus challenge. A significant difference in survival, compared with placebo, was observed for all the treatment groups (a). A significant difference in mean body weight was also observed for all treatment groups (b). ***P < 0.001.
Single-dose BTA938 protects mice from influenza virus when administered up to 7 days before challenge
Our previous in vivo experiments demonstrated that BTA938 was superior to zanamivir as a therapeutic option for the treatment of influenza following infection with different influenza viruses. To test the prophylactic potential of BTA938, the dimer was administered intranasally at a concentration of 10 mg/kg on three single occasions (−7, −3 or −1 days pre-virus challenge). Mice in groups of 10 (15 for placebo) were infected at day 0 with influenza A/California/04/2009 (H1N1)pdm09 virus. Each prophylactic treatment had its own control. Five mice were not infected and served as weight controls. Survival and body weight were monitored through day 21.
Remarkably, a single treatment administered 7 days before virus challenge protected 70% of the mice from death and a single treatment administered 1 or 3 days before challenge protected 90% of the mice (Figure 5a). Mice in all the treatment groups experienced weight loss (Figure 5b), although prevention of weight loss and improved weight gain following prophylactic treatment is a general indicator of reduced morbidity following infection.
Figure 5.
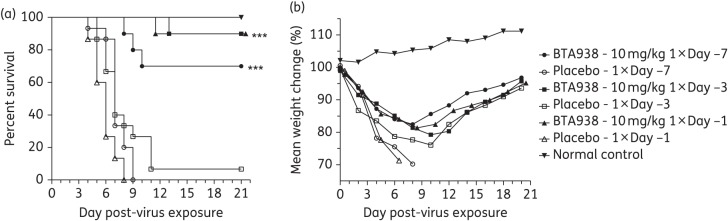
Effect of single-dose prophylactic treatment using BTA938 for an influenza A/California/04/2009 (H1N1)pdm09 virus infection. Survival and body weight were monitored following a single treatment with 10 mg/kg BTA938 on day −7, −3 or −1 pre-virus challenge. A significant difference in survival, compared with placebo, was observed for all prophylactic treatments (a). However, significant differences in mean body weights were not observed (b). ***P < 0.001.
BTA938 enhances intervirion aggregation at the cell surface as visualized by SEM
To test the hypothesis that the increased antiviral activity of BTA938 is a result of enhanced intervirion binding leading to more virus aggregation at the point of exit from the host cells, MDCK cells were infected with PR8 virus, followed by treatment with BTA938 or zanamivir. Infected cells without any treatment served as virus controls. At 10 h post-infection, MDCK cells were prepared for SEM in order to visualize the surface of the host cell and budding virions.
Micrographs of uninfected MDCK cells show the typical microfolds on the apical surface of the cell (Figure 6a). Following infection with PR8 virus, newly replicated virions were seen in different stages of the budding process from the host cell. Budding virions were relatively few and still attached to the microfolds (Figure 6b). In the presence of 1.5 μM zanamivir, the number of virions observed on the cell was greatly increased, suggestive of an inability to efficiently release from the cell surface (Figure 6c) compatible with the known function of the NA in the removal of receptors from the site of budding. The virions appeared to be packed into orderly arrays one layer deep surrounding the microfold projections. Some virions formed small clusters. In the presence of 0.5 μM BTA938, this aggregation appeared to be greatly enhanced, with vast numbers of virions covering the cell surface, several layers deep (Figure 6d). The results indicate a much greater degree of virus retention around the infected cell with the dimer compared with monomeric zanamivir. Although these data are not quantitative, they address the concern that the previous SEM data involving the aggregation of purified virions and purified NA ‘heads’ may not necessarily predict what occurs at the surface of the infected cell. Interestingly, BTA938 was slightly less potent than zanamivir or laninamivir in virus-based NA inhibition experiments (data not shown), suggesting that the enhanced aggregation seen at the cell surface is not a result of a substantially increased inhibition of NA alone. These data, considered together, suggest a dual mechanism of action, since NA inhibition alone cannot explain the potency seen in cell culture and in vivo.
Figure 6.

BTA938 aggregation of influenza virus particles on the surface of virus-infected MDCK cells. The surface of MDCK cells was visualized by SEM. (a) Uninfected control MDCK cells. (b) PR8-infected MDCK cells. (c) PR8-infected MDCK cells treated with 1.5 μM zanamivir. (d) PR8-infected MDCK cells treated with 0.5 μM BTA938.
Discussion
Surprisingly few antiviral agents have been approved over the last four decades for the treatment and prophylaxis of influenza virus infections. They include amantadine, rimantadine, oseltamivir and zanamivir. Of these four antiviral drugs, the adamantanes (amantadine and rimantadine) have already become ineffective due to widespread resistance among currently circulating influenza virus strains. This leaves only the NA inhibitors (oseltamivir and zanamivir) as the recommended therapeutic option.9 However, oseltamivir-resistant seasonal H1N1 viruses, and to a lesser degree pandemic 2009 H1N1 influenza viruses, have been reported worldwide. Emergence of drug resistance against antiviral compounds is a common problem, with some influenza virus strains developing resistance over a remarkably short time frame. This has particularly serious implications for treatment success during the early stages of an influenza epidemic when the timely production and delivery of vaccines against the new virus strain is a challenge.28
BTA938 has emerged as the lead compound in a drug discovery programme aimed at developing anti-influenza therapies based on the hypothesis that multivalent interactions of the sialidase analogue zanamivir with its binding partner, the viral NA, would increase the antiviral potential.20,21 In order to achieve multivalent binding, zanamivir molecules were linked through the C7 hydroxyl to generate dimeric, trimeric, tetrameric and polymeric zanamivir derivatives.20,21,29 The effect of linker length and functionality on the antiviral state was also explored and indicated that BTA938 with an unsubstituted 14-carbon alkyl linker connecting two zanamivir monomers was highly active against representative influenza A and B viruses in MDCK cells.20,21
Results from in vitro assays highlight the remarkable antiviral activity against a broad range of influenza viruses in vitro. Even so, high antiviral potency in cell-based assays does not necessarily translate into an equally successful protection/treatment of subjects against virus challenge in in vivo studies. Macdonald and colleagues20 tested the efficacy of BTA938 in a mouse model and showed that the zanamivir dimer significantly reduced virus titres in lungs infected with influenza A/Victoria/3/75 (H3N2) virus. These tests, however, only addressed the prophylactic potential of BTA938 since the compound was administered 7 days prior to infection. In addition, virus titre in the lung is not an absolute indicator of disease severity. Therefore, we used our established mouse infection model to follow the therapeutic and prophylactic effect of BTA938 on weight loss and survival in the context of infections caused by different influenza strains. However, we recognize that mouse lethality is also not an accurate measure of disease severity in the context of human infection. Results from our in vivo experiments showed that BTA938 was ∼10-fold more potent than zanamivir in protection against an influenza A/H1N1pdm09 virus infection and at least 3-fold more potent against an influenza A/Victoria/3/75 (H3N2) virus infection.
When zanamivir is used for the treatment of influenza virus infections in adults, 10 mg doses are administered by inhalation, twice a day, for 5 days.22 However, this dosing regimen may impede patient compliance. Reducing the frequency of inhalation is therefore a desirable feature of an antiviral drug. Macdonald and colleagues reported increased lung retention times for BTA938 in rats and suggested that a single low dose of the dimer would provide effective treatment and prophylaxis for influenza virus infections.20 In addition to studies completed by Macdonald et al. in the non-lethal mouse model,20 we evaluated this hypothesis in our lethal mouse model and demonstrated the validity of this assumption. A single 10 mg/kg dose of BTA938 administered 4 h post-infection was effective in protecting mice from death due to influenza A/H1N1pdm09 virus infection. A single dose of BTA938 was also successfully used in the treatment of an oseltamivir-resistant influenza A(H1N1) virus strain at an even lower dose (3 mg/kg). Moreover, 10 mg/kg doses of BTA938 proved to be highly effective in prophylactic treatments, although the degree of protection depended on the time span from drug administration to virus challenge.
Zanamivir, an analogue of sialic acid (SA), binds to the active site of influenza virus NA, a surface glycoprotein, and blocks its action. Influenza virus NA is important in the replication cycle of the virus, particularly at the point of exit of the virus from the cell by budding.30 In the absence of NA activity, the nascent virus particles can potentially bind back to SA-containing receptors on the cell from which they are exiting.31,32 In addition, the NA as well as the other surface glycoprotein HA have terminal SA on their carbohydrate side chains when newly synthesized. As the role of the HA is to bind to host glycoproteins with terminal SA side chains, nascent virions can potentially bind to each other unless the NA removes these terminal SA residues. The emerging virions can become trapped at the surface of dying cells, leading to inefficient release and a reduced number of virus particles to infect new cell targets.
The BTA938 zanamivir dimer, with an unsubstituted 14-carbon alkyl chain serving as the bridging element, associates with and inhibits the influenza NA, but available evidence suggests that it is not an intrinsically more potent enzyme inhibitor than zanamivir in NA assays. Rather, NA kinetic data indicate that modification at the C-7 position may actually impede the association of the zanamivir moiety with the NA active site.20 The calculated linker length is 22.8 Å assuming a fully extended linker.20 This distance could conceivably allow for head-to-head binding between NA sites on separate virions (intervirion binding). We hypothesized that if intervirion binding did occur, it might lead to cross-linking of separate virions and result in even more extensive virion aggregation at the cell surface, leading to superior antiviral properties.20,21 Evidence for the formation of high-molecular-weight virus aggregates was provided by electron micrographs of negatively stained, purified influenza virus particles incubated in the presence of the dimer and gel filtration experiments in which the dimer appeared to mediate the aggregation of purified NA.20 The effect, if any, of the dimer on virus emerging from infected cells was unknown. In addition, although negative staining has been a traditional and well-used approach for the visualization of virus particles,33–36 the information gained with this technique lacks spatial context in regard to the host cell.
We tested the hypothesis that the enhanced activity of the dimer was due to its ability to block the removal of viral receptors from around the site of budding, leading to virion attachment back to the host cell via the HA, coupled with an enhanced ability to promote intervirion aggregation to these trapped virions. Visualization of budding using SEM allowed us to view the process of virion trapping at the surfaces of infected cells in the presence of BTA938 or zanamivir. An enhanced aggregation of virus particles was clearly visible on the surface of infected MDCK cells that were treated with BTA938. The mechanism of enhanced aggregation is not fully elucidated but could conceivably be due to direct dimer-mediated intervirion binding. Alternatively, a dimer-mediated enhancement of NA inhibition may occur through inter-NA tetramer interactions on the surface of individual virions. These interactions could increase the ability of the dimer to block the cleavage of SA from the terminus of the carbohydrate side chains of nascent HA and NA, promoting clumping via HA–SA interactions. The available evidence does not allow us to distinguish between these possibilities. Nonetheless, these studies further support the hypothesis that the enhanced potency of BTA938 is likely to be a result of the enhanced aggregation of virus particles in the presence of dimeric zanamivir beyond that which can be attributed to the inherent potency of the compound as an inhibitor of NA in vitro. Arguably, this represents a dual mode of action compared with monomeric NA inhibitors: inhibition of NA and enhanced aggregation.
In conclusion, in vitro and in vivo experiments have clearly shown the high antiviral potential of BTA938 for the treatment of infections caused by a variety of influenza viruses. Moreover, we have proved that the administration of a single dose of BTA938 is sufficient for therapeutic and prophylactic protection in mice challenged with influenza A virus infections. The clinical advantages of a substantially more potent NA with an infrequent dosing profile have yet to be determined but may include enhanced efficacy and reduced exposure through the use of lower, infrequent doses. It may also offer advantages in the management of resistant viruses, should these emerge, and efficiencies in pandemic stockpile management.
Funding
This work was supported by the Virology Branch and the Respiratory Diseases Branch, Division of Microbiology and Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health under Contract Numbers N01-AI-30063 (awarded to Southern Research Institute, Birmingham, AL, USA), HHSN272201000039I/HHSN27200005/A37 and NIH grant number U01AI070263.
Transparency declarations
S. H., A. L., M. P. and S. P. T. are employees of Biota Holdings Limited, Australia. All other authors: none to declare.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgements
We gratefully acknowledge the careful review of the manuscript prior to submission by Dr Lorena E. Brown from the University of Melbourne.
References
- 1.CDC. Clinical Signs and Symptoms of Influenza. http://www.cdc.gov/flu/professionals/acip/clinical.htm. (18 March 2014, date last accessed)
- 2.Dugas AF, Rothman RE. Infectious disease. Antiviral agents for the treatment and chemoprophylaxis of influenza. Ann Emerg Med. 2011;58:299–303. doi: 10.1016/j.annemergmed.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Rothberg MB, Haessler SD, Brown RB. Complications of viral influenza. Am J Med. 2008;121:258–64. doi: 10.1016/j.amjmed.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson MG, Shay DK, Zhou H. Estimates of deaths associated with seasonal influenza—United States, 1976–2007. Morb Mortal Wkly Rep. 2010;59:1057–62. [PubMed] [Google Scholar]
- 5.WHO. WHO Influenza Fact Sheet No. 211, Revised March 2003 (Updated 19 October 2011). http://www.who.int/mediacentre/factsheets/2003/fs211/en/ (18 March 2014, date last accessed)
- 6.WHO. WHO Guidelines for Pharmacological Management of Pandemic (H1N1) 2009 Influenza and Other Influenza Viruses. http://www.who.int/csr/resources/publications/swineflu/h1n1_use_antivirals_20090820/en/ (18 March 2014, date last accessed)
- 7.Cheng VC, To KK, Tse H, et al. Two years after pandemic influenza A/2009/H1N1: what have we learned? Clin Microbiol Rev. 2012;25:223–63. doi: 10.1128/CMR.05012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Govorkova EA, McCullers JA. Therapeutics against influenza. Curr Top Microbiol Immunol. 2013;370:273–300. doi: 10.1007/82_2011_198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiore AE, Fry A, Shay D, et al. Antiviral agents for the treatment and chemoprophylaxis of influenza—recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 2011;60:1–24. [PubMed] [Google Scholar]
- 10.Da Dalt L, Calistri A, Chillemi C, et al. Oseltamivir-resistant pandemic (H1N1) 2009 treated with nebulized zanamivir. Emerg Infect Dis. 2010;16:1813–5. doi: 10.3201/eid1611.100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaur AH, Bagga B, Barman S, et al. Intravenous zanamivir for oseltamivir-resistant 2009 H1N1 influenza. New Engl J Med. 2010;362:88–9. doi: 10.1056/NEJMc0910893. [DOI] [PubMed] [Google Scholar]
- 12.Centers for Disease Control and Prevention. Update: influenza activity—United States and worldwide, May 20–September 22, 2012. Morb Mortal Wkly Rep. 2012;61:785–9. [PubMed] [Google Scholar]
- 13.Calatayud L, Lackenby A, Reynolds A, et al. Oseltamivir-resistant pandemic (H1N1) 2009 virus infection in England and Scotland, 2009–2010. Emerg Infect Dis. 2011;17:1807–15. doi: 10.3201/eid1710.110117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carr S, Ilyushina NA, Franks J, et al. Oseltamivir-resistant influenza A and B viruses pre- and postantiviral therapy in children and young adults with cancer. Pediatr Infect Dis J. 2011;30:284–8. doi: 10.1097/INF.0b013e3181ff863b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.CDC. Oseltamivir-resistant novel influenza A (H1N1) virus infection in two immunosuppressed patients—Seattle, Washington, 2009. Morb Mortal Wkly Rep. 2009;58:893–6. [PubMed] [Google Scholar]
- 16.Chan PA, Connell NT, Gabonay AM, et al. Oseltamivir-resistant 2009–2010 pandemic influenza A (H1N1) in an immunocompromised patient. Clin Microbiol Infect. 2010;16:1576–8. doi: 10.1111/j.1469-0691.2010.03212.x. [DOI] [PubMed] [Google Scholar]
- 17.Graitcer SB, Gubareva L, Kamimoto L, et al. Characteristics of patients with oseltamivir-resistant pandemic (H1N1) 2009, United States. Emerg Infect Dis. 2011;17:255–7. doi: 10.3201/eid1702.101724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storms AD, Gubareva LV, Su S, et al. Oseltamivir-resistant pandemic (H1N1) 2009 virus infections, United States, 2010–11. Emerg Infect Dis. 2012;18:308–11. doi: 10.3201/eid1802.111466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurt AC, Hardie K, Wilson NJ, et al. Characteristics of a widespread community cluster of H275Y oseltamivir-resistant A(H1N1)pdm09 influenza in Australia. J Infect Dis. 2012;206:148–57. doi: 10.1093/infdis/jis337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macdonald SJ, Watson KG, Cameron R, et al. Potent and long-acting dimeric inhibitors of influenza virus neuraminidase are effective at a once-weekly dosing regimen. Antimicrob Agents Chemother. 2004;48:4542–9. doi: 10.1128/AAC.48.12.4542-4549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macdonald SJ, Cameron R, Demaine DA, et al. Dimeric zanamivir conjugates with various linking groups are potent, long-lasting inhibitors of influenza neuraminidase including H5N1 avian influenza. J Med Chem. 2005;48:2964–71. doi: 10.1021/jm040891b. [DOI] [PubMed] [Google Scholar]
- 22.GlaxoSmithKline. Relenza® (Zanamivir) Inhalation Powder. http://us.gsk.com/products/assets/us_relenza_pil.pdf. (18 March 2014, date last accessed)
- 23.Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–7. [Google Scholar]
- 24.Tannock GA, Paul JA, Barry RD. Relative immunogenicity of the cold-adapted influenza virus A/Ann Arbor/6/60 (A/AA/6/60-ca), recombinants of A/AA/6/60-ca, and parental strains with similar surface antigens. Infect Immun. 1984;43:457–62. doi: 10.1128/iai.43.2.457-462.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ilyushina NA, Khalenkov AM, Seiler JP, et al. Adaptation of pandemic H1N1 influenza viruses in mice. J Virol. 2010;84:8607–16. doi: 10.1128/JVI.00159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finter NB. Dye uptake methods for assessing viral cytopathogenicity and their application to interferon assays. J Gen Virol. 1969;5:419–27. [Google Scholar]
- 27.Smee DF, von Itzstein M, Bhatt B, et al. Exacerbation of influenza virus infections in mice by intranasal treatments and implications for evaluation of antiviral drugs. Antimicrob Agents Chemother. 2012;56:6328–33. doi: 10.1128/AAC.01664-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bavagnoli L, Maga G. The 2009 influenza pandemic: promising lessons for antiviral therapy for future outbreaks. Curr Med Chem. 2011;18:5466–75. doi: 10.2174/092986711798194397. [DOI] [PubMed] [Google Scholar]
- 29.Watson KG, Cameron R, Fenton RJ, et al. Highly potent and long-acting trimeric and tetrameric inhibitors of influenza virus neuraminidase. Bioorg Med Chem Lett. 2004;14:1589–92. doi: 10.1016/j.bmcl.2003.09.102. [DOI] [PubMed] [Google Scholar]
- 30.Bouvier NM, Palese P. The biology of influenza viruses. Vaccine. 2008;26(Suppl 4):D49–53. doi: 10.1016/j.vaccine.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palese P, Tobita K, Ueda M, et al. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology. 1974;61:397–410. doi: 10.1016/0042-6822(74)90276-1. [DOI] [PubMed] [Google Scholar]
- 32.Palese P, Compans RW. Inhibition of influenza virus replication in tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA): mechanism of action. J Gen Virol. 1976;33:159–63. doi: 10.1099/0022-1317-33-1-159. [DOI] [PubMed] [Google Scholar]
- 33.Horne RW, Hobart JM, Markham R. Electron microscopy of tobacco mosaic virus prepared with the aid of negative staining-carbon film techniques. J Gen Virol. 1976;31:265–9. doi: 10.1099/0022-1317-31-2-265. [DOI] [PubMed] [Google Scholar]
- 34.Horne RW, Wildy P. Virus structure revealed by negative staining. Adv Virus Res. 1963;10:101–70. doi: 10.1016/s0065-3527(08)60698-3. [DOI] [PubMed] [Google Scholar]
- 35.Stannard LM, van der Riet FD, Moodie JW. The morphology of human immunodeficiency virus particles by negative staining electron microscopy. J Gen Virol. 1987;68:919–23. doi: 10.1099/0022-1317-68-3-919. [DOI] [PubMed] [Google Scholar]
- 36.Almeida JD, Waterson AP. A morphological comparison of Bittner and influenza viruses. J Hyg (Lond) 1967;65:467–74. doi: 10.1017/s0022172400046003. [DOI] [PMC free article] [PubMed] [Google Scholar]