

Abstract
Despite its narrow therapeutic window, lithium is still regarded as the gold standard comparator and benchmark treatment for mania. Recent attempts to find new drugs with similar therapeutic activities have yielded new chemical entities. However, these potential new drugs have yet to match the many bioactivities attributable to lithium's efficacy for the treatment of neuropsychiatric diseases. Consequently, an intense effort for re-engineering lithium therapeutics using crystal engineering is currently underway. We sought to improve the likelihood of success of these endeavors by evaluating the pharmacokinetics of previously unexplored lithium salts with organic anions (lithium salicylate and lithium lactate). We report that these lithium salts exhibit profoundly different pharmacokinetics compared to the more common FDA approved salt, lithium carbonate, in rats. Remarkably, lithium salicylate produced elevated plasma and brain levels of lithium beyond 48 hours post-dose without the sharp peak that contributes to the toxicity problems of current lithium therapeutics. These findings could be important for the development of the next generation of lithium therapeutics.
Introduction
One of the oldest psychiatric drugs in existence remains heavily utilized by clinicians today despite intense marketing of newer alternative drugs still under patent protection. This is because lithium has numerous bioactivities that remain unmatched by the alternatives. For example, lithium is the only drug that has consistently reduced suicidality in patients with neuropsychiatric disorders 1–3. It also exerts neuroprotective effects by increasing BDNF 4, 5 and attenuating the release of several inflammatory cytokines from activated microglia 6, 7. Perhaps the most highly studied bioactivities of lithium are GSK-3β inhibition 8 and inositol monophosphatase (IMPase) inhibition leading to cerebral inositol depletion 9, 10. These bioactivities have been widely regarded as the primary mechanisms of lithium therapy for its FDA-approved indication.
Recently, there have been efforts to find a lithium mimetic with improved safety 11, 12. It is our opinion that this use of the term “lithium mimetic” is somewhat misleading since none of these new chemical entities have matched lithium's polypharmacological mechanisms of action for the treatment of neuropsychiatric diseases. In particular, lithium therapeutics are deemed gold standard for treatment of mania, thus optimizing their safety and efficacy should have wide-ranging clinical applications.
Alternatively, others have used crystal engineering techniques to re-engineer lithium therapeutics by creating novel ionic cocrystals of lithium salts 7, 13, 14. We argue that cocrystallization represents a low risk, low cost approach with the most potential for achieving the desired therapeutic outcome for many reasons. For example, the active pharmaceutical ingredient (API) in this crystal engineering approach remains lithium, which is already FDA-approved with a long history of use in medicine. Also, the FDA has just issued a guidance for industry regarding the regulation of pharmaceutical cocrystals that includes an expedited pathway for their approval 15. Thus, the cost to bring a lithium cocrystal to market will likely be significantly lower than that of a new drug.
An important step in the crystal engineering of ionic cocrystals of lithium is the selection of the most appropriate parent lithium salt. One obvious consideration that has already been identified is that the anion of the lithium salt should be pharmaceutically acceptable 7. However, another important factor is pharmacokinetics. Often, lithium salts are assumed to dissociate following oral administration leading to very similar plasma and brain levels of lithium. In fact, one study compared lithium carbonate, lithium chloride, and lithium orotate in rats 16. This author found no differences in the uptake, distribution, and excretion of the lithium ion. Still, due to the complex nature of the pharmacokinetics of multi-component materials, we decided to evaluate the plasma and brain pharmacokinetics of two previously unexplored salts of lithium that seemed to be good candidates for crystal engineering endeavors: lithium salicylate and lithium lactate. Our findings are described in the report herein.
Results
Lithium Pharmacokinetics
Male Sprague Dawley rats weighing 200–250 grams were dosed via oral gavage with 4 mEq/kg elemental lithium as lithium salicylate or lithium lactate dissolved in deionized water (n=3 per time point per lithium salt). Blood and brain were collected and lithium was measured using atomic absorption spectroscopy (AAS). Plasma and brain lithium measurements are plotted as mean ± SEM in Figure 1.
Figure 1. Pharmacokinetic Curves.

Lithium measurements are plotted as mean ± SEM (* P<0.05, ** P<0.01, ***P<0.001).
Lithium lactate resulted in elevated lithium plasma levels at 2 hours but peaked at 24 hours post-dose and was eliminated rapidly. In contrast, lithium salicylate produced elevated lithium plasma levels through the first 48 hours and was eliminated slowly. Interestingly, both formulations produced elevated brain levels only at 24 and 48 hours post-dose. Table 1 shows some pertinent pharmacokinetic parameters in our experiment. However, these estimates should be used as preliminary indicators since only four carefully selected time points were utilized to limit the use of animals as much as possible. The plasma area under curve (AUC) for lithium lactate was higher than lithium salicylate. However, the brain AUC for lithium salicylate was slightly higher than lithium lactate. Because we utilized the same experimental protocol and time points for our pharmacokinetics study as previously used by Smith et al. for lithium carbonate 7, this allowed the determination of the relative bioavailability (Frel) of lithium salicylate and lithium lactate compared to lithium carbonate (Table 1). The relative bioavailability of both lithium salicylate and lithium lactate were lower than lithium carbonate. The plasma and brain Frel of lithium salicylate was .35 and .54, respectively. The plasma and brain Frel of lithium lactate was .45 and .54, respectively.
Table 1.
Pharmacokinetic Parameters
| Lithium Salicylate | Lithium Lactate | |||
|---|---|---|---|---|
| Plasma | Brain | Plasma | Brain | |
| TMAX (hour) | 24±0.0 | 48±0.0 | 24±0.0 | 24±0.0 |
| CMAX (ug/mL or ug/g) | 2.21±0.10 | 2.89±0.13 | 4.54±0.59 | 3.87±0.40 |
| AUC(0–72) (hour*μg/mL or /g) | 121.8±5.71 | 153.1±7.66 | 157.2±19.64 | 152.0±15.90 |
| Frel (vs. Lithium Carbonate) | .35 | .54 | .45 | .54 |
Discussion
There is a large disparity in regards to comparative studies of the pharmacokinetics of lithium salts in the peer-reviewed literature. Until now, only lithium chloride, carbonate, and orotate have been subjected to these types of studies 16. To our knowledge, the present study represents the first in vivo pharmacokinetic assessment of lithium salicylate and lithium lactate. Because both of these salts are considered to be pharmaceutically acceptable and are amenable for cocrystallization using crystal engineering techniques 7, such pharmacokinetic data will be critical in advancing lithium therapeutics.
Interestingly, we found that lithium salicylate exhibited an unexpected pharmacokinetic profile that is unlike any other lithium salt reported in the literature to date. The known toxicity issues of FDA approved lithium salts could be exacerbated by their pharmacokinetics given its narrow therapeutic window. We previously reported that lithium carbonate peaks rapidly and is eliminated within 48 hours 7. Comparatively, both of the lithium salts in our present study underperformed lithium carbonate from bioavailability standpoints. However, given that oral bioavailability is not a problem with lithium therapeutics17, 18 we don't anticipate that this discrepancy will disqualify either of these salts for development as drugs. In fact, we argue that the plateau plasma levels observed in this study of lithium salicylate could improve the safety of lithium therapy and, consequently, improve patient compliance. This is supported by previous investigators who suggested that an ideal lithium preparation would attenuate high blood level peaks and exhibit gradually declining blood concentrations 19. Encouragingly, this is precisely the pharmacokinetic profile that was produced by lithium salicylate in our study (Figure 1). Previous attempts at formulating proprietary controlled release lithium therapeutics have been somewhat successful at prolonging lithium plasma levels 20. Nonetheless, these formulations still produced the initial plasma spike attributable to toxicity problems observed in lithium therapy. We also found that although lithium salicylate produced comparatively lower plasma lithium exposure than lithium lactate, it produced better brain exposure. Thus, biodistribution also appears to be affected by the choice of anion. Future studies are required to explain the mechanisms behind these observed phenomenons.
Indeed, these pharmacokinetic differences were unexpected since both lithium salts were administered fully dissolved in an aqueous solution, eliminating the possibility of solubility-mediated effects. This would lead one to predict that the lithium pharmacokinetics would be similar for both salts. Since that was not the case, we hypothesize that the observed “plateau effect” and modulated brain biodistribution of lithium as lithium salicylate is likely due to absorption, distribution, metabolism, and/or elimination (ADME) effects from the salicylate anion. The precise mechanism for this is unclear. However, this could be due to the chemical modification of the physiological transporter(s) of lithium ions in vivo. For example, sodium ion transporters have similar permeability for both sodium and lithium ions 22. It's feasible that salicylate chemically modifies the sodium ion transporter, changing its permeability. Future studies should confirm that coadministration of salicylic acid and FDA-approved lithium salts produce similar pharmacokinetics to those observed for lithium salicylate in this contribution.
Because lithium is so effective at treating neuropsychiatric diseases such as bipolar disorder and suicidality1, 23, 24 it is still used despite known toxicity issues that require frequent blood monitoring by a clinician. We argue that finding a new molecule that is a true “lithium mimetic” is probably a lost cause and recognize that crystal engineering approaches like cocrystallization could solve the toxicity issues. The preliminary data presented here demonstrates that some currently available but understudied lithium salts (e.g. lithium salicylate) may also solve the toxicity issues of conventional lithium salts (lithium carbonate and lithium citrate). However, developing new lithium salts as drugs would require significant investment from a pharmaceutical company without composition of matter patent protection. Cocrystals are patentable25 which improves the likelihood of realizing a good return on the investment required to develop them as a new drug. Moreover, cocrystals of lithium salts might also offer improved efficacy since the coformers can be rationally selected to be synergistic as discussed in recent crystal engineering efforts 13, 14. Future studies should elucidate how the logical design of multi-component pharmaceutical materials can be used to improve the clinical performance of known APIs like lithium.
Experimental Section
Reagents and Materials
Lithium salicylate (≥98% purity) and lithium lactate (≥95% purity) were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA) and used as such without further purification. Chemical structures are shown in Figure 2. Both lithium salts were characterized using powder x-ray diffraction. This data is included as supplementary information.
Figure 2.
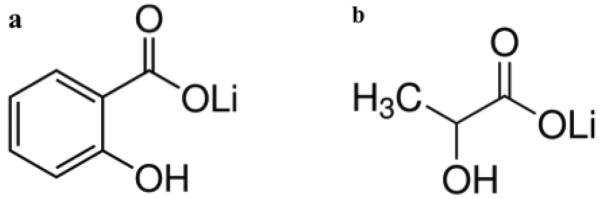
Chemical Structures of lithium salicylate (a) and lithium lactate (b).
Pharmacokinetics Studies
Previously described methodologies were used for the pharmacokinetics studies 7. Male Sprague Dawley rats weighing 200–250 grams were purchased from Harlan (Harlan Sprague Dawley Inc., Indianapolis, IN). The animals were housed at the Moffitt Cancer Center vivarium (Tampa, FL) with a 12 hour light-dark cycle. The rats were allowed to acclimate for a period of one week before any experiments are carried out. All experiments were conducted in accordance with USF IACUC approved protocols. They were allowed free access to food and water throughout the experiment. The rats were dosed via oral gavage with 4 mEq/kg elemental lithium as lithium salicylate or lithium lactate dissolved in deionized water. Animals in each treatment group were euthanized at 2, 24, 48, and 72 hours (n=3 per time point per lithium salt) and blood was collected by cardiac puncture and carefully perfused with a pressure-controlled pump to maintain microvasculature integrity before removing brain tissue. Blood was centrifuged at 1600 × g at room temperature for 10 minutes and plasma was separated. A 500 μl aliquot was diluted 10 fold in a 5% TCA and 10% isopropyl alcohol (IPA) solution, vortexed and allowed to sit for 10 minutes in order to precipitate proteins. These aliquots were centrifuged at 3000 × g for 30 minutes and the supernatant was transferred to clean tubes prior to measuring lithium content using atomic absorption spectroscopy (AAS). Brains were rinsed with PBS, weighed, and an equal volume of concentrated HNO3 was added. The brains were heated in this nitric acid solution for 1 hour, allowed to cool to room temperature, then centrifuged at 3000 × g for 1 hour. The supernatant was removed and diluted 10 fold in 10% IPA prior to measuring lithium content using AAS (Shimadzu AA-6200). Peak height measurements were carried out referring to values obtained for standards of known concentrations. Lithium measurements were plotted using GraphPad PRISM software (GraphPad Software, Inc.) as mean ± SEM in Figure 1. Two tailed t-tests were used to assess the statistical significance at each time point for the pharmacokinetic curves. The criterion for rejection of the null hypothesis was P<0.05. Phoenix WinNonlin® Version 6.3 (Pharsight Corporation, Mountain View, CA) was used to conduct a noncompartmental analysis of the pharmacokinetic data and generate the pharmacokinetic parameters in Table 1 as mean ± SEM. The reported parameters include CMAX, TMAX, area under curve (AUC), and relative bioavailability (Frel).
Conclusions
In this contribution, we described unexpected pharmacokinetic differences exhibited by two previously unexplored lithium salts in rats. That lithium salicylate produced steady plasma lithium levels out to 48 hours while attenuating the spike associated with the toxic side effects of current lithium therapeutics is significant to crystal engineering strategies for improving the safety and efficacy of lithium therapy. Future studies should be conducted to elucidate the mechanisms behind these pharmacokinetic observations.
Supplementary Material
Acknowledgements
This work was supported by royalty research funds awarded to DS for the sublicense of a CNS drug. The authors would like to thank Margaret Baldwin, Todd Casagni, and Ana Almonte for technical assistance during in vivo studies.
Footnotes
Conflicts of Interest AS and DS are inventors on patent applications pertaining to these technologies.
Electronic Supplementary Information (ESI) available: Powder X-ray Diffraction of Lithium Salicylate and Lithium Lactate. See DOI: 10.1039/b000000x/
Notes and References
- 1.Thies-Flechtner K, Muller-Oerlinghausen B, Seibert W, Walther A, Greil W. Pharmacopsychiatry. 1996;29:103–107. doi: 10.1055/s-2007-979553. [DOI] [PubMed] [Google Scholar]
- 2.Goodwin FK, Fireman B, Simon GE, Hunkeler EM, Lee J, Revicki D. Jama. 2003;290:1467–1473. doi: 10.1001/jama.290.11.1467. [DOI] [PubMed] [Google Scholar]
- 3.Cipriani A, Hawton K, Stockton S, Geddes JR. Bmj. 2013;346:f3646. doi: 10.1136/bmj.f3646. [DOI] [PubMed] [Google Scholar]
- 4.Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Psychopharmacology. 2001;158:100–106. doi: 10.1007/s002130100871. [DOI] [PubMed] [Google Scholar]
- 5.Leyhe T, Eschweiler GW, Stransky E, Gasser T, Annas P, Basun H, Laske C. J Alzheimers Dis. 2009;16:649–656. doi: 10.3233/JAD-2009-1004. [DOI] [PubMed] [Google Scholar]
- 6.Yuskaitis CJ, Jope RS. Cellular signalling. 2009;21:264–273. doi: 10.1016/j.cellsig.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith AJ, Kim SH, Duggirala NK, Jin J, Wojtas L, Ehrhart J, Giunta B, Tan J, Zaworotko MJ, Shytle RD. Mol Pharm. 2013 doi: 10.1021/mp400571a. DOI: 10.1021/mp400571a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein PS, Melton DA. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allison JH, Stewart MA. Nature: New biology. 1971;233:267–268. doi: 10.1038/newbio233267a0. [DOI] [PubMed] [Google Scholar]
- 10.Pollack SJ, Atack JR, Knowles MR, McAllister G, Ragan CI, Baker R, Fletcher SR, Iversen LL, Broughton HB. Proc Natl Acad Sci U S A. 1994;91:5766–5770. doi: 10.1073/pnas.91.13.5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh N, Halliday AC, Thomas JM, Kuznetsova OV, Baldwin R, Woon EC, Aley PK, Antoniadou I, Sharp T, Vasudevan SR, Churchill GC. Nature communications. 2013;4:1332. doi: 10.1038/ncomms2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gould TD, Manji HK. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2005;30:1223–1237. doi: 10.1038/sj.npp.1300731. [DOI] [PubMed] [Google Scholar]
- 13.Braga D, Grepioni F, Maini L, Capucci D, Nanna S, Wouters J, Aerts L, Quere L. Chemical communications. 2012;48:8219–8221. doi: 10.1039/c2cc33855f. [DOI] [PubMed] [Google Scholar]
- 14.Wouters J, Grepioni F, Braga D, Kaminski RM, Rome S, Aerts L, Quere L. CrystEngComm. 2013 DOI: 10.1039/C3CE41539B. [Google Scholar]
- 15.FDA U. D. o. H. a. H. Services; F. a. D. Administration; C. f. D. E. a. Research, editor. Apr, 2013.
- 16.Smith DF. British journal of pharmacology. 1976;56:399–402. doi: 10.1111/j.1476-5381.1976.tb07449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groth U, Prellwitz W, Jahnchen E. Clinical pharmacology and therapeutics. 1974;16:490–498. doi: 10.1002/cpt1974163part1490. [DOI] [PubMed] [Google Scholar]
- 18.Trautner EM, Morris R, Noack CH, Gershon S. The Medical journal of Australia. 1955;42:280–291. [PubMed] [Google Scholar]
- 19.Lippmann S, Evans R. Hospital & community psychiatry. 1983;34:113–114. doi: 10.1176/ps.34.2.113. [DOI] [PubMed] [Google Scholar]
- 20.Emami J, Tavakoli N, Movahedian A. Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques. 2004;7:338–344. [PubMed] [Google Scholar]
- 21.Zaitseva N, Newby J, Hull G, Saw C, Carman L, Cherepy N, Payne S. Crystal Growth & Design. 2009;9:3799–3802. [Google Scholar]
- 22.Hille B. The Journal of general physiology. 1972;59:637–658. doi: 10.1085/jgp.59.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baldessarini RJ, Tondo L, Hennen J. Ann N Y Acad Sci. 2001;932:24–38. doi: 10.1111/j.1749-6632.2001.tb05796.x. discussion 39–43. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin FK, Fireman B, Simon GE, Hunkeler EM, Lee J, Revicki D. Jama. 2003;290:1467–1473. doi: 10.1001/jama.290.11.1467. [DOI] [PubMed] [Google Scholar]
- 25.Almarsson Ö, Peterson ML, Zaworotko M. Pharmaceutical Patent Analyst. 2012;1:313–327. doi: 10.4155/ppa.12.29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.