

Abstract
Human butyrylcholinesterase (hBChE) is currently being developed as a detoxication enzyme for stoichiometric binding and/or catalytic hydrolysis of organophosphates. Herein, we describe the use of a molecular evolution method to develop novel hBChE variants with increased resistance to stereochemically defined nerve agent model compounds of soman, sarin, and cyclosarin. Novel hBChE variants (Y332S, D340H, and Y332S/D340H) were identified with an increased resistance to nerve agent model compounds that retained robust intrinsic catalytic efficiency. Molecular dynamics simulations of these variants revealed insights into the mechanism by which these structural changes conferred nerve agent model compound resistance.
Exposure to organophosphorous (OP) compounds in the form of pesticides or nerve agents is a significant public health concern, and pesticide exposure is one of the most common causes of poisoning throughout the world.1 OP nerve agents and pesticides inhibit human acetylcholinesterase (hAChE) in the central nervous system to disrupt neurotransmission. This can result in brain damage, loss of muscle control, or death.2 The current standard of treatment for nerve agent exposure involves concomitant administration of a cholinesterase (ChE) reactivator [e.g., an oxime, 2-pralidoxime (2-PAM)], a muscarinic receptor antagonist (atropine), and an anticonvulsant (diazepam).2 Even though this regime decreases mortality, the efficacy is limited because 2-PAM does not prevent toxicity associated with nerve agent exposure to the brain because 2-PAM does not cross the blood–brain barrier. Catalytic bioscavenger therapy, an attractive alternative prophylactic approach to nerve agent detoxication, involves administration of a detoxication enzyme that binds and/or hydrolyzes nerve agents before they reach hAChE in the brain. In principle, administration of peripheral bioscavengers could prevent irreversible brain damage. Human BChE (hBChE) is an ideal candidate enzyme for this type of therapy. hBChE is abundant in blood, does not require any cofactors, and is soluble and highly functional at the pH of plasma. Wild-type (WT) hBChE has been used with some success as a nerve agent detoxication scavenger in guinea pigs.3 However, the feasibility of the use of exogenously administered hBChE as a broad-scale peripheral blocker of OP toxicity is limited because the reaction of hBChE with a nerve agent is stoichiometric and irreversible and thus requires a large amount of highly pure hBChE for detoxication purposes. hBChE variants identified by rational design (i.e., hBChE G117H and E197Q) act as only low-level catalytic bioscavengers because of the decreased affinity of G117H and E197Q hBChE for nerve agents.4 Thus, elaboration of hBChE variants that show greater nerve agent resistance (i.e., variants that do not react with nerve agents but are functionally active cholinesterases) and/or nerve agent hydrolytic functional activity represents an unmet medical need to counter nerve agent exposure.
Previous studies reported the results of rationally designed hBChE variants for investigating the interaction between nerve agents and hBChE.5,6 Several hBChE variants were reported to be resistant to nerve agents.5−7 For example, hBChE variant G117H increased the rate of hydrolytic dephosphonylation of nerve agent–hBChE covalent adducts and functioned as a catalytic bioscavenger by converting reactive nerve agents to relatively inert products through a net hydrolysis reaction.5 An E197Q hBChE variant decreased the rate of enzyme aging, a process whereby nerve agent–enzyme adducts lose one or more of their alkyl moieties by spontaneous hydrolysis and are thereby rendered much less prone to reactivation. Site-directed mutagenesis studies of hBChE identified several residues that were important for nerve agent-induced hBChE aging, including D70 and W82.6 Also, hAChE residues W86, Y133, E202, F338, and E450 have been reported to be involved in the hAChE aging process.8 The molecular basis for efficient human ChE nerve agent hydrolytic functional activity is presumably attributed to a number of residues in the primary sequence of the protein that determine the rates of phosphorylation, dephosphorylation, and aging.8,9
Previously, we described a robust directed molecular evolution approach to identifying hBChE residues that conferred resistance to enzymatic inhibition by nerve agent model compounds.10 There are many potential mechanisms for hBChE resistance to nerve agent model compounds, including but not limited to hydrolysis, rapid reactivation, resistance to aging, or decreased binding affinity. Because use of authentic nerve agents is strictly regulated, nerve agent model compounds that have chemical and functional properties similar to those of authentic nerve agents were used.11 These nerve agent model compounds were used in molecular evolution studies to identify hBChE variants. hBChE resistance to nerve agent model compounds was determined on the basis of kinetic studies to identify OP-resistant variants.11 To facilitate kinetic studies, a previously described monomeric hBChE with a C-terminal histidine tag was used to permit protein expression, purification, and kinetics of hBChE variants.12 Monomeric hBChE showed kinetic parameters virtually identical to those of WT tetrameric hBChE.12 The molecular evolution method identified the previously described hBChE G117H variant that showed nerve agent resistance as well as novel variants G117N, G117R, E197C, and L125V that showed nerve agent model compound resistance.4
Herein, we describe the use of a molecular evolution method to develop novel hBChE variants with increased resistance to nerve agent model compounds. The method was flexible enough to pick up variants that worked via any of the possible resistance mechanisms described above.4 Recombinant protein expression of the hBChE variants identified permitted determination of the kinetics using stereochemically defined nerve agent model compounds of soman (GD), sarin (GB), and cyclosarin (GF). Novel hBChE variants with increased resistance to nerve agent model compounds that retained robust intrinsic catalytic efficiency were identified. On the basis of the unusual location of the amino acid variants within the protein, molecular dynamics simulations were undertaken and revealed insights into the mechanism by which these structural changes conferred nerve agent model compound resistance.
Materials and Methods
Biological and Chemical Reagents
Butyrylthiocholine (BTC) iodine and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) were purchased from Sigma-Aldrich (St. Louis, MO). Goat anti-rabbit antibodies conjugated with horseradish peroxidase (HRP) enzyme and Supersignal West Pico Chemiluminscent substrate were purchased from Thermo Fisher Scientific (Waltham, MA). Buffers and solvents were purchased from VWR (West Chester, PA) at the highest purity commercially available. Molecular biology reagents were purchased from Life Technologies (Carlsbad, CA) unless otherwise specified. Highly purified native hBChE, anti-hBChE polyclonal antibodies, echothiophate (ETP) iodide [2-(dithoxyphosphinyl)thio-N,N,N-trimethylethanaminium iodide], and the wild-type hBChE cDNA construct in pRC/CMV encoding the full-length wild-type hBChE G117H single mutant were generously provided by O. Lockridge (University of Nebraska Medical Center, Omaha, NE). Nerve agent model compounds (S)-2-[isopropoxy(methyl)phosphorylthio]-N,N-dimethylethanammonium chloride (SPGB3N), (SP)-2-[3,3-dimethylbutan-2-oxy(methyl)phosphorylthio]-N,N-dimethylethanammonium chloride (SPGD3N), and (S)-2-[cyclohexyloxy(methyl)phosphorylthio]-N,N-dimethylethanammonium chloride (SPGF3N) were prepared according to a previously published procedure.11,13,14 The nerve agent model compounds are toxic and must be handled with extreme care. Chemical waste containing nerve agent model compounds or other OPs were degraded by hydrolysis with overnight incubation with 2.5 M NaOH and 10% ethanol before being disposed.
hBChE Plasmid Construction and Adenovirus Production
Wild-type (WT) hBChE with an N-terminal signal sequence and a C-terminal histidine tag (amino acids 1–540, W541ΔH6) was cloned into the pENTR1a entry vector (Life Technologies) at the KpnI and XhoI restriction sites as described previously.12 The Y332S, D340H, and Y332S/D340H mutations were introduced into a hBChE W541ΔH6 pENTR1a construct by site-directed mutagenesis using mutagenic primer sequences listed in Table S1 of the Supporting Information. Following confirmation by DNA sequencing, hBChE variants were recombined into the pAD destination vector (Life Technologies).15 Adenovirus was generated by transfecting the hBChE W541ΔH6 pAD construct into HEK-293A cells using the ViraPower Adenoviral Expression System (Life Technologies). Subsequent viral amplification in HEK-293A cells yielded recombinant adenoviral stocks with titers ranging from 3.2 × 107 to 2.1 × 109 plaque-forming units/mL.
hBChE Protein Expression and Purification
CHO-cTA-CAR cells (ATCC, Manassas, VA) grown in suspension were infected with recombinant adenovirus at an MOI of 30 for 72 h. Medium supernatants were then clarified for purification by centrifugation (400g for 2.5 min). WT and variant hBChE were purified from clarified supernatants by nickel affinity chromatography (Qiagen, Valencia, CA) with running buffer [50 mM HEPES (pH 7.5) and 500 mM NaCl] and elution buffer [50 mM HEPES (pH 7.5), 500 mM NaCl, and 400 mM imidazole]. Imidazole of the elution buffer was removed by dialysis. To remove the imidazole in the elution buffer, highly purified hBChE variant proteins were dialyzed with 2 L (one buffer exchange) of phosphate-buffered saline (PBS) [50 mM potassium phosphate (pH 7.5) and 150 mM NaCl].
Quantification of WT hBChE and hBChE Variants by Western Blotting
Full-length tetrameric WT hBChE was used as a standard for the determination of protein concentrations of hBChE variants. Serial dilutions of WT standard hBChE (68 units/mL) were prepared and afforded final concentrations of 1–10 units/mL. Both the WT hBChE standard curve and hBChE variant samples were resolved on the same gel by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to nitrocellulose. Western blot analysis included sequential steps of blocking [PBS and Tween-20 (0.01%) (PBST) with 5% nonfat dry milk (NFDM), 1 h, 25 °C], primary antibody (i.e., rabbit anti-hBChE was generously provided by O. Lockridge, 1:1000 in a PBST/NFDM mixture, 16 h, 4 °C), secondary antibody [i.e., goat α-rabbit HRP (Jackson ImmunoResearch, West Grove, PA), 1:5000 in a PBST/NFDM mixture, 1 h, 4 °C], and detection with SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific). Images were scanned to a file, and band densitometry analysis was conducted using ImageJ (National Institutes of Health, Bethesda, MD). Linear regression analysis of WT hBChE afforded a best-fit line equation. Variant hBChE protein concentrations were determined using the density of the variant bands and the best-fit line equation from the WT hBChE standard curve (data not shown).
Substrate-Dependent Hydrolysis of Butyrylthiocholine Iodide (BTC) by hBChE Variants
hBChE functional activity was continuously measured spectrophotometrically (Tecan SpectraFluorPLUS microplate reader, Tecan Group Ltd.) with an Ellman assay.16 Briefly, BTC (Fluka, St. Louis, MO) was used as a substrate for WT and hBChE variants over a final concentration range of 0.01–100 mM. Incubations were conducted in PBS at 25 °C in the presence of 0.4 mM DTNB (Sigma-Aldrich). A highly purified hBChE enzyme (i.e., WT, Y332S, D340H, Y332S/D340H, or G117H) (3.5 × 10–3 IU) was added to initiate the incubation, and hydrolysis was continuously monitored by recording the absorbance at 405 nm. The functional activity was calculated from the molar extinction coefficient of 13600 M–1 cm–1.16 Observed rates of hydrolysis were fit to eq 1, which describes the activation of the enzyme (when b > 1) at relatively high BTC concentrations from the binding of the substrate to a secondary binding site27
| 1 |
that afforded best-fit kinetic values and relevant standard errors. For the purpose of analysis, b was fixed at 1.7 and Kss was constrained to be greater than zero.12
Resistance to Nerve Agent Model Compound Inhibition by hBChE Variants
hBChE variant activities were measured as discussed above with the following modifications. Butyrylthiocholine iodide (BTC) was used as a substrate for hBChE variants at a final concentration of 1 mM. Incubations were conducted in PBS at 25 °C in the presence of 0.2 mM DTNB. The functional activity was monitored and calculated as discussed above. Nerve agent model compounds [SPGD3N (soman model), SPGB3N (sarin model), and SPGF3N (cyclosarin model)] were used as freshly prepared solutions in DMSO.11 The final concentration of cosolvent in the inhibition mixtures was <5%. Control experiments showed no effect of <5% solvent on hBChE enzyme activity for BTC hydrolysis in the incubation (i.e., BChE in PBS at 25 °C in the presence of <5% DMSO). Inhibition was initiated by addition of an aliquot of the nerve agent model compound solution to 3.6 × 10–3 IU of highly purified hBChE. The mixtures were incubated at 25 °C, and at defined times, an aliquot of the reaction mixture was taken to determine the residual ChE activity at 25 °C as described above. For kinetic studies, five to seven concentrations of nerve agent model compound were used and at least four time points were taken for each concentration [for SPGB3N, WT (0.5, 1, 2.5, 5, and 10 μM), Y332S, D340H, and Y332S/D340H (5, 6.67, 8, 10, and 20 μM), and G117H (500, 750, 900, 1000, and 1500 μM); for SPGD3N, WT (1, 2, 5, 8, and 10 μM), Y332S, D340H, and Y332S/D340H (1, 1.33, 2, 5, and 10 μM), and G117H (500, 625, 750, and 1000 μM); for SPGF3N, WT (0.01, 0.0133, 0.02, 0.1, and 10 μM) and Y332S, D340H, and Y332S/D340H (500, 555, 750, 1000, and 2000 μM)]. Plots of the logarithm of the residual initial velocity versus incubation time afforded kapp (the apparent first-order rate constant for inhibition at a given concentration of nerve agent model compound). A replot of kapp versus the concentration of the nerve agent model compound afforded inhibition constant values (ki). For hyperbolic replots, nonlinear curve fitting to the equation kapp = k2[NA]/(KD + [NA]) (NA, nerve agent model compound) afforded both phosphorylation or phosphonylation rate constants (k2) and equilibrium dissociation constants (KD).9,11 Bimolecular rate constants (ki) were calculated from the k2/KD ratio. For linear replots, bimolecular rate constant (ki) values were determined by linear regression analyses of the slope.
Data Analysis
Data analyses, including statistical comparisons via a one-way analysis of variance (ANOVA) followed by a Dunnett’s multiple-comparison test, were conducted using GraphPad Prism version 5.01 (GraphPad Inc., San Diego, CA).
Docking Studies of Nerve Agent Model Compounds with hBChE Variants
Docking simulations of hBChE with nerve agent model compounds were conducted using AutoDock Vina.17 The structure of hBChE used for docking studies was taken from the crystal structure of hBChE [Protein Data Bank (PDB) entry 1P0I]18 after removal of the ligands of crystallization [i.e., butanoic acid, l-fucose, glycerol, 2-(N-morpholino)ethanesulfonic acid, and sulfate], water, and salts with a PyMol Molecular Graphics System (version 1.4.1, Schrödinger, LLC, San Diego, CA). The Y332S, D340H, and Y332S/D340H variants were also created in PyMol using the mutagenesis function. Ligand and protein docking parameters were assigned using an AutoDock Tools program. The search space encompassed the binding gorge and surrounding residues (including D340) with a volume of 31500 Å3. Nerve agent model compounds were designated as conformationally flexible in the docking simulations. Dockings used an exhaustiveness setting of 64 and were repeated to confirm the preferred binding mode in independent simulations.
Determination of the Modeling Parameters for the Nerve Agent Model Compound SPGB3N
The parameters for the topology of the SPGB3N ligand were obtained using the Automated Topology Builder (ATB).19,20 The validity of the charge distribution was confirmed by manually checking the model against density functional theory (DFT) charge distribution calculated using NWChem version 6.1.1.21−23 Minor adjustments were made to the charge groups to better match the conventions used in the GROMACS package (http://www.gromacs.org).
Molecular Dynamics (MD) Studies of hBChE Variants
Molecular dynamics simulations were conducted using GROMACS version 4.6.3 and the Gromos53a6 force field22,23 under periodic boundary conditions. The starting topology was built using the coordinates of the docked structure of hBChE (PDB entry 1P0I).18 A dodecahedral box was created around the protein 1.0 nm from the edge of the protein and filled with extended simple point charge water molecules. Sodium and chloride ions were added to produce an electroneutral solution at a final concentration of 0.15 M NaCl. The energy was minimized using a steepest gradient method to remove any close contacts before the system was subjected to a two-phase position-restrained MD equilibration procedure. The system was first allowed to evolve for 100 ps in a canonical NVT (N is the number of particles, V the system volume, and T the temperature) ensemble at 25 °C before pressure coupling was switched on and the system was equilibrated for an additional 100 ps in the NPT (P is the system pressure) ensemble at 1.0 bar. The production runs were conducted in an NPT ensemble at 25 °C and 1.0 bar for 50 ns with a step size of 2 fs. All bond lengths were constrained to their equilibrium values by using the LINCS algorithm. The neighbor list for the calculation of nonbonded interactions was updated every five time steps with a cutoff of 1.0 nm. A twin range cutoff of 1.0 nm was used for both Coulomb and Lennard-Jones interactions. The system was split into two groups, “Protein” and “Non-Protein”, which were coupled separately to a temperature bath using the V-rescale algorithm24 with a time constant of 0.1 ps while the pressure coupling was conducted using an isotropic Parrinello–Rahman barostat with a time constant of 2.0 ps.25,26 The PyMol Molecular Graphics System, version 1.4.1 (Schrödinger, LLC), was used to create structural models of the hBChE MD analysis.
Results
Identification of Nerve Agent Model Compound-Resistant hBChE Variants
To identify hBChE variants that showed resistance to inhibition of functional activity by nerve agent model compounds, we implemented a directed molecular evolution approach in a mammalian cell-based functional screen.10 Because work with actual nerve agents is precluded in most academic laboratories, we developed a series of nerve agent model compounds (Figure 1) that were used to identify variants of hBChE that were resistant to inhibition by these compounds.11 Initially, we designed and constructed six mutagenesis libraries that together encompassed structurally relevant regions of the active site areas and the peripheral anionic binding site (PAS) of hBChE.10 hBChE libraries were made by introduction of mutations through PCR using doped oligonucleotides, subcloning PCR library fragments into the shuttle vector pENTR1A, transfer of the mutation library from pENTR1A to an adenoviral expression vector (pAD) through recombination, and then packaging of the mutation libraries in pAD in recombinant AD particles through transfection as described previously.4 The sequence diversity of the hBChE libraries was analyzed by DNA sequencing. Sequencing of 50 representative library clones showed that the mutation rate was 3.8% in nucleotides and 1.8% in amino acids.4 One particular library of variants that encompassed residues 325–344 of hBChE was investigated for functional activity to identify hBChE variants that showed decreased rates of inhibition of nerve agent model compounds.4 Results of initial solid phase assays of the library were confirmed by liquid phase assays of hBChE functional activity.4 In a solid phase screen, bright yellow gel plugs were cored out as positive signals from an Ellman assay (i.e., in the presence of SPGD3N for 2–6 h, followed by incubation with 1 mM BTC and 1 mM DTNB). Hydrolysis of DTNB generated the yellow 2-nitro-5-thiobenzoate product, and candidate viral particles were identified and isolated. In confirmatory liquid phase hBChE functional assays, highly purified virus particles isolated from the yellow gel plugs were scaled up in sufficient quantity for hBChE functional assays. Briefly, virus particles were used to infect cells, and the expressed hBChE variants were examined for functional activity using a modified Ellman assay.16 Viral DNA encoding nerve agent model compound-resistant hBChE variants was sequenced to identify sites where mutagenesis was present. Using this approach, we identified hBChE variants Y332S, D340H, and Y332S/D340H from a hBChE library that encompassed residues 325–3444 (Figure S5 of the Supporting Information). The variants were resistant to inhibition by nerve agent model compounds of soman (i.e., SPGD3N), sarin (i.e., SPGB3N), and cyclosarin (i.e., SPGF3N) (Figure 1).
Figure 1.

Chemical structures of OP nerve agents and corresponding model compounds.
Characterization of WT and Variant hBChE Using Butyrylthiocholine (BTC) as a Substrate
The steady-state kinetic parameters for BTC hydrolysis in the presence of WT hBChE and hBChE variants Y332S, D340H, and Y332S/D340H were determined for each enzyme using an Ellman assay.16 The previously reported hBChE variant G117H was included for comparison,5 and the kinetic parameters are listed in Table 1. In this study, to facilitate hBChE protein purification, we used C-terminal histidine-tagged N-truncated hBChE that we have previously shown to be kinetically indistinguishable from full-length tetrameric WT hBChE.12 Kinetic data were fit to the Webb equation (eq 1) that described an initial hyperbolic substrate dependence for hydrolysis with a Michaelis constant (KM) followed by kinetic activation (when b > 1) upon saturation of the peripheral anionic site (PAS) with an apparent dissociation constant of Kss. Formation of the product was linearly dependent on WT and variant hBChEs. Product formation was also linearly dependent on BTC (from 5 μM to 50 mM) for the WT and variant hBChEs examined. For the hBChE enzymes examined, substrate-dependent activation was readily apparent as shown by the kinetic parameters listed in Table 1. On the basis of these observations, WT hBChE had a lower KM value for BTC relative to those of the hBChE variants Y332S (1.1-fold greater), D340H (1.9-fold greater), Y332S/D340H (5.7-fold greater), and G117H (2.4-fold greater). Compared to the KM value for WT hBChE, hBChE variant Y332S/D340H showed a statistically significantly greater KM (p < 0.05). There was no statistically significant difference (p > 0.05) between WT and variant hBChEs for the dissociation constant of BTC at the peripheral binding site (Kss) when the activation factor b was set to 1.7 as previously described (Table 1).27 However, the BTC hydrolysis constant (kcat) for WT hBChE was significantly lower than those of hBChE variants Y332S (2.2-fold greater; p > 0.05) and Y332S/D340H (2.6-fold greater; p > 0.05) but not those for the D340H (0.64-fold greater; p > 0.05) and G117H (0.58-fold greater; p > 0.05). Compared with those of WT hBChE and the previously characterized hBChE G117H variant, the catalytic efficiency (kcat/KM) of hBChE variants Y332S, D340H, and Y332S/D340H showed mixed results. The Y332S hBChE variant showed the highest catalytic efficiency, greater than those of both WT and G117H BChE (1.7- and 11-fold greater, respectively; p < 0.05). The D340H hBChE variant showed a catalytic efficiency lower than that of WT (2.9-fold lower; p < 0.05). However, the catalytic efficiency of D340H was still greater than that of G117H hBChE (2.3-fold greater; p < 0.05). The double variant Y332S/D340H hBChE apparently combined the effects of both single mutations and afforded a catalytic efficiency that was lower than that of WT (2.2-fold lower; p < 0.05) yet greater than that of G117H (3.0-fold greater; p < 0.05).
Table 1. Kinetic Parameters for Michaelis–Menten Kinetics of WT hBChE and Its Variantsa.
| hBChE | KMb (mM) | bc | Kssd (mM) | kcate (s–1) | kcat/KM (M–1 s–1) | R2 |
|---|---|---|---|---|---|---|
| wild type | 0.50 ± 0.03 | 1.7 | 0.26 ± 0.044 | 1.95 | 3931 | 0.99 |
| Y332S | 0.64 ± 0.45 | 1.7 | 0.17 ± 0.021 | 4.25 | 6682 | 0.99 |
| D340H | 0.93 ± 0.10 | 1.7 | 9.84 ± 0.597 | 1.25 | 1344 | 0.99 |
| Y332S/D340H | 2.84 ± 0.53 | 1.7 | 2.89 ± 0.900 | 5.1 | 1796 | 0.99 |
| G117H | 1.19 ± 1.06 | 1.7 | 5.70 ± 0.824 | 1.13 | 595 | 0.99 |
Enzymatic BTC hydrolysis ± the standard deviation, for three replicates monitored at room temperature in PBS (pH 7.5). The observed rates of hydrolysis were fit to the Webb equation (eq 1). The listed best fit values and fitting errors resulted from a single determination for each enzyme (18–24 data points). Statistical significance was determined using an ANOVA followed by a Dunnett’s multiple-comparison test.
Michaelis constant (KM).
The b value was constrained at a value of 2.2.
Substrate inhibition term (Kss).
Catalytic rate constant.
Evaluation of WT and Variant hBChEs for Resistance to Inhibition by SPGB3N, SPGF3N, and SPGD3N
In this work, enzyme inhibition studies with WT or variant hBChEs used nerve agent model compounds previously synthesized and characterized.11 These nerve agent model compounds have a 2-(N,N-dimethylamino)ethanethiol leaving group that in its protonated state (i.e., ammonium) mimics a thiocholine leaving group. The propensity of the leaving group in these nerve agent model compounds differs from those of highly reactive leaving groups of authentic agents, but otherwise, the model compounds recapitulate the properties of authentic agents. For example, the protein adduct formed through phosphonylation of S198 by the nerve agent model compounds is indistinguishable from the adduct formed with authentic nerve agents.28−30 The nerve agent model compounds described herein possess a center of chirality (i.e., R and S isomers at phosphorus) and stereoselectively inhibited ChEs with a stereoselectivity similar to that of the real agents.11 Other advantages of the nerve agent model compounds stem from their lack of volatility, water solubility, and increased chemical stability. The nerve agent model compounds described herein are also much less toxic than the authentic agents. For this study, we prepared SP enantiomers of nerve agent models of sarin (GB), cyclosarin (GF), and soman (GD) (Figure 1).11 The SP enantiomers of the nerve agent model compounds were approximately 50-fold more potent with respect to inhibition of hBChE hydrolysis of BTC than their corresponding RP enantiomers.11 To simplify the kinetics and conduct studies with the most biologically relevant nerve agent model compound, the SP enantiomers were used.
The kinetic parameters of phosphonylation by nerve agent model compounds SPGD3N, SPGB3N, and SPGF3N were examined in the presence of WT or variant hBChEs at concentrations from 1 to 10 μM for 60 min. After a 60 min preincubation, each enzyme/inhibitor mixture was diluted with an assay mixture containing BTC in PBS (pH 7.5). The remaining hBChE hydrolysis activity was measured using a modified Ellman assay.16 A plot of the percent remaining hBChE functional activity versus time gave a linear response, and the slopes of these plots afforded apparent rate constants (kapp values) (Figures S1–S3 of the Supporting Information). The kapp for inhibition was obtained from linear regression analysis of the semilogarithmic plots of hBChE activity versus incubation time.11 Replots of kapp versus the concentration of the nerve agent model compound gave linear correlations and allowed calculation of k2 and KD based on nonlinear curve fitting analysis11 (Figures S1–S3 of the Supporting Information). The inhibition constants (ki) for WT and variants were then calculated from k2/KD5 and are listed in Table 2.
Table 2. Kinetic Parameters for Inhibition of hBChE Variants by Nerve Agent Model Compoundsa.
| kib (μM–1 min–1) | k2c (min–1) | KDd (μM) | t1/2 (min)e | R2f | |
|---|---|---|---|---|---|
| SPGB3N | |||||
| wild type | 0.14 ± 0.08 | 1.17 ± 0.31 | 8.4 ± 3.9 | 3.8 ± 2.5g | 0.97 |
| Y332S | 0.01 ± 0.01 | 0.52 ± 0.09 | 50.9 ± 11 | 9.5 ± 3.0g | 0.99 |
| D340H | 0.03 ± 0.02 | 0.48 ± 0.04 | 19.0 ± 2.6 | 4.9 ± 1.3g | 0.99 |
| Y332S/D340H | 0.002 ± 0.001 | 0.22 ± 0.35 | 132.0 ± 23.3 | 58 ± 2.16g | 0.94 |
| G117H | 3.62 ± 3.03 | 0.02 ± 0.003 | 6202 ± 833 | NAh | 0.99 |
| SPGD3N | |||||
| wild type | 0.11 ± 0.05 | 0.58 ± 0.11 | 5.2 ± 2.2 | 4.0 ± 2.4g | 0.96 |
| Y332S | 0.02 ± 0.01 | 0.24 ± 0.16 | 10.9 ± 1.18 | 22.6 ± 1.57g | 0.85 |
| D340H | 0.04 ± 0.02 | 0.20 ± 0.08 | 5.4 ± 0.42 | 16.2 ± 1.07g | 0.84 |
| Y332S/D340H | 0.03 ± 0.01 | 0.03 ± 0.004 | 1.1 ± 0.5 | 38.9 ± 7.5g | 0.76 |
| G117H | 71.4 ± 5.59 | 0.19 ± 0.09 | 2613 ± 1500 | NAh | 0.97 |
| SPGF3N | |||||
| wild type | 2.03 ± 1.21 | 0.23 ± 0.01 | 0.11 ± 0.014 | 21.1 ± 8.97g | 0.99 |
| Y332S | 0.71 ± 0.35 | 0.04 ± 0.01 | 0.06 ± 0.015 | 43.0 ± 16.2g | 0.99 |
| D340H | 0.91 ± 0.45 | 0.05 ± 0.002 | 0.06 ± 0.005 | 48.6 ± 2.82g | 0.99 |
| Y332S/D340H | 0.31 ± 0.04 | 0.27 ± 0.05 | 0.88 ± 0.124 | 42.4 ± 9.85g | 0.92 |
| G117H | 4.3 ± 3.49 | 0.02 ± 0.02 | 3460 ± 7385 | NAh | 0.87 |
Values for k2 and KD were derived from the nonlinear curve fit of kapp over nerve agent model compound concentrations. Values reported represent best fit values ± the standard deviation derived from fitting 30–40 data points from a single experiment. Values for ki were calculated from k2/KD ratios. Statistical significance was evaluated by ANOVA followed by a Dunnett’s multiple-comparison test.
Bimolecular inhibition constant (ki).
Phosphonylation constant (k2).
Equilibrium dissociation constant (KD).
Half-life value for 50% inhibition of BTC hydrolysis.
Correlation constant for a nonlinear curve fit (R2).
Half-life values calculated for five concentrations of nerve agent model compounds.
Half-life value could not be calculated.
Compared to those of WT hBChE, hBChE variants Y332S and D340H had longer half-lives of inhibition and lower rates of inhibition for the nerve agent model compounds tested (Table 2). Compared to that of WT hBChE, the half-lives of inhibition (t1/2) of hBChE variants Y332S and D340H by the nerve agent model compounds were longer (i.e., 5.65- and 4.05-fold longer, respectively, for SPGD3N, 2.5- and 1.3-fold longer, respectively, for SPGB3N, and was 2- and 2.3-fold longer, respectively, for SPGF3N). However, the differences in half-lives between WT and variant hBChEs did not reach a statistically significant difference (p < 0.05) (Table 2). Compared to that of WT hBChE, Y332S and D340H hBChE variants showed statistically significantly lower k2 values for SPGD3N (2.4- and 2.9-fold lower, respectively; p < 0.05), SPGB3N (2.3- and 2.4-fold lower, respectively; p < 0.05), and SPGF3N (5.8- and 4.6-fold lower, respectively; p < 0.05) (Table 2). Compared to that of WT hBChE, hBChE variants Y332S and D340H showed significantly greater KD values for SPGB3N (6.1- and 2.3-fold greater, respectively; p < 0.05) but not SPGD3N (5.7- and 4.1-fold greater, respectively) and SPGF3N (2.0- and 2.3-fold greater, respectively) (Table 2). Compared to that of WT hBChE, the Y332S and D340H hBChE variants also showed significantly lower ki values for SPGD3N (5.5- and 2.8-fold lower, respectively; p < 0.05) and SPGB3N (14- and 4.7-fold lower, respectively; p < 0.05) but not SPGF3N (2.9- and 2.2-fold lower, respectively) (Table 2). The results suggested that single variants of hBChE (i.e., Y332S and D340H) conferred considerable resistance to inhibition with nerve agent model compounds.
The Y332S/D340H hBChE variant had a half-life for inhibition by the nerve agent model compounds significantly longer than those of WT hBChE [SPGD3N (9.7-fold longer; p < 0.05), SPGB3N (15-fold longer; p < 0.05), and SPGF3N (2-fold longer; p < 0.05)]. Compared to that of WT hBChE, the Y332/D340H variant showed significantly lower k2 rate constants for inhibition by SPGD3N (i.e., 19-fold lower; p < 0.05) and SPGB3N (5.3-fold lower; p < 0.05) but not by SPGF3N (i.e., 1.2-fold lower). Similar to those of the single hBChE variants Y332S and D340H, the affinity (KD) of the Y332S/D340H hBChE double mutant was significantly lower than that of WT hBChE [i.e., 4.7-fold lower for SPGD3N (p < 0.05), 16-fold lower for SPGB3N (p < 0.05), or 8-fold lower for SPGF3N (p < 0.05)]. Relative to that of WT hBChE, slower rates of inhibition (ki) of Y332S/D340H hBChE by SPGD3N (4-fold lower; p < 0.05), SPGB3N (70-fold lower; p < 0.05). and SPGF3N (6.6-fold lower; p < 0.05) were observed. Compared to Y332S/D340H, the G117H hBChE variant was the most resistant to nerve agent model compound inhibition and had the lowest inhibition rate constant for SPGD3N (649-fold lower; p < 0.05) and SPGB3N (26-fold lower; p < 0.05). These data are consistent with previously published results.11 However, the half-life (t1/2) for hBChE inhibition by nerve agent model compounds SPGB3N, SPGD3N, and SPGF3N could not be calculated for G117H hBChE because hydrolysis activity decreased by only <15% during the assay. Overall, the results showed that the Y332S/D340H variant conferred significantly greater resistance to inhibition by nerve agent model compounds than WT hBChE as well as the Y332S and D340H single-variant hBChEs. We also compared the resistance of WT hBChE and hBChE Y332S/D340H variants to inhibition by ecothiophate iodide (ETP). The hBChE Y332S/D340H variant showed a rate of inhibition (ki) (29-fold lower; p < 0.05) lower than that of WT hBChE. The resistance of hBChE Y332S/D340H to inhibition by the nerve agent model compounds was confirmed by the resistance to ETP. The Y332S/D340H hBChE variant showed a resistance to SPGB3N greater than that of ETP (2.4-fold greater; p < 0.05).
Studies of Docking of SPGB3N to WT, Y332S, D340H, and Y332S/D340H hBChE Variants
To investigate the binding of nerve agent model compounds to hBChE, we docked the SPGB3N nerve agent model compound to the crystal structure of WT hBChE and Y332S, D340H, and Y332S/D340H hBChE variants. For WT hBChE, energy-minimized binding studies placed SPGB3N in the active site of the enzyme with the phosphorus of the nerve agent model compound 4.0 Å from the oxygen atom of the active site serine [i.e., S198 (Figure 2)]. The large 2-(N,N-dimethylammonium)ethyl substituent of SPGB3N extended into the relatively large pocket proximal to W82 of hBChE that is the choline binding site. A distance of 4.2 Å between the ammonium nitrogen atom of the nerve agent model compound and the nearest aromatic carbon suggested the presence of stabilizing cation−π interactions at W82. The 2-propyl substituent of SPGB3N that mimicked the sarin side chain was directed to the acyl binding pocket. The preference for docking the relatively large 2-(N,N-dimethylammonium)ethyl and 2-propyl substituents to these sites forced the methyl and oxo substituents of the SPGB3N phosphonate to be oriented toward the opening of the gorge. Docking the SPGB3N model compound in the active site of WT hBChE and the Y332S, D340H and Y332S/D340H variants resulted in almost no change in the preferred binding mode of the ligand. The root-mean-square deviation (rmsd) of SPGB3N docked to either WT hBChE or the Y332S/D340H variant was 0.09 Å. The ligand structures were overlaid (i.e., depicted in Figure 2), which showed the similarity of the poses that were docked. Together, the docking results showed that SPGB3N adopted a binding mode that was consistent with other choline-containing substrates of hBChE, although the distance between the active site serine and the nerve agent phosphorus atom suggested that this precomplex was not reactant-like for the phosphonylation reaction. As expected from docking to static proteins, hBChE variant residues on the periphery of the protein such as D340H, or even Y332S, had a minimal effect on the binding orientation of the ligand in the active site.
Figure 2.
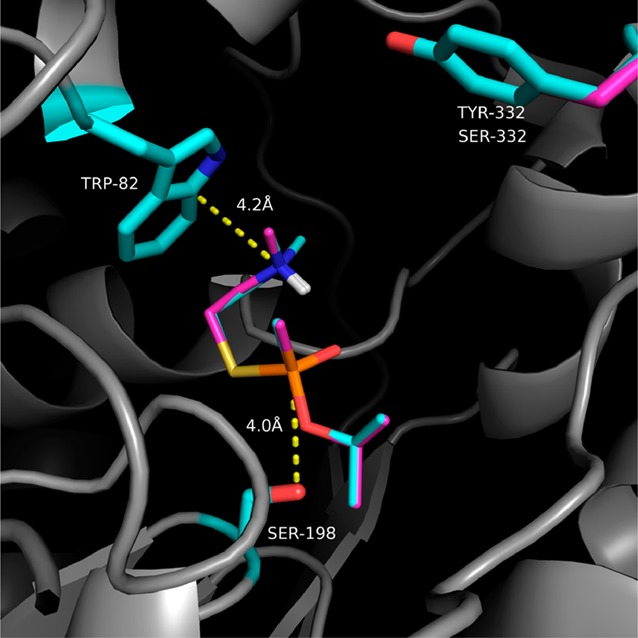
Overlay of SPGB3N–hBChE and SPGB3N–Y332S/D340H docked complexes. Dotted lines indicate distances (in angstroms) between indicated atoms. Side chains of select amino acid residues in the protein are colored cyan (WT) or pink (Y332S/D340H). Likewise, the SPGB3N ligand in the WT complex is colored cyan, and the SPGB3N ligand in the Y332S/D340H complex is colored pink. Non-carbon atoms are colored blue (nitrogen), red (oxygen), yellow (sulfur), and white (polar hydrogens).
Molecular Dynamics Structural Modeling of WT, Y332S, D340H, and Y332S/D340H hBChE Variants
As described above, studies of docking of SPGB3N to WT or variant hBChE showed a very similar binding mode. The distance between variant residues Y332 and D340 and the active site S198 was 17 Å for the S198–Y332 pair and 25 Å for the S198–D340 pair in the crystal structure of hBChE (PDB entry 1P0I).18 Docking studies predicted a very similar mode of binding of the nerve agent SPGB3N to all variants of hBChE examined, suggesting that the geometry of the binding site was not directly affected by these relatively distal mutations. Therefore, a MD approach was undertaken to investigate the effect of the Y332S, D340H, and Y332S/D340H hBChE variants on the overall structure of hBChE and the possible mechanism by which resistance to nerve agent model compounds was conferred. The WT and three hBChE variants (D340H, Y332S, and D340H/Y332S) were simulated, each of them with and without SPGB3N docked in their active sites. This resulted in a total of eight 50 ns trajectories. After the initial relaxation over the first few nanoseconds, the global protein size of gyration values remained stable and confirmed that the protein structures examined were stable under the simulation conditions and that its global (i.e., globular) form was retained. However, evaluation of the root-mean-square deviation (rmsd) of the protein backbone atoms (C, CA, and N) among variants and WT hBChE showed marked differences (Figure 3). Comparison of WT hBChE to the WT hBChE–SPGB3N complex showed lower average rmsd values in the complex. This suggested that for the WT enzyme, the ligand stabilized the conformation that was observed in the crystal structure (Figure 3A,B). The Y332S hBChE variant also showed a lower average rmsd for protein backbone atoms when bound to SPGB3N (Figure 3A). In contrast, for D340H and D340H/Y332S hBChE variants, this trend was reversed and lower rmsd values were observed for the unbound structures than for the WT complex (i.e., WT, 3.5 Å; WT with SPGB3N, 2.3 Å; Y332S, 3.0 Å; Y332S with SPGB3N, 2.3 Å; D340H, 2.5 Å; D340H with SPGB3N, 3.3 Å; Y332S/D340H, 2.6 Å; Y332S/D340H with SPGB3N, 3.7 Å) (Figure 3A,B). Thus, D340H and Y332S/D340H hBChE variants showed a difference in the dynamics and possibly substrate recognition of these variants compared to those of WT or Y332S hBChE variants. The Y332S/D340H hBChE–SPGB3N complex showed even greater structural deviation than WT hBChE in the absence of SPGB3N, reaching a 4 Å average backbone rmsd for the variant complex after 50 ns. However, in the absence of SPGB3N, the Y332S/D340H hBChE variant showed a backbone rmsd similar to that of the WT hBChE complex (Figure 3B).
Figure 3.
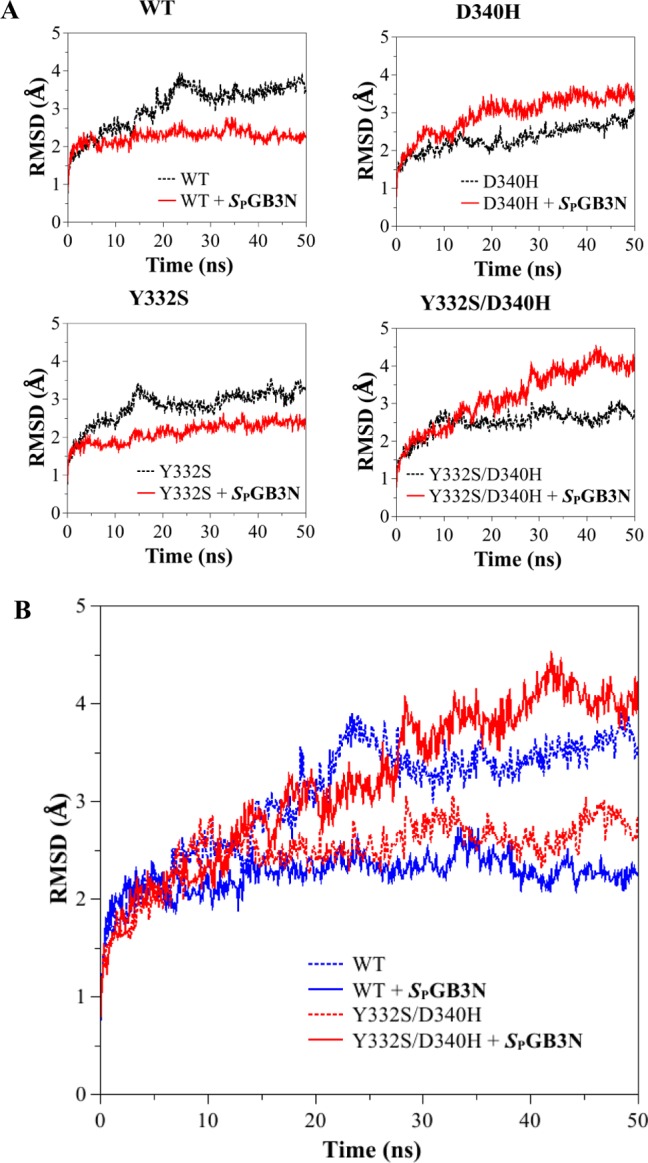
(A) Root-mean-square deviation (rmsd) of the backbone atoms (carbonyl carbon, C; α-carbon, CA; nitrogen, N) from the starting structure (PDB entry 1P0I) as a function of time (nanoseconds), for MD simulations of the WT and hBChE variants. MD simulations were used to compare the starting structure as a function of time for WT and Y332S, D340H, and Y332S/D340H hBChE variants in the absence (black line) and presence (red line) of SPGB3N. (B) Plot of rmsd vs time of all the backbone atoms (C, CA, and N) from the starting structure as a function of time for MD simulations of WT hBChE (blue) and the Y332S/D340H hBChE variant (red) in the presence (solid lines) or absence (dotted lines) of SPGB3N bound to the active site.
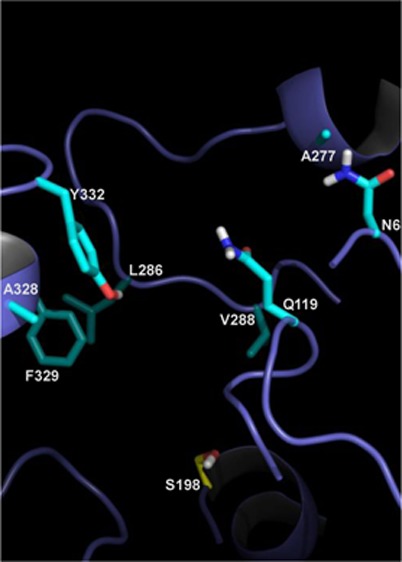
An rmsd analysis of hBChE MDs was conducted on selected regions of hBChE to further help define a mechanism of resistance among Y332S, D340H, and Y332S/D340H hBChE variants to inhibition by nerve agent model compounds. Recent studies indicated that several residues in the vicinity of the BChE active site form a door that has a major modulatory effect on the hBChE activity and its substrate selectivity.31 This “main door” consists of residues V288, Q119, L286, N68, and A277 on one side and residues A328, P329, and Y332 on the other side of the binding gorge of hBChE.31 We investigated the aperture of the “main door” in novel hBChE variants by computing the time-averaged distances between all 15 pairs of residues on opposite sides of the “main door” (Figure 4). In most cases, the values varied synchronously and indicated that the sides of the “main door” acted mostly as rigid surfaces (Figure S4 of the Supporting Information). The aperture, the average of the 15 distances, was therefore used as a single metric for the extent of the “main door” opening and closing. The time-averaged aperture gave a value for the WT hBChE that was 15.0 Å for the unbound enzyme that decreased to 12.9 Å after it formed a complex with SPGB3N (Table 3). In contrast, the D340H hBChE variant aperture showed essentially the opposite trend. For D340H, in the absence of the ligand, the distance was determined to be 14.0 Å, but in the presence of a ligand, the distance increased to 15.1 Å (Table 3). This suggested that the presence of SPGB3N in the active site prevented the door from closing as observed for WT hBChE. Analysis of the Y332S hBChE variant indicated that the “main door” of Y332S in the presence of ligand had a distance similar to that of the WT hBChE “main door” without a ligand (i.e., 15.0 Å for unbound WT and 14.7 Å for SPGB3N-bound Y332S) (Table 3). However, in the absence of ligand, the Y332S “main door” aperture was only 12.8 Å (Table 3). The largest deviation from that of WT hBChE was observed for the double mutant, Y332S/D340H hBChE, that had a 16.3 Å “main door” aperture in the presence of SPGB3N (Table 3). In the absence of SPGB3N, the “main door” of the Y332S/D340H variant value remained equally high (15.8 Å aperture). Taken together, both the Y332S and D340H hBChE variants introduced marked changes to the dynamics of the “main door” in the presence or absence of a ligand, and the effect of the Y332S/D340H hBChE amino acid variant on structural distortions from WT hBChE was even larger and was unaffected by ligand binding (Table 3 and Figure 5). Thus, compared to that of WT hBChE, differences in the “main door” aperture for D340H, Y332S, and Y332S/D340H hBChE variants in the presence or absence of nerve agent model compounds emerged as a probable mechanism for resistance for these variants.
Figure 4.
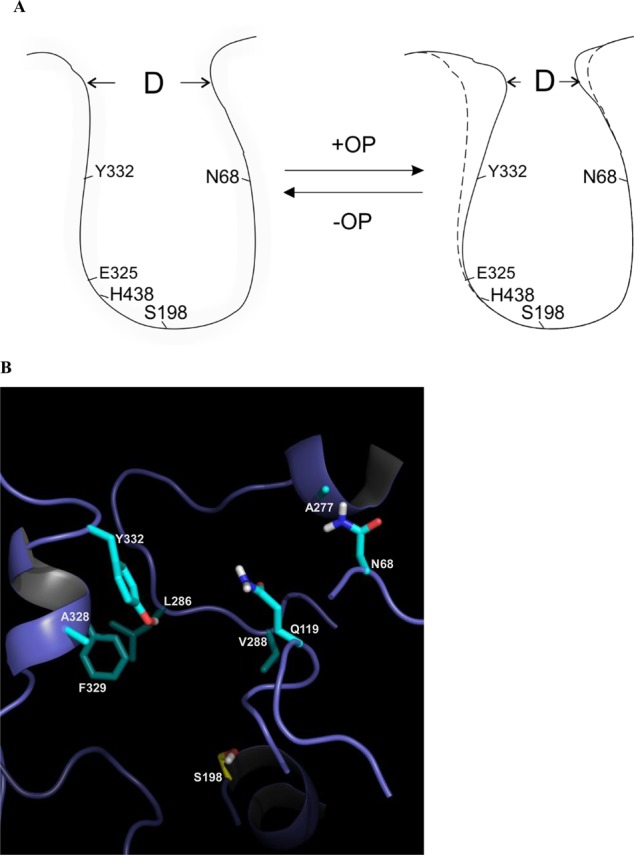
(A) Cartoon of WT hBChE gorge and “main door” apertures in the presence or absence of nerve agent model compounds. hBChE active site residues (i.e., S198, E325, and H438) are indicated. D is the distance of the “main door” aperture. (B) Structural model of WT hBChE with eight residues in the gorge highlighted (i.e., N68, Q119, A277, L286, V288, A328, F329, and Y332). Key amino acids in the protein are colored cyan. Non-carbon atoms are colored blue (nitrogen), red (oxygen), yellow (sulfur), and white (polar hydrogens) in the depiction.
Table 3. Average Sizes of the “Main Door” in WT hBChE and Y332S, D340H, and Y332S/D340H Variants Alone or Complexed with SPGB3N.
| average size (Å) | standard deviation | |
|---|---|---|
| WT | 15.0 | 0.7 |
| WT complex | 12.9 | 0.4 |
| Y332S | 12.8 | 0.6 |
| Y332S complex | 14.7 | 0.5 |
| D340H | 14.0 | 0.9 |
| D340H complex | 15.1 | 0.8 |
| Y332S/D340H | 15.8 | 0.8 |
| Y332S/D340H complex | 16.3 | 0.9 |
Figure 5.

(A) Plot of rmsd vs time of the “main door” backbone atoms (C, CA, and N) from the starting structure as a function of time for MD simulations of WT hBChE (blue) and the Y332S/D340H hBChE variant (red) in the presence (solid lines) or absence (dotted lines) of SPGB3N bound to the active site. (B) Overlay of structural models of noncomplexed WT, Y332S, and D340H hBChE (B1) and SPGB3N-complexed WT, Y332S, and D340H hBChE (B2) from MD simulations. For panels B1 and B2, side chains and secondary structure in the protein are colored cyan (WT) or pink (Y332S/D340H). Non-carbon atoms are colored blue (nitrogen), red (oxygen), yellow (sulfur), or white (polar hydrogens) in the depictions. (B1) Structural model of the atom alignment of WT and Y332S/D340H hBChE in the absence of SPGB3N after MD simulation for 50 ns. (B2) Structural model of atom alignment of WT and Y332S/D340H hBChE in the presence of SPGB3N after MD simulation for 50 ns. The SPGB3N in the WT complex is colored cyan, and the SPGB3N in the Y332S/D340H complex is colored pink.
Discussion
Research efforts have been directed toward identifying hBChE variants that are resistant to organophosphorus compounds that could provide a basis for new catalytic bioscavenger therapies.3,9 hBChE variants that are resistant to OP or nerve agent model compounds have been developed on the basis of the protein structure of hBChE.5,6,9,31,32 For example, rational design was used to identify hBChE variant G117H/E197Q, which is resistant to the PR/SCR, PSCS, and PRCS stereoisomers of soman with apparent rate constants of 0.006, 0.077, and 0.13 min–1, respectively.5 However, the rate of soman hydrolysis catalyzed by G117H or E197Q hBChE variants is not efficient enough to be useful for clinical applications. Rational design of new OP-resistant variants may be limited by the quality of the hBChE X-ray structures. It may be that X-ray crystal structures do not take into account the dynamic nature of the protein structure.33 In addition, target residues of mutagenesis are often restricted to the active site or vicinity and may not encompass important peripheral residues where the effect of structural distortions may not be as apparent or would require a laborious process of mutagenesis and iterative testing.33 For example, the G117H hBChE variant introduces a relatively large histidine side chain into the oxyanion binding region of the active site of the WT enzyme. This is a major structural distortion that may confer resistance to nerve agent model compounds and disrupt enzymatic function of the enzyme itself.
By contrast, the method described herein used a highly efficient adenoviral cloning method to construct diverse mutant libraries. In addition, a robust mammalian cell protein expression system was used to express hBChE variants because hBChE has post-translational modifications (i.e., glycosylation) required for optimal functional activity.9 Six libraries were designed to probe various regions of hBChE, including the gorge or substrate binding sites, the active site residues of the catalytic triad (S198-E325-H438), and the PAS/omega loop and acyl pocket regions (Figure S5 of the Supporting Information).4 From a functional screen, several nerve agent model compound-resistant variants were identified. The presence of G116, G117, and E197 variants discovered from both rational and molecular evolution approaches shows the importance of the approach to identifying residues in nerve agent model compound resistance.4 Amino acid 117 is part of the hBChE oxyanion hole that can stabilize the substrate or putative transition state during ester hydrolysis. Herein, several variants at amino acid residue 117 were also identified, including known variants G117N and G117R that confer resistance. Reportedly, G117H shows a stronger resistance to nerve agent model compounds than G117N and -R because it forms two hydrogen bonds with the substrate instead of one as in the case of G117N and -R,4,5,10 and this may facilitate hydrolysis of covalently adducted organophosphate esters. We also previously reported novel variants L125V and N68D that are resistant to nerve agent model compounds.4 The increased KM values for BTC hydrolysis for L125V and N68D hBChE variants as compared to G117H indicate L125V and N68D variants may have a different mechanism of nerve agent model compound resistance. The L125 and N68 residues of hBChE are near the entry site to the substrate binding gorge, possibly influencing the opening of the gorge and accessibility and binding of the substrate. These variants are unlikely to be directly involved in catalytic degradation of covalently adducted organophosphorous esters observed for G117H hBChE. As further exemplified in this study, our approach has proven to be broadly useful for identifying hBChE variants resistant to nerve agent model compounds and is capable of finding combinatorial mutations (e.g., Y332S/D340H variant) that afford marked resistance.4
Proposed Mechanisms of Resistance to Nerve Agent Model Compounds
A comparison of WT and hBChE variants by protein backbone rmsd analysis revealed unusual features. For WT hBChE and the Y332S variant, the presence of SPGB3N in the active site stabilized the conformation found in the crystal structure of PDB entry 1P0I,18 whereas the unbound D340H and Y332S/D340H hBCHE variants showed an rmsd similar to that of the crystal structure of PDB entry 1P0I. This suggests that the D340H and Y332S/D340H hBChE variant residues could, to some extent, imitate the effects of the ligand binding on the overall structure. For these two variants, the binding of ligand caused an increase in rmsd that likely reflected the need for additional structural distortion to accommodate the substrate. Such global rmsd analysis provided a strong indication that the variants were different in their dynamics and possibly in their substrate recognition. However, a more focused rmsd analysis provided an opportunity to examine the marked differences in the kinetic properties of hBChE WT and Y332S, D340H, and Y332S/D340H variants observed experimentally.
As recently described, hBChE residues V288, Q119, L286, N68, A277, A328, F329, and Y332 of the “main door” influence the activity of hBChE by determining the accessibility of the substrate and active sites31 (Figure 4). On the basis of these observations and molecular dynamics simulations described herein, residues at the opening of the hBChE gorge were generally among those residues most distorted from the WT structure. Thus, alteration of the “main door” could be the basis for significant resistance to nerve agent model compounds observed for hBChE variants. Investigation of the aperture of the “main door” by molecular dynamics showed a potential new mechanism of resistance for Y332S, D340H, and Y332S/D340H hBChE variants. The results show that the aperture for WT hBChE was larger in the unbound enzyme than in the enzyme with a nerve agent model compound bound in its active site. This suggests that amino acids 332 and 340 play an integral part in the function of hBChE because decreasing the “main door” size after the binding of the substrate could increase the stability of the precomplex between nerve agent model compounds and hBChE. The D340H hBChE variant showed the opposite “main door” behavior between SPGB3N-bound and unbound configurations: the SPGB3N–hBChE form showed a larger aperture that suggested that the “main door” was prevented from closing.. In principle, this could lead to a lower level of nerve agent model compound resistance by providing a nerve agent model compound with an increased likelihood of dissociating from the active site before reaction with an active site serine (S198) occurs. The analysis of the Y332S hBChE variant suggested yet another mechanism for nerve agent model compound resistance. In this case, the “main door” is similar in size to the WT hBChE “main door” when SPGB3N is bound. However, in the absence of SPGB3N, the “main door” of WT hBChE has the smallest aperture calculated among hBChE variants examined (i.e., Y332S, D340H, and Y332S/D340H). The smaller aperture of the “main door” of the Y332S hBChE variant could restrict the entry of substrates, especially larger ones, into the active site. Compared to WT hBChE, the largest difference of the “main door” aperture was observed for the double variant, Y332S/D340H hBChE. The Y332S/D340H hBChE variant “main door” aperture was larger than the WT hBChE “main door” aperture independent of the presence of SPGB3N in the Y332S/D340 active site. In this case, the “main door” may be nonfunctional and may permit SPGB3N to escape the hBChE substrate binding site before reaction occurs with the active site serine. The plausibility of this proposal is strengthened by prior reports that have identified the role of various alternative entries or exits for hBChE34 or hAChE kinetics.35−37 Also, for hAChE, the aromatic residues lining the substrate binding gorge have been reported to guide substrates to the active site deep within the gorge.35 In addition to being part of the “main door”, residue 332 of hBChE is also part of the peripheral anionic binding site for hBChE substrates. Because ETP is a positively charged inhibitor, this difference in resistance between ETP and SPGB3N is potentially due to interaction of the positively charged choline with the peripheral anionic binding site as well as differences in the “main door” aperture distances.
We observed that the net effect on enzymatic function in the hBChE variants reported herein was substrate-dependent. For example, the kcat for BTC hydrolysis by the Y332/D340H hBChE variant is 2-fold greater than that of the WT enzyme, while the ki for inhibition of the Y332/D340H variant by SPGB3N was 70-fold lower than that of the WT enzyme. Thus, structural modifications of the “main door” appear to permit modulation of enzymatic function that does not necessarily lower function for other substrates. This is further supported by studies of the G117H hBChE variant that has a modified active site and lower rates of BTC hydrolysis and inhibition by SPGB3N, at least as supported by these two specific examples. Furthermore, molecular docking of SPGB3N to the hBChE variants was very similar among variants (Y332S, D340H, and Y332S/D340H) and WT hBChE. Accordingly, the resistance conferred by the Y332S, D340H, and Y332S/D340H hBChE variants is less likely to arise from direct modulation of the structure of the active site catalytic triad (i.e., S198, E325, and H438), and this notion is supported by the large distances between the variant residues and the active site S198.
Novel variants of hBChE (i.e., Y332S, D340H, and Y332S/340H) that are resistant to nerve agent model compounds were discovered using a molecular evolution–functional screening approach. As shown by kinetic data and molecular dynamics studies, these hBChE variants showed a unique mechanism of nerve agent model compound resistance. The results show the utility of this method for identifying hBChE variants that are resistant to NAs with unique mechanisms of action.
Acknowledgments
We thank Robert R. Reddy, Beilin Wang, Nora Barakat, and Longkuan Xiang for hBChE library screening and variant purification, Eric Ralph for assistance with hBChE variant enzyme kinetic experiments, Sigeng Chen for figure preparation, and Cynthia B. Gilley, Mary MacDonald, and Karl Okolotowicz for chemical synthesis of nerve agent model compounds.
Glossary
Abbreviations
- OP
organophosphorous
- AChE
acetylcholinesterase
- BChE
butyrylcholinesterase
- hBChE
human BChE
- ChE
cholinesterase
- ETP
echothiophate
- GB
sarin
- GD
soman
- GF
cyclosarin
- GA
tabun
- ATC
acetylthiocholine
- BTC
butyrylthiocholine iodide
- DTNB
5,5-dithiobis(2-nitrobenzoic acid)
- AD
adenovirus
- pAD
AD plasmid vector
- PCR
polymerase chain reaction
- MOI
multiplicity of infection
- CHO
Chinese hamster ovary
- WT
wild type
- CMV
cytomegalovirus
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- PBS
phosphate-buffered saline
- HRP
horseradish peroxidase
- SPGB3N
(S)-2-[isopropoxy(methyl)phosphorylthio]-N,N-dimethylethanammonium chloride
- SPGD3N
(SP)-2-[3,3-dimethylbutan-2-oxy(methyl)phosphorylthio]-N,N-dimethylethanammonium chloride
- SPGF3N
(S)-2-[cyclohexyloxy(methyl)phosphorylthio]-N,N-dimethylethanammonium chloride.
Supporting Information Available
Plots of residual hBChE activity versus incubation time with SPGB3N, SPGD3N, and SPGF3N for WT and variant hBChE (Figures S1–S3), replots of kapp versus inhibitor concentration for the incubations of WT and variant hBChE (Figures S1–S3), a plot of the distance between the center of mass of pairs of residues on each side of the hBChE “main door” as a function of time for molecular dynamics simulations of WT hBChE in the presence or absence of SPGB3N bound to the active site (Figure S4), and a table of primer sequences used to construct the variant hBChEs (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This research was supported by National Institutes of Health Grants U01NS058038 and U54NS058183.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Eddleston M.; Buckley N. A.; Eyer P.; Dawson A. H. (2008) Management of acute organophosphorus pesticide poisoning. Lancet 371, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro N. B.; Watson A. P.; Ambrose K. R.; Griffin G. D. (1990) Treating exposure to chemical warfare agents: Implications for health care providers and community emergency planning. Environ. Health Perspect. 89, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz D. E.; Clarkson E. D.; Schulz S. M.; Cerasoli D. M. (2010) Butyrylcholinesterase as a therapeutic drug for protection against percutaneous VX. Chem.-Biol. Interact. 187, 249–252. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen S.; Ralph E. C.; Dwyer M.; Cashman J. R. (2012) Identification of human butyrylcholinesterase organophosphate-resistant variants through a novel mammalian enzyme functional screen. J. Pharmacol. Exp. Ther. 343, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockridge O.; Blong R. M.; Masson P.; Froment M. T.; Millard C. B.; Broomfield C. A. (1997) A single amino acid substitution, Gly117His, confers phosphotriesterase (organophosphorus acid anhydride hydrolase) activity on human butyrylcholinesterase. Biochemistry 36, 786–795. [DOI] [PubMed] [Google Scholar]
- Masson P.; Froment M. T.; Bartels C. F.; Lockridge O. (1997) Importance of aspartate-70 in organophosphate inhibition, oxime re-activation and aging of human butyrylcholinesterase. Biochem. J. 325(Part 1), 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel M.; Quinn D. M.; Nair H. K.; Silman I.; Sussman J. L. (1996) The X-ray structure of a transition state analog complex reveals the molecular origins of the catalytic power and substrate specificity of acetylcholinesterase. J. Am. Chem. Soc. 118, 2340–2346. [Google Scholar]
- Shafferman A.; Ordentlich A.; Barak D.; Stein D.; Ariel N.; Velan B. (1996) Aging of phosphylated human acetylcholinesterase: Catalytic processes mediated by aromatic and polar residues of the active centre. Biochem. J. 318(Part 3), 833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokugras A. N. (2003) Butyrylcholinesterase: Structure and physiological importance. Turk Biyokim. Derg. 28, 54–61. [Google Scholar]
- Zhang J.; Chen S.; Ralph E. C.; Dwyer M.; Cashman J. R. (2012) Identification of human butyrylcholinesterase organophosphate-resistant variants through a novel mammalian enzyme functional screen. J. Pharmacol. Exp. Ther. 343, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat N. H.; Zheng X.; Gilley C. B.; MacDonald M.; Okolotowicz K.; Cashman J. R.; Vyas S.; Beck J. M.; Hadad C. M.; Zhang J. (2009) Chemical synthesis of two series of nerve agent model compounds and their stereoselective interaction with human acetylcholinesterase and human butyrylcholinesterase. Chem. Res. Toxicol. 22, 1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph E. C.; Xiang L.; Cashman J. R.; Zhang J. (2011) His-tag truncated butyrylcholinesterase as a useful construct for in vitro characterization of wild-type and variant butyrylcholinesterases. Protein Expression Purif. 80, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisiak J.; Ralph E. C.; Zhang J.; Cashman J. R. (2011) Amidine-oximes: Reactivators for organophosphate exposure. J. Med. Chem. 54, 3319–3330. [DOI] [PubMed] [Google Scholar]
- Kalisiak J.; Ralph E. C.; Cashman J. R. (2012) Nonquaternary reactivators for organophosphate-inhibited cholinesterases. J. Med. Chem. 55, 465–474. [DOI] [PubMed] [Google Scholar]
- Gateway Technology Manual (2014) pp 1–74, Life Technologies, Carlsbad, CA. [Google Scholar]
- Ellman G. L.; Courtney K. D.; Andres V. Jr.; Feather-Stone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Lockridge O.; Masson P.; Fontecilla-Camps J. C.; Nachon F. (2003) Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 278, 41141–41147. [DOI] [PubMed] [Google Scholar]
- Malde A. K.; Zuo L.; Breeze M.; Stroet M.; Poger D.; Nair P. C.; Oostenbrink C.; Mark A. E. (2011) An automated force field topology builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 7, 4026–4038. [DOI] [PubMed] [Google Scholar]
- Canzar S.; El-Kebir M.; Pool R.; Elbassioni K.; Malde A. K.; Mark A. E.; Geerke D. P.; Stougie L.; Klau G. W. (2013) Charge group partitioning in biomolecular simulation. J. Comput. Biol. 20, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiev M.; Bylaska E. J.; Govind N.; Kowalski K.; Straatsma T. P.; Van Dam H. J. J.; Wang D.; Nieplocha J.; Apra E.; Windus T. L.; de Jong W. A. (2010) NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477–1490. [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–448. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. (2005) GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1719. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 1–7. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. (1980) Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 45, 1196–1205. [Google Scholar]
- Rahman A.; Parrinello M. (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7191. [Google Scholar]
- Radic Z.; Pickering N. A.; Vellom D. C.; Camp S.; Taylor P. (1993) Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 32, 12074–12084. [DOI] [PubMed] [Google Scholar]
- Grigoryan H.; Schopfer L. M.; Thompson C. M.; Terry A. V.; Masson P.; Lockridge O. (2008) Mass spectrometry identifies covalent binding of soman, sarin, chlorpyrifos oxon, diisopropyl fluorophosphate, and FP-biotin to tyrosines on tubulin: A potential mechanism of long term toxicity by organophosphorus agents. Chem.-Biol. Interact. 175, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Schopfer L. M.; Nachon F.; Froment M. T.; Masson P.; Lockridge O. (2007) Aging pathways for organophosphate-inhibited human butyrylcholinesterase, including novel pathways for isomalathion, resolved by mass spectrometry. Toxicol. Sci. 100, 136–145. [DOI] [PubMed] [Google Scholar]
- Li H.; Schopfer L.; Spaulding R.; Thompson C. M.; Lockridge O. (2005) Identification of organophosphate-reactive proteins by tandem mass spectrometry. Chem.-Biol. Interact. 157–158, 383–384. [DOI] [PubMed] [Google Scholar]
- Fang L.; Pan Y.; Muzyka J. L.; Zhan C. (2011) Active Site Gating and Substrate Specificity of Butyrylcholinesterase and Acetylcholinesterase: Insights From Molecular Dynamices Simulations. J. Phys. Chem. B 115, 8797–8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard C. B.; Lockridge O.; Broomfield C. A. (1995) Design and expression of organophosphorus acid anhydride hydrolase activity in human butyrylcholinesterase. Biochemistry 34, 15925–15933. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U. T.; Pohl M. (2001) Improved biocatalysts by directed evolution and rational protein design. Curr. Opin. Chem. Biol. 5, 137–143. [DOI] [PubMed] [Google Scholar]
- Suárez D.; Field M. J. (2005) Molecular dynamics simulations of human butyrylcholinesterase. Proteins: Struct., Funct., Bioinf. 59, 104–117. [DOI] [PubMed] [Google Scholar]
- Ripoll D. R.; Faerman C. H.; Axelsen P. H.; Silman I.; Sussman J. L. (1993) An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. U.S.A. 90, 5128–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodek S. T.; Clark T. W.; Scott L. R.; McCammon J. A. (1997) Molecular Dynamics of Acetylcholinesterase Dimer Complexed with Tacrine. J. Am. Chem. Soc. 119, 9513–9522. [Google Scholar]
- Enyedy I. J.; Kovach I. M.; Brooks B. R. (1998) Alternate Pathways for Acetic Acid and Acetate ion Release from Acetylcholinesterase: A Molecular Dynamics Study. J. Am. Chem. Soc. 120, 8043–8050. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.