

Abstract
Multiple endocrine neoplasia type 1 (MEN1) is an inherited tumor syndrome that includes susceptibility to pancreatic islet tumors. This syndrome results from mutations in the MEN1 gene, encoding menin. Menin acts as an oncogenic co-factor for MLL-fusion protein-mediated histone H3 lysine 4 methylation, but the precise basis for how menin suppresses gene expression and proliferation of pancreatic beta cells remains poorly understood. Here we show that menin ablation enhances Hedgehog (Hh) signaling, a pro-proliferative and oncogenic pathway, in murine pancreatic islets. Menin directly interacts with protein arginine methyltransferase 5 (PRMT5), a negative regulator of gene transcription. Menin recruits PRMT5 to the promoter of the Gas1 gene, a crucial factor for binding of Sonic hedgehog (Shh) ligand to its receptor PTCH1 and subsequent activation of the Hh signaling pathway, increases repressive histone arginine dimethylation (H4R3m2s) and suppresses Gas1 expression. Notably, MEN1 disease-related menin mutants have reduced binding to PRMT5, and fail to impart the repressive H4R3m2s mark at the Gas1 promoter, resulting in its elevated expression. Pharmacological inhibition of Hh signaling significantly reduces proliferation of insulinoma cells, and expression of Hh signaling targets including Ptch1, in MEN1 tumors of mice. These findings uncover a novel link between menin and Hh signaling whereby menin/PRMT5 epigenetically suppress Hh signaling, revealing it as a target for treating MEN1 tumors.
Keywords: Menin, PRMT5, Hh signaling, Epigenetics, GDC-0449
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an inherited tumor syndrome, with development of tumors in several endocrine organs including pancreatic islets (1–4). The gene mutated in this syndrome, MEN1, encodes a nuclear protein, menin (5, 6). Menin interacts with diverse proteins to regulate a variety of cellular functions including control of gene transcription (7, 8). Target-based therapy against MEN1 syndrome is currently lacking, yet highly desirable.
Epigenetic regulation of gene expression via histone methylation is crucial for the regulation of onset and maintenance of various cancers (9). Menin can positively or negatively affect gene expression. It upregulates expression of anti-proliferative genes, such as cyclin dependent kinase inhibitors (CDKIs) p18 and p27, partly via up-regulating MLL (mixed lineage leukemia)-mediated histone H3 lysine 4 (H3K4) methylation (10, 11). However, it is not yet well understood how menin represses gene transcription and signaling pathways.
The Hedgehog (Hh) signaling pathway regulates diverse biological processes ranging from embryonic development to cell cycle and tumorigenesis (12). With assistance from accessory proteins, such as GPI-anchored cell surface proteins GAS1, BOC, or CDO (13–15), Hh ligands bind to the cell surface receptor Patched (PTCH1), resulting in the release of PTCH1-mediated repression of the cell membrane protein Smoothened (SMO) (12). Activated SMO enters the primary cilia and promotes the dissociation of GLI proteins from SuFu, Fused and Cos2 complex (16), resulting in nuclear translocation of transcription factors GLI1 and GLI2 and transcription of target genes including pro-proliferative genes (12). Concomitantly, GLI3 proteolysis to generate the repressive form, GLI3R, is inhibited (17). Constitutively activated Hh signaling triggers the development of tumors, such as medulloblastomas and basal cell carcinoma (BCC) (18, 19). It is not yet clear whether menin influences Hh signaling during the development of the MEN1 tumor. Here we show that menin potently suppresses Hh signaling at least partly via PRMT5-mediated suppression of GAS1, and Men1 mutations lead to enhanced Hh signaling.
Materials and Methods
Plasmids and Cell Culture
Lentiviral constructs expressing Prmt5 shRNAs were obtained from Open Biosystems. Lentiviral packaging plasmids, pMD2G and pAX2G, were purchased from Addgene. Retroviral plasmids expressing Flag-tagged or mutant menin have been described elsewhere (20). GST-PRMT5 fragments were generated using the site-directed mutagenesis kit (Stratagene). Men1-null MEF cells complemented with wild type or mutant menin (20), and 293 cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) medium supplemented with 10 % fetal bovine serum (FBS) and 1% Pen/Strep.
Immunoprecipitation and Immunoblotting
Nuclear extract (NE) was isolated as described previously (21). Immunoprecipitation and immunoblotting were performed using standard techniques. Details can be found in Supplementary Material. Antibodies used were anti-menin (Bethyl, A300-105A), anti-PRMT5 (Abcam, ab31751 and ab109451), anti-MEP50 (Bethyl, A301-562A), anti-PRMT7 (Abcam, ab126965), anti-FLAG (Sigma, F3165), and anti-β-Actin (Sigma, A5441). Rabbit antibodies to GAS1 are described elsewhere (22).
RNA Extraction and Quantitative Real Time PCR (qRT-PCR)
Total RNA was extracted from cultured cells with Trizol and an RNeasy extraction kit from Qiagen. Details and primer sequences can be found in Supplementary Material.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed as previously described using a Quick ChIP kit from Imgenex (23). Details, and primer sequences can be found in Supplementary Material. Antibodies used for ChIP were anti-menin (Bethyl, A300-105A), anti-PRMT5 (Abcam, ab31751), anti-Histone H4 Symmetric Di-Methyl R3 (Abcam, ab5823), anti-Histone H3 (ab1791).
Mice
All laboratory mice were maintained on a 12 hr light-dark cycle in the animal facility at the University of Pennsylvania. All experiments on mice in our research protocol were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania and were performed in accordance with relevant institutional and national guidelines and regulations. Men1l/l;CreER, Men1l/l; RipCre and Men1l/l;Pdx1CreER mice were generated as described previously (24). Genotyping of mice was performed by PCR on mouse-tail DNA.
Excision of the Floxed Men1 Promoter Using Tamoxifen (TAM)
Men1l/l;Cre-ER and Men1l/l; pdx1Cre-ER and their littermate controls were fed TAM (MP Biomedicals) at 200 mg/kg of body weight per day for two consecutive days, followed by one day off and then for another two consecutive days as described previously (20).
Physiological Measurements
GDC-0449 was obtained from ChemieTek (Indianapolis). Blood glucose levels were assayed from tail vein blood by a glucose meter (OneTouch, Lifescan). Blood serum insulin levels were measured by ELISA using a mouse insulin kit (Crystal Chem).
Detection of mRNA Levels in Islets of Mice Pancreas
Islets were individually isolated from mouse pancreas by collagenase digestion and separated using a Ficoll gradient as previously described (11). The islets were digested in Trizol, and RNA was extracted using a RNeasy mini kit (Invitrogen). 50 ng RNA was reverse transcribed into cDNA using the Omniscript RT kit from Qiagen.
Immunofluorescence
Images were captured using a Nikon Eclipse E800 fluoresescence microscope equipped with a CCD digital camera. Total insulin staining area was quantified using Metamorph software (Molecular Devices Corporation, Sunnyvale, CA). Antibodies used for immunostaining were insulin (Abcam, ab7842) and BrdU (Accurate Chemical and Scientific, OBT0030G). Secondary antibodies used were FITC (Abcam, ab6904) and Alexa fluor 546 (Invitrogen, A11035).
PRMT5 and Menin Binding Assay
His-Menin (in pET28a vector) and GST-PRMT5 (from pGEX-4T1 vector) were expressed in BL21 Codon Plus (Stratagene). Details and antibodies used can be found in Supplemental Material.
Histone Methyltransferase Assay
Cells were lysed in Tween-20 buffer (50 mM Hepes-KOH, pH 8, 150 mM NaCl, 2.5 mM EGTA, 1 mM EDTA, 0.1 % Tween 20, 1 mM PMSF, 1 mM DTT) supplemented with a cocktail of protease inhibitors, and menin complexes were collected using anti-Flag M2 beads (Sigma). Beads were washed in Tween-20 buffer, and then incubated with 2.5 μCi S-adenosyl-L-(methyl-3H) methionine (SAM) (Amersham Pharmacia) and 1μg recombinant Histone H4 (NEB) in a total volume of 25 μl of methyltransferase buffer (50 mM Tris-HCl (pH 8.0), 50 mM NaCl, and 1 mM PMSF) for 2 hours at 30 °C. The reaction mixture was resolved on SDS-PAGE, and the gel was soaked in Amersham Amplify solution (GE Healthcare) for 30 mins, and subsequently exposed to film at −80 °C for autoradiography.
Statistical Analyses
Statistical analyses were performed by using Graphpad Prism (version 5.0; Graphpad Software). The data are presented as the mean ± s.d. of n determinations unless noted otherwise. A two-tailed student’s t test was used for measuring statistical differences.
Results
Menin represses expression of Gas1, a crucial co-factor for Hedgehog signaling
Our previous microarray analysis showed that Men1 excision in mouse embryonic fibroblasts (MEFs) up-regulated expression of Gas1 (21), and recent reports show that GAS1 plays a crucial role in enhancing Hedgehog (Hh) signaling in cultured cells and during embryonic development (13–15). We found that ectopic menin expression in menin-null MEFs reduced the mRNA and protein levels of GAS1 (Fig. 1A). Quantitative RT-PCR (qRT-PCR) showed that complementing the menin-null cells with menin suppressed sonic hedgehog ligand (Shh)-induced expression of Gli1 (Fig. 1B), an effector of Hh signaling (17). Our results suggest that menin suppresses Hh signaling.
Figure 1.

Menin regulates GAS1 and Hedgehog signaling. (A) Quantitative RT-PCR (qRT-PCR) and immunoblotting showing expression of Gas1 mRNA and protein in menin-null MEFs complemented with either vector or wild type (WT) menin. Ponceau S is included as a loading control. (B) qRT-PCR showing expression of Gli1 mRNA in menin-null MEFs complemented with either vector or WT menin cultured in varying concentrations of Sonic Hedgehog-conditioned medium (Shh-CM). Error bars indicate ± s.d.
Menin directly interacts with PRMT5, a repressive histone H4 arginine 3 (H4R3) methyltransferase, and co-elutes with the PRMT5 complex
As menin interacts with proteins like MLL to up-regulate gene expression (11, 25), we sought to identify transcriptional repressive partner(s) of menin that may down-regulate the expression of Gas1. Ion-exchange chromatography showed that the majority of menin from nuclear extracts eluted in the range of 200–300 mM NaCl (Fig. 2A). The menin-containing fractions were subjected to affinity purification with anti-Flag M2-conjugated beads. Menin-interacting proteins were eluted and separated by gel electrophoresis, followed by silver staining (Fig. 2B). Mass spectrometry analysis of the purified proteins showed that PRMT5 and its associated factor MEP50 were among the major eluted proteins (Fig. 2B). PRMT5 symmetrically dimethylates protein arginine residues, such as histone H4 arginine 3 and histone H3 arginine 8, resulting in repression of gene expression (26, 27).
Figure 2.

Menin interacts directly with PRMT5 to methylate histone H4. (A) Immunoblotting of menin in nuclear extract (NE) from HEK293 cells expressing Flag-menin fractionated by anion-exchange chromatography using a Q-Sepharose column eluted with varying concentrations of NaCl. (B) Silver staining of menin-containing fractions after affinity purification using anti-Flag M2 beads. Visible bands were excised for identification by mass spectrometry. * = PRMT5 peptide fragments identified by mass spectroscopy; 77DWNTLIVGK, 228AAILPTSIFLTNKK, 241KGFPVLSK, 334YSQYQQAIYK, 369GPLVNASLR. (C) Immunoblotting showing menin (top), PRMT5 and MEP50 (bottom) in NE of HEK293 cells ectopically expressing Flag-menin immunoprecipitated with indicated antibodies. (D, E) Endogenous interaction between menin and PRMT5 in HEK293 cells immunoprecipitated with two independent anti-menin (D) or anti-PRMT5 antibodies (E). (F) Immunoblotting for menin, PRMT5 and MEP50 in gel filtration chromatography fractions of NE from HEK293 cells ectopically expressing Flag-menin. (G) Immunoblotting of His-menin expressed in E. coli immunoprecipitated by GST-PRMT5 immobilized to glutathione beads. (H) Biotinylated PRMT5 peptides of various lengths were immobilized on streptavidin beads, and the binding of His-menin was identified by immunoblotting. (I) Histone methyltransferase (HMT) assay using anti-menin immunoprecipitates from 293T cells transfected with Flag-menin and/or Myc-PRMT5 incubated with 3H-AdoMet and histone H4. Autoradiography for 3H-methylated histone H4 (middle); Coomassie stain of H4 is included as a loading control. (J) Immunoblotting of menin and PRMT5 in cells used for HMT assay in I. Ponceau S is included as a loading control.
Reciprocal co-immunoprecipitation (co-IP) using lysates from HEK293 cells ectopically expressing Flag-tagged menin, followed by Western blotting, confirmed the interaction between menin and PRMT5 (Fig. 2C). Furthermore, MEP50 was also detected in the anti-menin immunoprecipitate (Fig. 2C). Importantly, in HEK293 cells expressing only endogenous menin and PRMT5, immunoprecipitation with two independent menin antibodies effectively pulled down PRMT5 (Fig. 2D). Similarly, immunoprecipitation with anti-PRMT5 antibody pulled down menin (Fig. 2E), demonstrating interaction of the endogenous proteins. Gel filtration chromatography showed that while menin eluted at several peaks, the fractions eluting at ~500 kDa contained PRMT5 and MEP50 (Fig. 2F, lanes 5–6), suggesting that menin/PRMT5/MEP50 co-exist in a complex. GST pull-down with proteins expressed and purified from E. coli showed that GST-PRMT5 directly interacted with His-tagged menin (Fig. 2G). Further analysis of GST-PRMT5 fragments of various lengths (Supplementary Fig. S1A) showed that amino acid (aa) residues 1-210 bound to menin (Supplementary Fig. S1B, Lane 3). Deletional analysis of PRMT5 fragment 1-210 indicated that aa 1-93 was sufficient for binding to menin (Supplementary Fig. S1C, Lane 4). A biotinylated PRMT5 peptide (aa 23-51 of PRMT5) conjugated to avidin beads was sufficient for pulling down menin (Fig. 2H), indicating that menin binds to the N-terminus of PRMT5.
Menin pulls down PRMT5-associated histone H4 methyltransferase activity
To test whether immunoprecipitated menin pulls down histone methylating activity mediated by PRMT5, 293T cells were transfected with MEN1 and/or PRMT5 cDNA, followed by IP with an anti-menin antibody. Histone methyltransferase (HMT) assay with the immunoprecipitated complex, 3H-SAM and recombinant histone H4 showed that menin pulled down a histone H4 methylating activity, and this activity was enhanced further when both menin and PRMT5 were ectopically expressed (Fig. 2I, lane 4 vs. 3), thus indicating that menin-associating PRMT5 is enzymatically functional. As controls, menin and PRMT5 were expressed as expected (Fig. 2J).
Microarray analysis reveals common targets of menin and PRMT5 including Gas1, and shows correlation with the Hedgehog (Hh) pathway signature
We performed cDNA microarray analysis with primary pancreatic islets from control Men1l/l or Men1l/l mice expressing a tamoxifen-inducible Cre under control of the ubiquitous Ubc9 promoter (Men1l/l; Ubc9CreER) 14 days post Men1 excision (24). Excision of Men1 in pancreatic islet cells was confirmed by co-immunostaining for menin and insulin (Supplementary Fig. S2). Gene set enrichment analysis (GSEA) (28) of genes enriched upon Men1 excision in pancreatic islets, in an unbiased search, matched a group of genes up-regulated in the Prmt5-knockdown NIH3T3 subset including Gas1 (27) (Supplementary Fig. S3A). Moreover, the leading-edge subset of the associated significant gene sets included Gas1 (Supplementary Fig. S3A). Since GAS1 is actively involved in promoting pro-proliferative Hh signaling, these findings suggest that menin and PRMT5 functionally interact to suppress a common set of genes including Gas1 to modulate Hh signaling.
Ablation of all three Hh co-receptors, GAS1, CDO and BOC abrogates Shh-dependant neural progenitors (14), and a complete loss of Hh-dependant CGNP proliferation (13). Knockdown of Gas1 in MEFs inhibits Shh-induced gene expression (15). From our microarray analysis of isolated islets from control and Men1Δ/Δ mice, GSEA indicated that there was a modest increase in the Hedgehog signaling gene set (Supplementary Fig. S3B). These findings suggest that menin may effectively dampen, but not completely block Hh signaling.
PRMT5 is required for suppressing Gas1 expression, and for optimal histone H4 arginine 3 symmetric dimethylation (H4R3m2s) at the Gas1 promoter
We knocked down Prmt5 in MEFs using shRNA’s, and qRT-PCR showed that two independent shRNA clones reduced expression of Prmt5 (Fig. 3A), and moderately increased the mRNA levels of Gas1 (Fig. 3B). Consistently, reduced PRMT5 expression correlated with increased expression of GAS1 at the protein level, as shown by Western blotting (Fig. 3C). To determine the impact of Prmt5 knockdown on H4R3m2s, catalyzed by PRMT5 (27), at the Gas1 promoter, we performed chromatin immunoprecipitation (ChIP) assay and found that PRMT5 bound to the Gas1 promoter, and PRMT5 knockdown moderately reduced PRMT5 binding (Fig. 3D). Consistently, PRMT5 knockdown also reduced H4R3m2s levels at the Gas1 promoter (Fig. 3D, and Supplementary Fig. S4A). As a control, no difference in binding was observed at the actin promoter (Supplementary Fig. S4B). We cultured both control and PRMT5 knockdown cells in the presence or absence of Shh ligand and found that PRMT5 knockdown markedly increased Shh-induced expression of Gli1 and Ptch1 (Figs. 3E and 3F), both targets of Hh signaling (17). Together, these results suggest that PRMT5 plays a crucial role in suppressing Hh signaling, at least in part by repressing GAS1 and increasing the repressive H4R3m2s mark at the promoter.
Figure 3.

Loss of PRMT5 results in elevated Gas1 levels and enhanced Hedgehog (Hh) signaling. (A) Quantitative RT-PCR (qRT-PCR) showing reduction of Prmt5 mRNA in MEFs expressing two independent Prmt5-targeting shRNA clones compared to MEFs expressing scrambled control shRNA. (B) qRT-PCR showing expression of Gas1 mRNA levels in MEFs expressing shRNA targeting Prmt5. (C) Immunoblotting for PRMT5 and GAS1 in MEFs expressing either control or Prmt5-targeting shRNA’s. Ponceau S is included as a loading control. (D) Chromatin immunoprecipitation (ChIP) with antibodies against PRMT5 and histone H4 arginine 3 symmetric dimethylation (H4R3m2s) at the Gas1 promoter. ChIP amplicon, Gas1 (−780 bp / −609 bp). (E–F) MEFs expressing control or two independent shRNA clones targeting Prmt5 were cultured in either control or Sonic hedgehog conditioned medium (Shh-CM) and Gli1 (E) and Ptch1 (F) mRNA levels were quantitated by qRT-PCR. Error bars indicate ± s.d.
Menin binds to the promoter of Gas1 and recruits PRMT5 to increase H4R3m2s
To determine whether menin directly regulates expression of Gas1, we performed ChIP assay with menin-expressing or menin-null cells, and showed that menin bound to the Gas1 promoter (Fig. 4A). Consistently, PRMT5 binding and H4R3m2s at the Gas1 promoter were also markedly reduced in Men1-null cells (Fig. 4A), indicating that menin recruits PRMT5 to methylate H4R3 at this target gene.
Figure 4.

Menin recruits PRMT5, and its associated histone modification mark, H4R3m2s, to the Gas1 promoter. (A) Chromatin immunoprecipitation (ChIP) with antibodies against menin, PRMT5, and histone H4 arginine 3 symmetric dimethylation (H4R3m2s) at the Gas1 promoter in Men1-null MEFs complemented with either empty vector or wild-type (WT) menin. ChIP amplicon, Gas1 (−780 bp / −609 bp). (B) Quantitative RT-PCR (qRT-PCR) showing expression of Gas1 mRNA in menin-null MEFs complemented with empty vector, WT menin or MEN1 disease related mutants, L22R and A242V. Levels of ectopic WT and mutant menin expression are shown, and immunoblotting for β actin is included as a loading control. (C) ChIP with antibodies against menin and H4R3m2s at the Gas1 promoter in Men1-null MEFs complemented with vector, WT menin or MEN1 disease related mutants, L22R and A242V. ChIP amplicon, Gas1 (−780 bp / −609 bp). (D) Co-immunoprecipitation of PRMT5 in menin immunoprecipitates from 293T cells ectopically expressing PRMT5, and either WT or MEN1 disease related menin mutants (top panel). Anti-menin immunoprecipitates from cells above were incubated with 3H-AdoMet and histone H4 for Histone Methyltransferase (HMT) assay, and 3H-methylated histone H4 was detected by autoradiography (third panel). Coomassie stain of H4 is included as a loading control (bottom panel). Error bars indicate ± s.d.
To determine whether MEN1 disease-related menin point mutations affect Gas1 expression, we complemented menin-null MEFs with either wild type (WT) or mutant forms of menin, and showed that WT and mutant menin were expressed at comparable levels based on Western blotting (Fig. 4B). However, while WT menin expression reduced Gas1 mRNA levels, mutant menin lost or partially lost their ability to repress Gas1 expression (Fig. 4B). ChIP assay showed that the menin mutants largely retained their ability to bind to the Gas1 promoter, but failed to impart the H4R3m2s mark at the promoter (Fig. 4C, and Supplementary Fig. S4C). As a control, no changes in menin binding or histone H4R3 methylation were observed at the actin promoter (Supplementary Fig. S4D).
To determine whether the mutations affect the ability of menin to interact with PRMT5, we ectopically expressed menin and PRMT5 in 293 cells (Supplementary Fig. S5), and observed that the interaction between menin and PRMT5 was reduced considerably in the menin mutants upon comparison to WT (Fig. 4D, top panel). Furthermore, HMT assay with the immunoprecipitated menin complex, 3H-SAM, and recombinant histone H4 showed that the histone H4 methylating activity was dramatically reduced in the mutant menin immune complexes compared to WT menin (Fig. 4D, third panel). Collectively, these results suggest that menin-mediated repressive H4R3 methylation (H4R3m2s) at the Gas1 promoter plays a role in suppressing MEN1 tumorigenesis.
Men1 excision in primary islets results in increased Hh signaling
To determine whether Men1 is also crucial for regulating Gas1 expression and Hh signaling, we isolated primary islets from control Men1l/l and Men1l/l;Rip-cre mice (8 mos. old), and qRT-PCR showed that Men1 excision resulted in undetectable Men1 mRNA levels (Fig. 5A), but increased mRNA levels of Gas1 (Fig. 5B), Gli1 (Fig. 5C) and Ptch1 (Fig. 5D), indicative of enhanced Hh signaling. These results suggest that menin plays a crucial role in vivo in pancreatic islets in repressing the expressions of Gas1, Gli1 and Ptch1, thereby suppressing Hh signaling.
Figure 5.

Loss of Men1 correlates with activated Hedgehog signaling in Men1-null mice. (A–D) Pancreatic islets were isolated from 8 mo. old Men1l/l; RipCre and control Men1l/l mice (n = 4 mice) and qRT-PCR was used to quantitate the mRNA levels of Men1, p < 0.0001 (A), Gas1, p < 0.0044 (B), Gli1, p = 0.0538 (C) and Ptch1, p = 0.0258 (D). Error bars indicate ± s.d.
Treatment of Men1-excised mice harboring insulinomas by a pharmacological SMO inhibitor reduces proliferation of insulinoma cells, and inhibits Hh signaling
Effective treatment for MEN1 tumors is not yet available. An Hh signaling inhibitor, GDC-0449, is proven safe and effective for treating human basal cell carcinoma and medulloblastomas (29). Since the mouse MEN1 tumor model largely phenocopies the human MEN1 tumor syndrome (30, 31), we treated 8-mo. old Men1l/l; Rip-cre mice, which developed insulinomas, with either control vehicle or GDC-0449 for 4 weeks (Fig. 6A), and found that GDC-0449 treatment significantly reduced the number of BrdU positive cells in insulinomas (Figs. 6B and 6C), by approximately 60 % (Fig. 6D). The marked impact of inhibiting Hh signaling on suppressing neuroendocrine tumors is underscored by the common practice of using the mitotic index (proliferation) as a common parameter for grading and prognosis of human neuroendocrine tumors (32). GDC-0449 treatment did not affect the area of the insulinoma (Supplementary Fig. S6A), likely because of the limited duration of the treatment and/or relative slow growth of neuroendocrine tumors. However, the treatment reduced the level of blood insulin, an indicator of secreted insulin from insulinomas (Supplementary Fig. S6B). This is consistent with the reported observation that mutation of SMO in adult mice reduces the production of insulin in islets (33). Moreover, qRT-PCR showed that expression of Ptch1, a Hh signaling target, was reduced in isolated islets from Men1l/l; pdxCreER mice treated with GDC-0449 (Fig. 6E) compared to vehicle-treated mice, indicating that GDC-0449 was effective in suppressing Hh signaling. We cannot rule out that the SMO inhibitor also, at least, partially inhibits Hh signaling from stromal cells surrounding the Men1-deleted tumor cells that act in a paracrine fashion. Nevertheless, our findings clearly indicate that GDC-0449 effectively suppresses proliferation of MEN1 tumor cells in vivo, link menin to suppression of Hh and uncovers the Hh signaling pathway as a promising target for improving therapy against neuroendocrine tumors.
Figure 6.
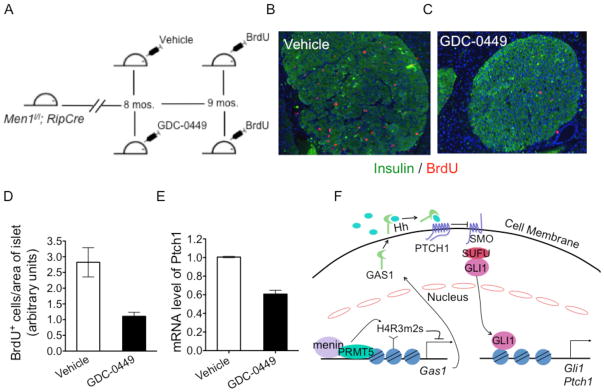
Inhibition of Hedgehog (Hh) signaling in Men1-excised mice results in decreased islet cell proliferation. (A) Scheme for inhibition of the Hedgehog (Hh) pathway with GDC-0449 in Men1-excised mice. (B–C) Immunofluorescence for BrdU and insulin in pancreas of Men1-excised mice gavaged with either vehicle (B), or GDC-0449 (C) for four weeks at a dose of 100 mg/kg b.i.d. (D) Quantitation of BrdU incorporation in islets of mice above (same as B, C). (E) Quantitative RT-PCR (qRT-PCR) for Ptch1 mRNA in islets of Men1l/l;pdxCreER mice gavaged for 10 days with either vehicle or GDC-0449 at a dose of 100 mg/kg b.i.d., p = 0.0010. (F) A model for menin-PRMT5-mediated inhibition of Hh signaling through epigenetic regulation of GAS1. Error bars indicate ± s.d.
Discussion
While menin regulates cell cycle genes and represses cell proliferation (34), it has been under-explored as to what signaling pathways are dysregulated by menin mutations. Our findings suggest that normally menin “gates” the Hh signaling threshold, at least partly, by repressing Gas1 to prevent overt activation of Hh signaling in islet cells. Men1 mutations in some islet cells may stochastically trigger overt Hh signaling, which can induce expression of pro-proliferative genes such as cyclin D1 (35), thereby increasing cell proliferation. Consistently, expression of Gli1 and Ptch1, an indicator of activated Hh signaling, was increased in primary islets from Men1-excised mice. The link between menin and Hh signaling represents a new mechanism for menin-mediated tumor suppression (Fig. 6F).
We found that menin directly interacts with PRMT5, which catalyzes repressive H4R3m2s (27). Both menin and PRMT5 directly bound to the promoter of Gas1, a component of the Hh signaling pathway. Menin is crucial for recruitment of PRMT5 to and for effective methylation of H4R3 at the Gas1 promoter (Fig. 4A), a novel means for menin-mediated repression of gene expression. However, it must be noted that it remains unclear how menin is recruited to the Gas1 promoter, and whether menin directly binds the Gas1 promoter DNA to repress Gas1 expression. In addition to PRMT5, the repressive histone H4R3m2s mark is catalyzed by another type II PRMT enzyme, PRMT7 (36, 37). However, we found that only PRMT5 was detected in the menin immunoprecipitates while PRMT7 did not co-immunoprecipitate with menin (Supplementary Fig. S7). This clearly indicates that the menin-dependant, histone H4R3m2s mark at the Gas1 promoter can be attributed to PRMT5, and not PRMT7.
Our data shows that menin immunoprecipitates possess HMT activity towards recombinant histone H4, and that this activity is lost in certain disease-related Men1 mutations resulting from compromised interaction with PRMT5 (Fig. 4D). It has previously been reported that menin immunoprecipitates have HMT activity towards histone H3 and not histone H4 (25), while we clearly observed HMT activity towards recombinant histone H4 in our studies. It is possible that the IP conditions used by Hughes et. al. preferentially retains MLL activity that methylates H3K4. Alternatively, the MLL enzymatic activity towards histone H3 might be more robust than the PRMT5 activity towards histone H4 in the menin immunoprecipitates. This would result in preferential methylation of histone H3 when a mixture of core histones is used. In contrast, purified recombinant histone H4 alone was used in our study, thus favoring observation of histone H4 methylation. Nevertheless, our data clearly show that menin interacts with PRMT5, and the associated histone arginine methylation activity is necessary for repressing Gas1 and the canonical Hh signaling pathway.
While multiple factors have been previously reported to affect Hh signaling, such as PKA-mediated phosphorylation of GLI proteins and proteolysis of GLI proteins (16), it has been poorly understood as to whether and how epigenetic factors affect Hh signaling. We found that both menin and PRMT5 are crucial for PRMT5-mediated H4R3m2s at the Gas1 promoter. PRMT5-mediated H4R3m2s suppresses gene expression of beta globin genes in hematopoietic cells partly through recruiting DNMT3A via increasing repressive mark H4R3m2s at the promoter to silence the gene by promoter DNA methylation (38). Our results reveal the PRMT5-mediated epigenetic mechanism in repressing Hh signaling and suppression of tumorigenesis.
Our studies involving MEN1 disease-related menin mutants showed that certain point mutants failed to suppress the expression of Gas1 (Figs. 4B and 4C), suggesting that menin-mediated suppression of Gas1 and canonical Hh signaling may be relevant to its function in suppressing MEN1 tumor syndrome. Furthermore, inhibition of Hh signaling with the SMO inhibitor GDC-0449 markedly reduces tumor cell proliferation in a MEN1 mouse model (Fig. 6D), indicating that canonical Hh signaling plays a significant role in proliferation of Men1-excised islet cells. It is noteworthy that GDC-0449 inhibition of SMO may have additional effects besides restoring the role of menin in repressing Gas1, as inhibition of Hh signaling in a transgenic mouse model in which the artificial Rip-driven SV40 T antigen promotes development of insulinoma, represses growth of the transgene-induced islet tumors (39). However, we found, for the first time, that Men1 mutation in beta cells in vivo leads to enhanced Hh signaling, which may contribute to increasing beta cell proliferation.
The normal role of Hh signaling in regulating pancreatic islets and beta cells is complex, influenced by temporal and spatial factors (33). Hh ligand stimulates production of insulin in cultured beta cells (40), while knock-out of SMO in pancreatic epithelial cells initially reduces the size of islets during embryonic development and decreases total insulin production in adult mice (33). Hh ligand expression or Hh signaling is also increased in human gastrointestinal neuroendocrine tumors or in mouse small-cell lung cancer (41, 42). Our results show that Men1 ablation leads to increase in expression of Hh target genes in pancreatic islets, and the SMO inhibitor suppresses proliferation of MEN1 tumors. However, MEN1 deletion in mice does not result in the typical Hh pathway-related tumors such as medulloblastoma. These findings are consistent with the notion that menin/PRMT5 modulates (but does not completely block) Hh signaling partly through reducing Gas1 expression. It is likely that menin does so in endocrine cells like beta cells because menin is more abundantly expressed in beta cells (24). Similarly, it has been shown that Men1 excision in the liver has no affect on proliferation (43), thus highlighting the tissue-specific effect of Men1 ablation. Although PRMT5 can promote tumorigenesis in multiple tissues via regulating various partners such as p53 (44), Cyclin D1 (45) and Jak2 (46), it may partner with menin to dampen Hh signaling in certain endocrine cells. In aggregate, our findings, for the first time, reveal that MEN1 mutation leads to enhanced Hh signaling via a previously unknown epigenetic mechanism, and pharmacological inhibition of this pathway significantly reduces proliferation of Men1-mutated pancreatic tumors in mice. As over 40 % of pancreatic neuroendocrine tumors harbor somatic mutations in the MEN1 gene (47), Hh antagonists may be valuable to improve therapy for neuroendocrine tumors with MEN1 mutation.
Supplementary Material
Acknowledgments
We thank Dr. J. A. Diehl for myc-PRMT5 plasmid construct and stimulating discussions, Dr. Chaoxing Yuan at Penn Proteomics facility for mass spectrometry analysis and Dr. Allen Bale for critically reading the manuscript. We thank our colleagues for critically reading the manuscript and thank Shivani Sethi for editing the manuscript.
Grant Support
This work was supported in part by grants from the NIH (R01-CA-113962 and R01-DK085121 to XH, R01 DK084963 to CMF) and Caring for Carcinoid Foundation-AACR Grant Care for Carcinoid Foundation (11-60-33XH).
Footnotes
Conflict of Interest: No conflicts of interests are declared by any of the authors.
Author Contribution
Z.F. performed the HMT assays, D.V.I. and A.T. carried out menin-PRMT5 interaction studies. G.J., C-M.F., and T.C. assisted in experimental design and discussion. B.G. performed all other experiments described in the manuscript. B.G. and X.H. conceived the research and wrote the manuscript.
References
- 1.Libe R, Bertherat J. Molecular genetics of adrenocortical tumours, from familial to sporadic diseases. Eur J Endocrinol. 2005;153:477–87. doi: 10.1530/eje.1.02004. [DOI] [PubMed] [Google Scholar]
- 2.Burgess JR, Nord B, David R, et al. Phenotype and phenocopy: the relationship between genotype and clinical phenotype in a single large family with multiple endocrine neoplasia type 1 (MEN 1) Clin Endocrinol (Oxf) 2000;53:205–11. doi: 10.1046/j.1365-2265.2000.01032.x. [DOI] [PubMed] [Google Scholar]
- 3.Farrell WE, Azevedo MF, Batista DL, et al. Unique gene expression profile associated with an early-onset multiple endocrine neoplasia (MEN1)-associated pituitary adenoma. J Clin Endocrinol Metab. 2011;96:E1905–14. doi: 10.1210/jc.2011-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marx SJ. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat Rev Cancer. 2005;5:367–75. doi: 10.1038/nrc1610. [DOI] [PubMed] [Google Scholar]
- 5.Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–7. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 6.Lemmens I, Van de Ven WJ, Kas K, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997;6:1177–83. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- 7.Balogh K, Racz K, Patocs A, Hunyady L. Menin and its interacting proteins: elucidation of menin function. Trends Endocrinol Metab. 2006;17:357–64. doi: 10.1016/j.tem.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol. 2007;265–266:34–41. doi: 10.1016/j.mce.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milne TA, Hughes CM, Lloyd R, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–54. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karnik SK, Hughes CM, Gu X, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–64. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–12. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izzi Luisa CF. Boc and Gas1 Each Form Distinct Shh Receptor Complexes with Ptch1 and Are Required for Shh-Mediated Cell Proliferation. Developmental Cell. 2011;20:788–801. doi: 10.1016/j.devcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen BL, Song JY, Izzi L, et al. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell. 2011;20:775–87. doi: 10.1016/j.devcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinelli DC, Fan CM. Gas1 extends the range of Hedgehog action by facilitating its signaling. Genes Dev. 2007;21:1231–43. doi: 10.1101/gad.1546307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, McMahon AP, Allen BL. Shifting paradigms in Hedgehog signaling. Curr Opin Cell Biol. 2007;19:159–65. doi: 10.1016/j.ceb.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9:873–86. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- 18.Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–51. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 19.Taipale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–9. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 20.Schnepp RW, Chen YX, Wang H, et al. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–15. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.La P, Schnepp RW, CDPA, CS, Hua X. Tumor suppressor menin regulates expression of insulin-like growth factor binding protein 2. Endocrinology. 2004;145:3443–50. doi: 10.1210/en.2004-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee CS, Buttitta L, Fan CM. Evidence that the WNT-inducible growth arrest-specific gene 1 encodes an antagonist of sonic hedgehog signaling in the somite. Proc Natl Acad Sci U S A. 2001;98:11347–52. doi: 10.1073/pnas.201418298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YX, Yan J, Keeshan K, et al. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci U S A. 2006;103:1018–23. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Gurung B, Wu T, Wang H, Stoffers DA, Hua X. Reversal of preexisting hyperglycemia in diabetic mice by acute deletion of the Men1 gene. Proc Natl Acad Sci U S A. 2010;107:20358–63. doi: 10.1073/pnas.1012257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–97. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 26.Friesen WJ, Wyce A, Paushkin S, et al. A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem. 2002;277:8243–7. doi: 10.1074/jbc.M109984200. [DOI] [PubMed] [Google Scholar]
- 27.Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol. 2004;24:9630–45. doi: 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Res. 2003;63:4836–41. [PubMed] [Google Scholar]
- 31.Crabtree JS, Scacheri PC, Ward JM, et al. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Mol Cell Biol. 2003;23:6075–85. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strosberg J, Nasir A, Coppola D, Wick M, Kvols L. Correlation between grade and prognosis in metastatic gastroenteropancreatic neuroendocrine tumors. Hum Pathol. 2009;40:1262–8. doi: 10.1016/j.humpath.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Lau J, Hebrok M. Hedgehog signaling in pancreas epithelium regulates embryonic organ formation and adult beta-cell function. Diabetes. 2010;59:1211–21. doi: 10.2337/db09-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu T, Zhang X, Huang X, Yang Y, Hua X. Regulation of cyclin B2 expression and cell cycle G2/m transition by menin. J Biol Chem. 2010;285:18291–300. doi: 10.1074/jbc.M110.106575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–67. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JH, Cook JR, Yang ZH, et al. PRMT7, a new protein arginine methyltransferase that synthesizes symmetric dimethylarginine. J Biol Chem. 2005;280:3656–64. doi: 10.1074/jbc.M405295200. [DOI] [PubMed] [Google Scholar]
- 37.Karkhanis V, Wang L, Tae S, Hu YJ, Imbalzano AN, Sif S. Protein arginine methyltransferase 7 regulates cellular response to DNA damage by methylating promoter histones H2A and H4 of the polymerase delta catalytic subunit gene, POLD1. J Biol Chem. 2012;287:29801–14. doi: 10.1074/jbc.M112.378281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao Q, Rank G, Tan YT, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009;16:304–11. doi: 10.1038/nsmb.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fendrich V, Wiese D, Waldmann J, et al. Hedgehog inhibition with the orally bioavailable Smo antagonist LDE225 represses tumor growth and prolongs survival in a transgenic mouse model of islet cell neoplasms. Ann Surg. 2011;254:818–23. doi: 10.1097/SLA.0b013e318236bc0f. discussion 23. [DOI] [PubMed] [Google Scholar]
- 40.Thomas MK, Rastalsky N, Lee JH, Habener JF. Hedgehog signaling regulation of insulin production by pancreatic beta-cells. Diabetes. 2000;49:2039–47. doi: 10.2337/diabetes.49.12.2039. [DOI] [PubMed] [Google Scholar]
- 41.Shida T, Furuya M, Nikaido T, et al. Sonic Hedgehog-Gli1 signaling pathway might become an effective therapeutic target in gastrointestinal neuroendocrine carcinomas. Cancer Biol Ther. 2006;5:1530–8. doi: 10.4161/cbt.5.11.3458. [DOI] [PubMed] [Google Scholar]
- 42.Fendrich V, Waldmann J, Esni F, et al. Snail and Sonic Hedgehog activation in neuroendocrine tumors of the ileum. Endocr Relat Cancer. 2007;14:865–74. doi: 10.1677/ERC-07-0108. [DOI] [PubMed] [Google Scholar]
- 43.Scacheri PC, Crabtree JS, Kennedy AL, et al. Homozygous loss of menin is well tolerated in liver, a tissue not affected in MEN1. Mamm Genome. 2004;15:872–7. doi: 10.1007/s00335-004-2395-z. [DOI] [PubMed] [Google Scholar]
- 44.Durant ST, Cho EC, La Thangue NB. p53 methylation--the Arg-ument is clear. Cell Cycle. 2009;8:801–2. doi: 10.4161/cc.8.6.7850. [DOI] [PubMed] [Google Scholar]
- 45.Aggarwal P, Vaites LP, Kim JK, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010;18:329–40. doi: 10.1016/j.ccr.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu F, Zhao X, Perna F, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19:283–94. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.