

Abstract
Guanine nucleotide (G)-protein coupled receptor (GPCR) linked cell signaling cascades are initiated upon binding of a specific agonist ligand to its cell surface receptor. Linking multiple heterologous ligands that simultaneously bind and potentially cross-link different receptors on the cell surface is a unique approach to modulate cell responses. Moreover, if the target receptors are pre-selected, based on analysis of cell specific expression of a receptor combination, then the linked binding elements may provide enhanced specificity of targeting to the cell type of interest; i.e., only to cells that express the complementary receptors. Two receptors whose expression is relatively specific, as a combination, to the insulin secreting β-cell of the pancreas, are the sulfonylurea-1 (SUR1) and the glucagon-like peptide-1 (GLP-1) receptors. A heterobivalent ligand was assembled of the active fragment of GLP-1 ([Phe12, Arg36] 7-36 GLP-1) and glibenclamide,a small organic ligand to the SUR1. The synthetic construct was labelled with Cy5 or Europium chelated in DTPA to evaluate binding to β-cell lines using fluorescence microscopy or time-resolved saturation and competition binding assays, respectively. Once the ligand binds to β-cells, it is rapidly capped and presumably removed from the cell surface via endocytosis. The bivalent ligand had an affinity ~3 fold higher than monomeric Europium labelled GLP-1, likely due to cooperative binding to the complimentary receptors on the βTC3 cells. The high affinity binding was lost in the presence of either unlabelled monomer demonstrating that interaction with both receptors is required for the enhanced binding at low concentrations. Importantly, bivalent enhancement was accomplished in a cell system with physiological levels of expression of the complementary receptors, indicating that this approach may be applicable for β-cell targeting in vivo.
Keywords: binding assay, lanthanide-based time-resolved fluorescence, molecular dynamics, peptidomimetics, solid-phase synthesis
Introduction
Pancreatic β-cell loss and dysfunction underlies the presentation of overt diabetes. This deficiency in function provides a rationale to develop novel therapeutic strategies. Moreover, to determine the success of these strategies, and to better understand diabetic pathology, reliable methods are required for assessment of β-cell regeneration or destruction. In particular, the ability to monitor changes in β-cell mass (BCM) and β-cell function is critical to assess the developing disease state and therapeutic responses.[1–3] A primary limitation for the analysis and therapy of β-cells is low specificity of current agents. It has been proposed that selection and simultaneous binding to multiple receptors expressed on a cell surface, using multivalent ligands, will improve the selectivity of cell targeting.[4–8]
Multivalent ligands are composed of multiple copies of the same (homo) or unique (hetero) ligands connected via linkers or a scaffold.[9] Dynamic modelling suggests that multivalent interactions can lead to high affinity and specific binding,[5–7,10,11] which may be exploited to target contrast and therapeutic agents.[12,13] In previous studies,[11,12,14] we demonstrated that the structure of the linker/scaffold used to combine the binding elements has considerable influence on binding affinities.[12] Multivalent compounds must present ligands simultaneously to their cognate receptors with minimal possible entropic penalty. Although rigid linkers have low internal entropy, they may not be ideal. Indeed, melanocyte stimulating hormone (MSH) ligands connected with short rigid phenyl, biphenyl, and terphenyl linkers did not enhance binding affinity at the hMC4R.[15,16] Conversely, high flexibility may present a high conformational entropic cost. Interestingly, some investigators concluded that the flexibility of the linker does not have significant effect on the avidity.[17] Therefore, flexible linkers with lengths longer than optimal distance may still be effective. In addition, linkers must have good physicochemical properties (e.g. solubility, low nonspecific binding affinity) and low toxicity.
Based on modelling (MacroModel 9.1) using Monte Carlo conformational searches and Molecular Dynamic (MD) simulations,[14,18,19] we chose short flexible ethylene glycol spacers (PEGO) in combination with semi-rigid Pro-Gly (PG) repeats as linkers for the heterobivalent ligand. The secondary structure of the Pro-Gly repeats was found to form a structure typical for Type II polyproline helices with a pitch of 5 Å per 7 residues in its lowest conformation. However, MD simulation showed that the pitch could be extended up to 12 Å per turn. This observation suggests that these repeats might promote a population of low-to-medium energy conformers that could broaden the helix to reach longer distances.[12] Both components of the linker (PEGO and PG) fit well into a modular solid phase synthesis scheme wherein ligands or linker units are systematically built on the solid support. The combination of PEGO and PG repeats also allows linkers to be constructed with various lengths and rigidities.
Using this approach for linking binding domains, we prepared a series of melanocyte stimulating hormone (MSH), δ-opioid, and cholecystokinin (CCK) analogs. Studies with these multivalent ligands demonstrate that enhanced binding affinity and selectivity can be obtained via multivalent interactions, which also greatly enhance targeting specificity [7,14,20]and may potentially provide unique therapeutic properties. In vivo selectivity was recently demonstrated using a mouse model wherein bilateral flank tumors expressing only one (control) or both targets (the melanocortin (MC1R) and cholecystokinin (CCK2R) receptors), were injected with a Eu-labelled MSH(7)/CCK(6) ligand. The fluorescent signal was highly retained only in tumors expressing both complimentary receptors (~13 fold compared to MC1R expressing tumors and ~6 fold compared to CCK2R expressing tumors).[13]
In order to produce a potentially β-cell specific therapeutic agent using a multivalent approach, a target receptor pair, which as a combination is relatively specific to the β-cell, was required. Based on genetic profiling studies,[21,22] and previous in depth binding analysis of potential ligands, the glucagon-like peptide-1 receptor (GLP-1R) and the sulfonylurea-1 receptor (SUR1) were identified as a receptor target pair that is relatively specific to β-cells.[23–26] The GLP-1R, a class B GPCR, has been difficult to target with small molecule drugs. However, GLP-1R agonists have been modified to produce several stable analogs, such as clinically proven Exenatide or Liraglutide.[27–30] Partial peptide truncation to residues 7-36 is permitted without loss of activity, and the remaining N- and C-termini are both required for activity. Furthermore, a lysine in position 26 can be modified with long fatty chains or flexible oligoethyleneglycol without loss of function.[31] With this information in hand, we designed and synthesized a prospective β-cell specific targeting and therapeutic agent based on GLP-1 and glibenclamide (SUR1 ligand).
Results and Discussion
Presented are the design, synthesis and initial evaluation of a heterobivalent ligand targeted to two heterologous receptors expressed on the surface of pancreatic β-cells in islets of Langerhans. The GLP-1 and SUR1 receptors were chosen as targets, because as a combination they represent a relatively specific β-cell target.[21–23,25,32]
Ligand Synthesis
A GLP-1 peptide with truncation to residues 7-36 was used as the starting backbone. The lysine in position 26 was modified with a flexible oligoethyleneglycol [31] to allow conjugation of the second component, glibenclamide (the SUR1 ligand).
Glibenclamide does not contain a functional group suitable for direct attachment to GLP-1. Therefore, glibenclamide was modified by carboxylate (compound 6, Scheme 1) to allow solid-phase conjugation to resin-bound GLP-1 peptide. From structure- activity relation analyses, glibenclamide is relatively inert to modification in its benzamide ring, allowing even hydrophilic carbohydrates to be conjugated without loss of activity.[24]The glibenclamide carboxy-derivative 6 was prepared in six synthetic steps starting with commercially available methyl 5-chloro- salicylate in overall yield (35%) and high purity (96% by HPLC).
Scheme 1.
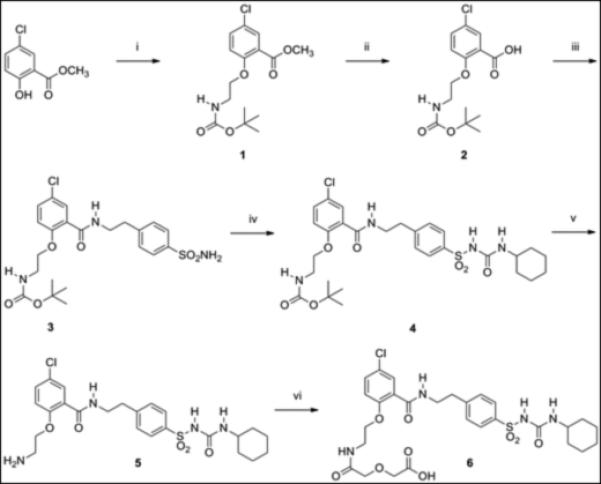
Synthesis of glibenclamidecarboxy-derivative 6.(i) Boc-glycinol, PPh3, DEAD, THF, 0 °C to room temp. (ii) 2.0M NaOH, methanol, room temp. (iii) (a) ethyl chloroformate, NMM, DMF, 0 °C. (b) 4-(2-aminoethyl)benzenesulfonamide, DMF, 0 °C to room temp. (iv) cyclohexylisocyanate, CuI, DMF, room temp. (v) TFA, room temp. (vi) diglycolic anhydride, DMF, room temp.
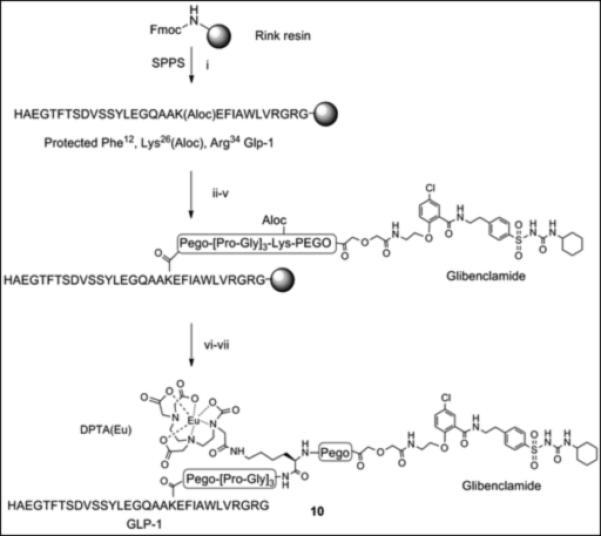
The glibenclamidecarboxy derivative 6 fit well in our modular solid-phase synthetic scheme. This modular approach provides flexibility in heterovalent ligand design and synthesis allowing for repetitive tuning based on analysis of initial construct binding properties. The glibenclamidecarboxy derivative 6 was conjugated to the resin-bound GLP-1 via lysine side chain in position 26 as shown in Scheme 2. Briefly, the GLP-1 resin intermediate 7, prepared from Rink amide resin by Fmoc/tBu solid phase synthesis, was treated with Pd(0) to remove the Aloc group from lysine in position 26 while maintaining Boc/tBu permanent protection. The free amine of the lysine side chain was sequentially coupled with linkers (PEGO and Pro-Gly repeats), cysteine or glycine, followed by PEGO and then with derivative 6 as illustrated in Scheme 2. The resin 8 was treated with TFA scavenger cocktail, and the crude thiol intermediate was purified by HPLC and characterized by high-resolution Mass Spec. A cysteine residue was conjugated with a maleimide derivative of Cy5 to form Cy5 labelled GLP-1/Glb 9 (Cy5-GLP-1/Glb). A Europium labelled GLP-1/Glb 10 (Eu-GLP-1/Glb) was prepared similarly as 9; the cysteine in Scheme 2 was substituted with Lys(Aloc), the orthogonal Aloc protection was removed and free ε-lysine functionalized with DTPA (Scheme 3). DTPA-GLP-1/Glb was cleaved off the resin and labelled with Europium as described.[4] Similarly, a non-labelled GLP-1/Glb compound 11 (cysteine was substituted with glycine), and Europium-labelled GLP-1 12 (Eu-GLP-1) was prepared in high yield and purity.
Scheme 2.
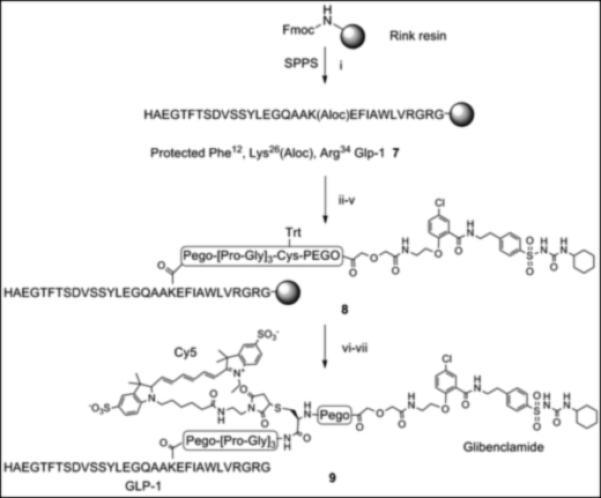
Synthesis of Cy5 labelled GLP-1/Glb 9. (i) Fmoc/tBu solid phase synthesis, ε-amine of Lys26 protected with Aloc. (ii) Pd(0) tetrakis Ph3P, N,N-dimethyl barbituric acid, DCM, 30 min., room temp. (iii) Fmoc-PEGO, DIC, DCM, room temp. (iv) Fmoc/tBu solid phase synthesis, Pro-Gly repeats and Cys(Trt). (v) 6, DIC, HOBt, room temp, overnight. (vi) TFA, scavenges, 4 hrs, room temp. (vi) Cy5-maleimide, DMF, room temp.
Scheme 3.
Evaluation of β-cell Binding
The β-cell line βTC3 was chosen as a platform for analysis of ligand binding, because mRNA for both of the complimentary receptors are expressed (Figure S4). Protein expression was verified by western blotting with receptor specific antibodies (Figure S3). Conversely, the INS-1F line expressed relatively little GLP-1R. Receptor number was evaluated using population based saturation binding analysis with an estimated 80,000 SUR and 250,000 GLP-1R expressed per cell (Figure S2)
The Cy5-labelled GLP-1/Glb 9 was used to evaluate single cell binding using fluorescence microscopy (Figure 1). The bound ligand caps on the cell surface (Figure 1B) and is moved into the cell within 3 min. Moreover, cells expressing both receptors bind significant ligand at concentrations where little binding is observed to cells that express only one of the complimentary receptors (i.e., SUR1; Figure 2).
Figure 1.
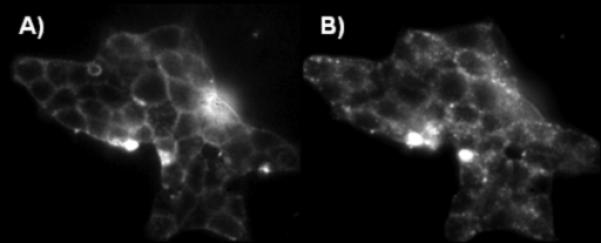
Live βTC3 cells expressing the GLP-1R and SUR1 receptor pair bind 25 nm Cy5-labeled GLP-1/Glb 9. Binding at 2 A) and 5 B) min following addition of Cy-5-GLP-1/Glb 9.
Figure 2.
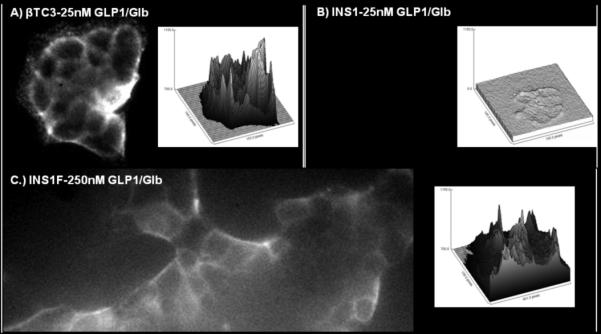
Binding of Cy5-GLP-1/Glb 9 to βTC3 A) and INS-1F cells B & C). Cells were incubated with 25 nm ligand for 1 min then washed with images acquired 2 min following the wash. Surface plots to the right of each image show fluorescence intensity (y-axis) at each pixel within the image. At 25 nm ligand, fluorescence is high on the βTC3 cells A) but low for the INS-1F B), while at 250 nm ligand binding to the INS-1F cells is greatly increased C). Since the INS-1F cells only express the SUR1, the observation of binding only at 250 nm indicates that both receptors are required to bind significant ligand at lower concentrations; i.e., 25 nm.
Eu-GLP-1/Glb 10 binding was measured in the presence of individual monomeric ligands to further evaluate the requirement of bivalent interactions for high affinity binding. At negligible concentrations of monomers (fM), binding of GLP-1/Glb (at 15 nM) was significant, but in the presence of 1.6 nM of either monomer alone, GLP-1/Glb 10 binding was reduced substantially (Figure 3). This finding indicates that high affinity binding is dependent on bivalent interactions, and that the component monomers effectively compete bivalent binding at relatively low concentrations. Previously we demonstrated that a heterobivalent ligand composed of α-MSH and CCK-6 bound with enhanced affinity to cells over-expressing receptors in the range of 1 million copies per cell. Importantly, the present studies show that the heterobivalent Cy5-GLP-1/Glb 9 binds to cells that express a naturally occurring number of receptors (~100,000 copies per cell, Figure S2), indicating that the ligand can ‘cross-link’ these receptors at physiologically relevant receptor levels.
Figure 3.
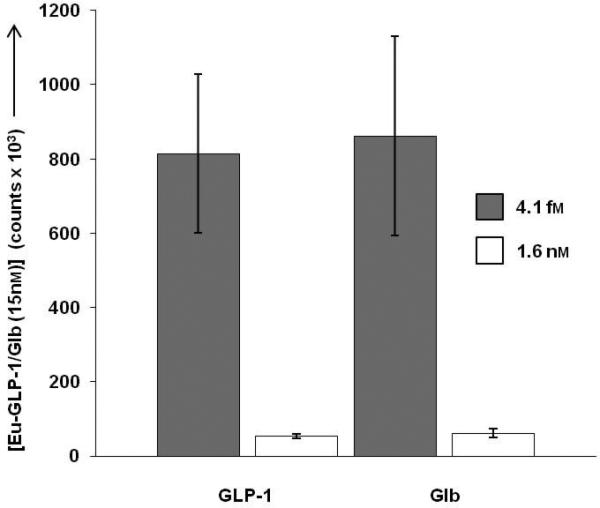
Competition of 15 nm Eu-GLP-1/Glb 10 by unlabelled monomers in live βTC3 cells, measured by time-resolved fluorescence assays.[33] Eu-GLP-1/Glb 10 binding was measured on cell populations in the presence of 4.1 fm and 1.6 nm unlabeled GLP-1 or Glb. Incubations were 1 hr. When the concentration of either monomer is increased to levels where receptor binding occurs, bivalent binding is lost demonstrating that the high avidity interactions are dependent on binding of both moieties of Eu-GLP-1/Glb 10 to the complimentary receptors.
Estimation of Binding Constants for the Bivalent GLP-1/Glb
Shown in Figure 4 are the total and non-specific binding curves for the Eu-GLP-1 and Eu-GLP-1/Glb 10 ligands. The presence of Glb in the bivalent ligand caused an elevated non-specific binding (compared to Eu-GLP-1), and an overall linear signal increase with increasing ligand concentration. However, the signal:noise ratio remained sufficient for evaluation of concentration dependent specific binding up to, approximately, 100 nM ligand.
Figure 4.
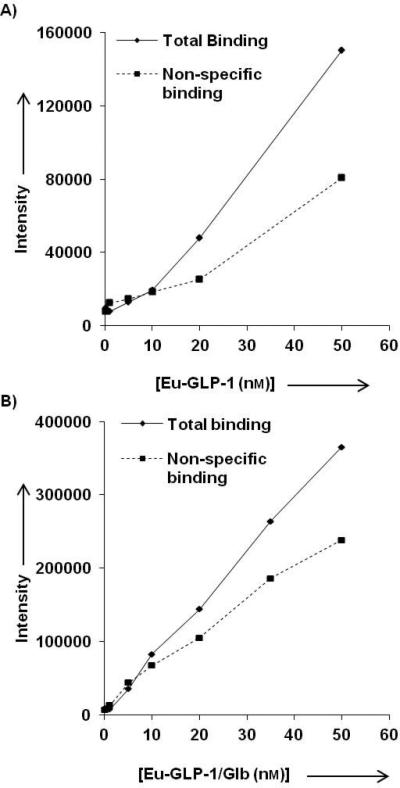
Total and non-specific binding A) Eu-GLP-1 12 and B) for Eu-GLP-1/Glb 10. Non-specific binding was determined at each concentration by competing the Eu-labeled ligands with 300 nm of their respective unlabelled ligands (GLP-1 or GLP-1/Glb 11). The non-specific binding of the Eu-GLP-1/Glb 10 increases linearly with increasing ligand concentration.
Figure 5 shows the specific binding curve for Eu-GLP-1 12 compared to that of the bivalent Eu-GLP-1/Glb 10, generated from the data presented in Figure 4. Eu-GLP-1 12 exhibits a smooth binding profile with a Kd near 20 nM. For the bivalent Eu-GLP-1/Glb 10 construct, a clear shift to the right is observed indicating a decreased binding affinity. However, there is significant binding observed at concentrations below 20 nM that is masked by the high level of binding observed at 75 nM - 100 nM. We interpret the ‘high affinity’ binding to be related to bivalent interactions that are limited by the available SUR-1, while the low affinity site is related to binding of the GLP-1 domain within the bivalent construct to spare GLP-1R, since this receptor is expressed at substantially higher copy number per cell than the SUR-1 (Figure S2). The apparent high affinity binding of Eu-GLP-1/Glb10 was evaluated in detail by focusing on ligand concentrations within a narrow range below 50 nM (Figure 6).
Figure 5.
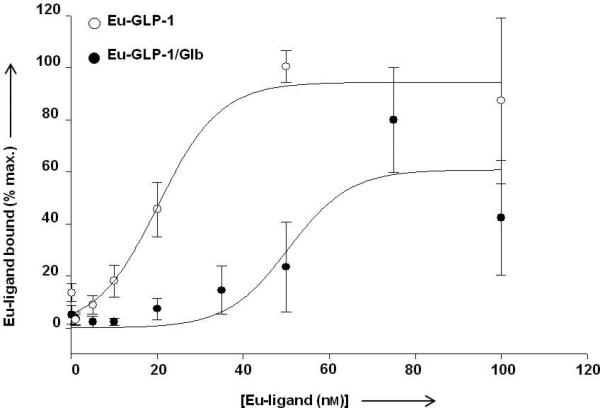
Saturation binding analysis of Eu-GLP-1 12 and Eu-GLP-1/Glb 10. To obtain estimated binding constants, data were fit using a sigmoidal relation. The data for Eu-GLP-1 12 match the fit with an R2 value of 0.99 and a Kd = 20.2 nm, while Eu-GLP-1/Glb 10 data fit with an R2= 0.90 and a Kd = 50.5 nm. Data are expressed as a percent maximum binding observed within individual experiments with an n= 6 for Eu-GLP-1 12 at concentrations below 75 nm and an n=3 for 75 nm and 100 nm. n=4 for Eu-GLP-1/Glb 10. For Eu-GLP-1 12, maximal binding was consistently observed at either 75 or 100 nm depending on the experiment.
Figure 6.
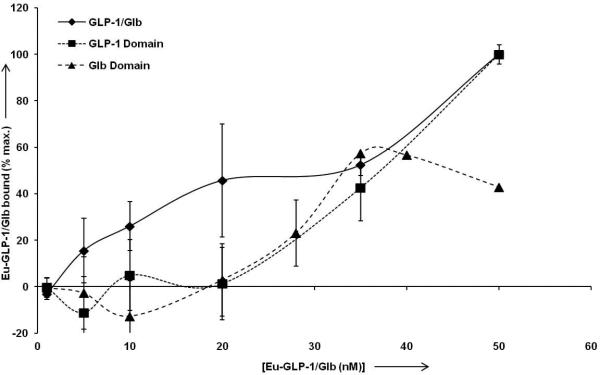
High affinity binding of Eu-GLP-1/Glb 10. For normalization of data between experiments, and for optimal display of ‘high affinity’ binding, the data are normalized to the signal observed at 50 nm Eu-labelled ligand for each individual experiment. Analysis of the Eu-GLP-1/Glb 10 (diamonds) curve between 0.1 and 35 nm indicates high affinity binding with a Kd = 10.0 nm (+/− 1.4 nm, R2= 0.97). Competition with unlabelled 300 nm Glb blocks high affinity binding (below 35 nm, solid triangles), and thereby provides analysis of the GLP-1 binding domain within the Eu-GLP-1/Glb 10. Curve fitting indicates a Kd =57.1 nm (+/− 2.5 nm, R2= 0.998) for the GLP-1. Conversely, incubation with unlabeled GLP-1 provides analysis of the Glb domain within the Eu-GLP-1/Glb10 which is estimated to bind with a Kd = 30.5 nm (+/− 3.8 nm, R2 = .83).
Data in Figure 5 suggest that a ‘high affinity’ binding mode for Eu-GLP-1/Glb 10 may exist below 50 nM that presumably is due to bivalent interactions with both complementary receptors. Fitting the data between 0.1 to 35 nM Eu-GLP-1/Glb 10 provided an estimate of the bivalent binding constant with half maximal saturation at ~10 nM. Eu-GLP-1/Glb 10 binding in the presence of unlabelled Glb (300 nM) or GLP-1 (500 nM) also was analyzed to estimate the respective binding constants for the GLP-1 and Glb domains within the Eu-GLP-1/Glb 10. Competition with unlabelled Glb blocked the high affinity binding (Figure 6), without effect on the curve shape at higher Eu-GLP-1/Glb 10 concentrations (not shown). The binding constant of the GLP-1 domain within the bivalent construct was interrogated by competition with unlabelled Glb which shifted the estimated binding constant to ~60 nM. Similarly, in the presence of saturating unlabelled GLP-1 (300 nM), the innate binding of the Glb domain within Eu-GLP-1/Glb 10 is interrogated, and a binding constant of ~ 30 nM was obtained.
Functional Activity
To determine if the ligand retains functional activity, we evaluated the effects of Eu-GLP-1/Glb 11 on insulin secretion under basal (1 mM) and glucose stimulated (20 mM) conditions. For these studies, we used the INS-832/3 β-cell line, because previously they were shown to exhibit incretin mediated potentiation of glucose stimulate insulin secretion (GSIS). [38,39] As seen in Figure 7, there was no effect of Eu-GLP-1/Glb 11 on basal insulin secretion, but there was a substantial potentiation of GSIS. A high concentration of Eu-GLP-1/Glb 11 was chosen in order to evaluate the effectiveness of each element within Eu-GLP-1/Glb 11 on secretion. Clearly, the GLP-1 domain remains functional, however the absence of activation of secretion by the Glb domain at basal glucose suggests a potential attenuation of the Glb signalling response. These initial findings indicate a need for further evaluation of signalling potential.
Figure 7.
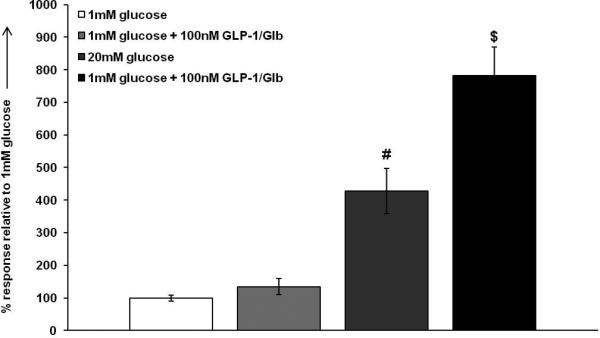
100 nm GLP-1/Glb 11 potentiates insulin secretion. 20 mm glucose causes a ~4 fold induction in insulin secretion in INS 832/3 cells. At non-stimulatory glucose, GLP-1/Glb 11 had no significant effect on basal secretion. However, GLP-1/Glb 11 substantially potentiates glucose stimulated insulin secretion in the INS 832/3 cell line by ~6 fold ( #= p<0.05 between 1 mm glucose and 20 mm glucose and $= p<0.05 between 20 mm glucose and 20 mm glucose + 100 nm GLP-1/Glb).
Conclusion
A heterobivalent ligand consisting of GLP-1 and Glb, linked by a combination of PEGO and Pro-Gly repeats, was synthesized to specifically target pancreatic β-cells. Initial analyses of ligand binding indicate that this ligand cross-links the complimentary surface receptors (i.e., GLP-1R and SUR1), that as a combination are relatively specific for β-cells. To our knowledge, this is the first time that cross-linking of multiple receptors using a heterobivalent ligand has been accomplished in a cell system without over-expression of cognate receptors. This finding is especially important for potential use under physiological conditions, such as for measurement of β-cell mass, and the development of targeted therapeutics.
Detailed binding analysis indicates that the ligand exhibits two binding domains with apparent Kd's of ~10 and 60 nM. The relatively high affinity ‘site’, dependent on bivalent interactions (Figures 2, 3 & 6), is limited in magnitude by the relatively low levels of SUR-1 expression, such that most binding occurs at higher concentrations (> 30 nM) where the ligand binds in monovalent mode with its GLP-1 domain (Figure 5 and 6). When Eu-GLP-1/Glb10 binding was evaluated in the presence of either on the unlabeled monomers, the high affinity binding was lost, and the innate affinity of the individual elements within the ligand were interrogated. The affinity of the GLP-1 domain was verified at ~ 60 nM, while the Glb domain was found to have a Kd of ~ 30 nM. Therefore, the binding affinity of the Eu-GLP-1/Glb10 in bivalent mode was shifted approximately 3 fold from the highest affinity monovalent binding element in this analysis. The estimate of 30 nM for the Glb domain is likely conservative due to of the low signal to noise at the SUR-1 (low receptor expression), and the significant non-specific binding of the Eu-GLP-1/Glb 10 (Figure 4), which limits identification of an absolute saturation point. Neverlethess, Eu-GLP-1/Glb 10 exhibits relative high affinity binding, and by vertue of the need for expression of both receptors for the high affinity binding mode (Figure 2, 3 and 6), the ligand shows, in this case, β-cell specificity.
In our recent work, we have demonstrated that large relative shifts in multivalent ligand binding affinity can be obtained when the cognate elements within the ligand have low affinity. For example, multimerization of a low affinity analog of melanocyte stimulating hormone (MSH-4), Kd ~ 3 mM) exhibits a 10 fold enhancement in affinity with dimerization and nearly 100 increase as a trimer [20]. Similar though less profound observations were made with a heterodimer synthesized from CCK-6 and MSH-7.[14] Therefore, the specificity for β-cell targeting, based on shifts in binding sensitvity between monomeric and multimeric modes, may be improved using lower affinity building blocks.
Here we demonstrate that the GLP-1/Glb retains an ability to potentiate glucose stimulates insulin secretion (GLP-1 dependent), but not activate secretion by itslef (Glb domain) at the concentration used (Figure 7). As affinities of the inherent ligands used to generate the multivalents decrease, the effect on signaling may be altered even tough they bind at low concentrations. Again, our initial data with the MSH-4 multimers suggest that signaling sensitivity tracts the measured ‘apparent‘ binding affinity,[20] so this may not be an issue as we move forward from prototype ligands such as the Eu-GLP-1/Glb10 presented here.
Experimental Section
General Synthesis
All chemical reactions were conducted under Ar atmosphere using oven-dried glassware. All chemicals were obtained from commercial sources and used without further purification. 1HNMR spectra were recorded on a Bruker-DRX-300 MHz instrument with chemical shifts reported relative to TMS (0.0 ppm) and residual DMSO (2.50 ppm). Proton-decoupled 13C NMR spectra were referenced to CDCl3 (77.0 ppm) as well as DMSO (39.51 ppm). Low resolution mass spectra were obtained on AGILENT (HP) MDS 1100 using AP-ESI. High resolution mass spectra (HRMS) were recorded on a JEOL HX110A instrument. Melting points were measured using a Thomas Hoover capillary melting point apparatus and are uncorrected.
N-α-Fmoc-protected amino acids, HBTU, and HOBt were purchased from SynPep (Dublin, CA) or from Novabiochem (San Diego, CA). Rink amide Tentagel S and R resins were acquired from Rapp Polymere (Tubingen, Germany). For the Nα-Fmoc-protected amino acids, the following side chain protecting groups were used: Arg(Ng-Pbf); Gln(Nam-Trt); Glu(O-tBu); Asp(O-tBu); His(Nim-Trt); Ser(O-tBu); Thr(O-tBu); S; Trp(Ni-Boc) and Nα-Boc-Tyr(O-tBu). An Fmoc-protected version of PEGO (1-(9H-fluoren-9-yl)-3,19-dioxo-2,8,11,14,21-pentaoxa-4,18-diazatricosan-23-oic acid) was obtained from Novabiochem (San Diego, CA). The structure is depicted in Figure S1. Reagent grade solvents, reagents, and acetonitrile for HPLC were acquired from VWR (West Chester, PA) or Aldrich-Sigma (Milwaukee, WI), and were used without further purification unless otherwise noted. The solid-phase synthesis was performed in fritted syringes using a Domino manual synthesizer obtained from Torviq (Niles, MI). DEAD (6.97 g, 40 mmol) was added to a mixture of methyl 5-chlorosalicylate (7.46 g, 40 mmol), Boc-glycinol (6.13 g, 38mmol) and triphenylphosphine (10.5 g, 40 mmol) in anhydrous THF (200 mL) at 0 °C then was stirred at this temperature for 30 min, and at room temperature overnight. The reaction mixture was concentrated under reduced pressure and the concentrated residue was diluted with hexane (400 mL). The resulting precipitate was filtered and concentrated under reduced pressure.
Synthesis of methyl-2-(2’-(tert-butoxycarbonylamino)ethoxy)-5-chlorobenzoate (1)
The residue was purified by silica gel column chromatography using a mixture of Hexane and ethyl acetate (4:1=v:v) to give methyl-2-(2’-(tert-butoxycarbonylamino)ethoxy)-5-chlorobenzoate (1, 12.5 g) a colorless oil (80 % isolated yield).
Colorless oil; 1H NMR (300 MHz, CDCl3) δ 1.45 (s, 9H), 3.56 (q, J = 5.25 Hz, 2H), 3.92 (s, 3H), 4.10 (t, J = 5.05 Hz, 2H), 5.51 (brs, 1H), 6.91 (d, J = 8.88 Hz, 1H), 7.41 (dd, J = 8.86, 2.74 Hz, 1H), 7.80 (d, J = 2.73 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 28.4, 39.8, 52.3, 69.1, 79.4, 115.4, 121.5, 125.9, 131.5, 133.4, 156.0, 157.0, 165.3; MS (ESI) m/z Calcd for C15H20ClNO5Na (M+Na)+ 352.1, obsd 352.0.
Synthesis of 2-(2’-(tert-butoxycarbonylamino)ethoxy)-5-chlorobenzoic acid (2)
To a solution of compound 1 (9.99 g, 30.3 mmol) in methanol (60 mL) was added an aqueous solution of 2N NaOH (30.3 mL, 60.6 mmol) and the resulting mixture was stirred at room temperature for 2 hours. The reaction mixture was diluted with 10 % aqueous Na2CO3 (100 mL) and washed with diethyl ether (200 mL). The aqueous layer was acidified to pH 3 by addition of 1 M citric acid and extracted with ethyl acetate. The organic layer was washed with H2O, dried over anhydrous MgSO4, filtered and concentrated. Recrystallization using a mixture of chloroform and hexane gave 2-(2’-(tert-butoxycarbonylamino)ethoxy)-5-chlorobenzoic acid (2, 8.75 g) as a white solid at 91 % isolated yield.
White solid, mp = 118-120 °C; 1H NMR (300 MHz, DMSO-d6) δ 1.37 (s, 9H), 3.28 (q, J = 5.96 Hz, 2H), 4.04 (t, J = 6.00 Hz, 2H), 6.85 (t, J = 5.43 Hz, 1H), 7.17 (d, J = 8.96 Hz, 1H), 7.53 (dd, J = 8.89, 2.80 Hz, 1H), 7.61 (d, J = 2.76 Hz, 1H), 12.9 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 28.2, 39.8, 67.9, 77.9, 116.1, 123.5, 124.2, 129.9, 132.5, 155.6, 156.1, 166.0; MS (ESI) m/zCalcd for C14H18ClNO5Na (M+Na)+ 338.1, obsd 338.0.
Synthesis of 4-[β-(2-(2’-(tert-butoxycarbonylamino)ethoxy)-5-chlorobenzene-carboxamido)ethyl]benzenesulfonamide (3)
To a mixture of compound 2 (1.58 g, 5.0 mmol) and N-methylmorpholine (0.556 g, 5.5 mmol) in DMF (15 mL) was added a solution of ethyl chloroformate (0.597 g, 5.5 mmol) in DMF (10 mL) at 0 °C. After 30 min. of stirring at this temperature, a solution of 4-(2-aminoethyl)benzenesulfonamide (1.05 g, 5.25 mmol) in DMF (15 mL), pre-cooled at 0 °C, was added. The resulting mixture was stirred at 0 °C for 30 min. and allowed to warm-up to room temperature overnight. The reaction was quenched by an addition of saturated NaHCO3 aqueous solution and extracted with ethyl acetate. The organic layer was washed with H2O, dried over anhydrous MgSO4, filtered and concentrated. Recrystallization using the mixture of ethyl acetate and hexane gave4-[β-(2-(2’-(tert-butoxycarbonylamino)-ethoxy)-5-chlorobenzenecarboxamido)ethyl] benzenesulfonamide (3, 2.40 g) as a white solid at 96 % isolated yield.
White solid, mp = 164-165 °C; 1H NMR (300 MHz, DMSO-d6) δ 1.36 (s, 9H), 2.95 (t, J = 7.48 Hz, 2H), 3.32 (m, 2H), 3.57 (q, J = 6.87, 2H), 4.08 (t, J = 4.78 Hz, 2H), 7.15-7.20 (m, 2H), 7.31 (s, 3H), 7.44 (d, J = 8.20 Hz, 2H), 7.50 (dd, J = 8.86, 2.82 Hz, 1H), 7.74-7.77 (m, 3H), 8.40 (t, J = 5.48 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 28.2, 34.8, 39.8, 68.8, 78.1, 115.2, 124.1, 124.7, 125.8, 129.1, 130.1, 131.9, 142.1, 143.6, 155.2, 156.1, 163.2; MS (ESI) m/z Calcd for C22H28ClN3O6SNa (M+Na)+ 520.1, obsd 520.1
Synthesis of N-{4-[β-(2-(2’-(tert-butoxycarbonylamino)ethoxy- 5-chlorobenzene-carboxamido)ethyl]benzenesulfonnyl]-N’-cyclohexylurea (4)
To a mixture of compound 3 (3.00 g, 6.0 mmol) and CuI (0.229 g, 1.2 mmol) in DMF (25 mL) was added a solution of cyclohexylisocyanate (1.13 g, 9.0 mmol) in DMF (5 mL). The resulting mixture was stirred in the dark overnight then cyclohexylisocyanate (0.377g, 3.0 mmol) in DMF (2 mL) was added, then this mixture was again stirred in the dark for 24 hr. The reaction was quenched by an addition of saturated NaHCO3aqueous solution and extracted with ethyl acetate. The organic layer was washed with saturated NaHCO3 aqueous solution, dried over anhydrous MgSO4, filtered and concentrated. Recrystallization using ethyl acetate and hexane gave N-{4-[β-(2-(2’-(tert-butoxycarbonylamino)-ethoxy)-5-chlorobenzenecarbox-amido) ethyl]benzenesulfonnyl]-N’-cyclohexyl urea (4, 3.05 g) as white solid at 81 % isolated yield.
White solid, mp = 141-143 °C; 1H NMR (300 MHz, DMSO-d6) δ 0.95-1.20 (m, 6H), 1.35 (s, 9H), 1.45-1.70 (m, 4H), 2.96 (t, J = 7.09 Hz, 2H), 3.20-3.40 (m, 3H), 3.58 (q, J = 6.54 Hz, 2H), 4.04-4.10 (m, 2H), 6.33 (brs, 1H), 7.13-7.19 (m, 2H), 7.41-7.52 (m, 3H), 7.76-7.83 (m, 3H), 8.39 (t, J = 5.70 Hz, 1H), 10.3 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 24.2, 25.0, 28.1, 32.3, 34.8, 39.8, 48.1, 68.8, 78.0, 115.1, 124.2, 124.6, 127.3, 129.1, 130.0, 131.8, 138.4, 145.0, 150.8, 155.1, 156.1, 163.2; MS (ESI) m/z Calcd for C29H40ClN4O7S (M+H)+ 623.2, obsd 623.2.
Synthesis of N-{4-[β-(2-(2’-aminoethoxy)-5-chlorobenzene carboxamido)ethyl]-benzenesulfonyl]-N’-cyclohexylurea (5)
Compound 4 (3.05 g, 4.89 mmol) was dissolved in TFA (20 mL) and the resulting mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated under reduced pressure and the residue was subjected to recrystallization using diethyl ether to give N-{4-[β-(2-(2’-aminoethoxy)-5-chlorobenzene-carboxamido)ethyl] benzenesulfonyl]-N’-cyclohexylurea (5,2.90 g) as a TFA salt with a 93% isolated yield.
White solid, mp = 126-128 °C; 1H NMR (300 MHz, DMSO-d6) δ1.02-1.72 (m, 10H), 2.95 (t, J = 7.31 Hz, 2H), 3.17-3.30 (m, 3H), 3.54 (q, J = 6.70 Hz, 2H), 4.28 (t, J = 4.55 Hz, 2H), 6.52 (d, J = 7.81 Hz, 1H), 7.23 (d, J = 9.74 Hz, 1H), 7.47 (d, J = 8.13 Hz, 2H), 7.50-7.57 (m, 2H), 7.85 (d, J = 8.08 Hz, 2H), 7.90-8.15 (brs, 2H), 8.44 (t, J = 5.53 Hz, 1H), 10.5 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 24.2, 25.0, 32.3, 34.4, 38.6, 40.2, 48.1, 66.4, 116.5, 125.3, 126.8, 127.3, 129.1, 129.4, 131.4, 138.3, 145.0, 150.5, 154.1, 164.6; MS (ESI) m/zCalcd for C24H32ClN4O5S (M+H)+ 523.2, obsd 523.2.
Synthesis of 2-(2-(2-(4-chloro-2-(4-(N-(cyclohexylcarbamoyl) sulfamoyl)phenethyl-carbamoyl)phenoxy)ethylamino)-2-oxoethoxy)acetic acid (6)
To a mixture of compound 5 (1.05 g, 1.64 mmol), diglycolic anhydride (0.464 g, 4 mmol) in DMF (20 mL) was added TEA (1.01 g, 10 mmol) and the resulting mixture was stirred at room temperature for overnight. The reaction was quenched by addition of saturated NaHCO3 solution and extracted with ethyl acetate. The aqueous layer was acidified to pH 2 using a 1N HClsolution and extracted with ethyl acetate. The organic layer was washed with H2O, dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by silica gel column chromatography using a 20 % methanol in dichloromethane. The compound was further purified by recrystallization using diethyl ether to give 2-(2-(2-(4-chloro-2-(4-(N-(cyclohexylcarbamoyl) sulfamoyl)-phenethylcarbamoyl)phenoxy)-ethylamino)-2-oxoethoxy) acetic acid (6,700 mg) to give a white solid (700 mg) with a 67 % isolated yield.
White solid, mp = 139-140 °C; 1H NMR (300 MHz, DMSO-d6) δ 1.04-1.70 (m, 10H), 2.94 (t, J = 7.03 Hz, 2H), 3.26-3.30 (m, 1H), 3.54-3.62 (m, 4H), 4.03 (s, 2H), 4.09-4.18 (m, 4H), 6.36 (d, J = 7.73 Hz, 1H), 7.19 (d, J = 8.97 Hz, 1H), 7.39-7.59 (m, 3H), 7.72-7.90 (m, 3H), 8.20 (t, J = 5.90 Hz, 1H), 8.37 (t, J = 5.87 Hz, 1H), 10.4 (brs, 1H), 12.7 (brs, 1H); 13C NMR (75 MHz, DMSO-d6) δ 24.2, 25.0, 32.3, 35.0, 37.5, 40.0, 48.1, 67.7, 68.5, 70.0, 115.0, 123.9, 124.7, 127.3, 129.2, 130.2, 131.9, 138.1, 145.3, 150.5, 155.2, 163.1, 169.7, 171.4; HRMS (ESI) m/z Calcd for C28H36ClN4O9S (M+H)+ 639.1886, obsd 639.1892.
Solid-phase Synthesis
Ligands were prepared by solid-phase synthesis summarized in Scheme 2 on Rink Amide Tentagel resin (2 g, 0.23 mmol/g) using a Fmoc/tBu synthetic strategy and standard DIC and HBTU activation [7,13,19]. Nα-Fmoc amino acid was double coupled using preactivated 0.3 M HOBt esters and HCTU/2,4,6-collidine. After the GLP-1 sequence assembly {Boc-His(Boc)-Ala-Glu(O-tBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(O-tBu)-Val-Ser(tBu)-Ser(tBu)-Tyr(tBu)-Leu-Glu(O-tBu)-Gly-Gln(Nam-Trt)-Ala-Ala-Lys(Aloc)-Glu(O-tBu)-Phe-Ile-Ala-Trp(Ni-Boc)-Leu-Val-Arg(Ng-Pbf)-Gly-Arg(Ng-Pbf)-Gly-Rink resin}, the resin was washed, then dried under vacuum. Dry resin (10 μmol aliquot) was placed into a 2 mL reactor for synthesis of individual ligands. For heterobivalent ligand, the synthesis continued by stepwise assembly of repetitive Pro-Gly linker units, then attachment of Cys(Trt), Lys(Aloc), or Gly, another PEGO, and Glibenclamidecarboxy derivative 6 (Scheme 1). The resin was cleaved with TFA-scavengers and the crude products were purified.
Solid-phase Synthesis of Protected GLP-1-resin (7,Scheme 2)
The Rink resin was washed with DMF, and the Nα-Fmoc protecting group was removed with 1:10 piperidine in DMF (1 × 2 min and 1 × 20 min). The resin was washed successively with DMF, DCM, DMF, a solution of 0.05 mM solution of Bromophenol Blue in 0.2 M HOBt in DMF, then DMF. The Nα-Fmoc amino acid was coupled using pre-activated 0.3 M HOBt esters in DMF-DCM mixture (3 equiv of Nα-Fmoc amino acid, 3 equiv of HOBt or HOCt, and 3 equiv of DIC). The resin slurry was stirred for 2 h or until the bromophenol test became negative [34]. If the test failed, the resin was washed with DMF and the amino acid was coupled again by the HCTU/2,4,6-lutidine procedure (0.3 M solution of 3 equiv of Nα-Fmoc amino acid, 3 equiv of HCTU, and 6 equiv of 2,4,6-lutidine in DMF) for 3 h or by a preformed symmetric anhydride ( 3 equiv of Nα-Fmoc amino acid and 1.5 equiv of DIC in a 1:1 DMF-DCM mixture) until Kaiser test was negative. If the couplings did not result in a negative Kaiser test, the resin was washed with DMF, and the free amino groups were capped with 50% Ac2O in pyridine for 10 minutes. After GLP-1 coupling sequentially to the Rink amide resin, the resin was washed with DCM, dried under vacuum, and then stored in refrigerator.
Solid-phase Synthesis of Cy5 labelled GLP-1/ Glibenclamideheterobivalent ligand (9) (Scheme 2)
An aliquot of the dry protected GLP-1 resin 7 (10 μmol, from previous step) was placed in a 2 mL syringe reactor. The resin was swelled in DCM for an hour. The Aloc side chain of the lysine in position 26 was deprotected by Pd[0] under argon and oxygen-free solvents as follows: dissolve 0.5% w/v of tetrakis(triphenylphosphine) palladium(0) [Pd(0)TPP4] and 3% w/v of dimethylbarbituric acid (DMBA) in DCM (10 mL per 1 g of resin). Treat the resin with the deprotection reagent mixture (2 X 30 min). Wash the resin with DCM (3X), DMF (3X), 5% DIEA in DMF (3X), DMF (2X), 1% sodium diethyldithiocarbamatetrihydride in DMF (2X, 5 min; this step removes the resin-bound palladium), DMF (2X), 5% piperidine in DMF (2X), DMF (3X), 0.2 M HOBt in DMF (2X), DMF (2X), and DCM (3X).The Fmoc-PEGO spacer was introduced using HBTU coupling. The resin was washed with DMF and Gly and Pro were attached alternatively as needed to synthesize the [Pro-Gly]3 repeats and Cys(TRT) using the Fmoc protocol (Scheme 2). The resin was coupled with the second PEGO spacer as described above. The resin was treated with activated compound 6 (0.2 M solution, 2 equiv.), HBTU (1.95 equiv.), DIEA (4 equiv.) in DMF overnight. The final resin was washed thoroughly with DCM then cleaved. A cleavage cocktail (10 mL per 1 g of the resin) consisting of CF3CO2H (91%), H2O (3%), EDT (3%), and PhSMe (3%) was injected into the resin and the mixture was agitated at room temperature for 5 h. The solution was filtered, the resin was washed with CF3CO2H (2 × 3 min), the liquid phases were collected and concentrated under a stream of nitrogen, and the product was precipitated using cold Et2O. The crude thiol-intermediate was washed three times with cold Et2O, lyophilized, purified, and characterized as described above. The lyophilizate was dissolved in minimal DMSO. A 1 μmol aliquot was treated with Cy5-maleinimide (1.3 equiv., Invitrogen) overnight. The coupling conversion was verified by HPLC. When Cy5 conjugation complete, the reaction was quenched by 1.0 M acetic acid, was lyophilized and purified by HPLC. Mass spectra and HPLC characterization data are provided in the supplemental information (Table S1).
Solid-phase Synthesis of Eu-GLP-1/Glb (10)
A resin bound intermediate protected Phe12, Lys(Aloc)26, Arg36 GLP-1 (Scheme 3) was treated with Pd(0) to remove Aloc protecting group from lysine in position 26. The solid phase synthesis continued on ε-amine of lysine with attachments of Fmoc-PEGO, Fmoc-Gly, Fmoc-Pro, Fmoc-Lys(Aloc), Fmoc-PEGO, and compound 6 as described for 9 (Scheme 2). The resin was washed with DMF and DCM. The resin was as treated with Pd(0) to remove Aloc protecting group then washed with DCM and DMSO. Attachment of DTPA of was performed using preformed HOBt activation (3 equiv. DTPA anhydride and 6 equiv. HOBt, Scheme 3). Briefly, DTPA anhydride (3 equiv.) and HOBt (6 equiv.) in DMSO were heated until dissolved (50-60°C) then stirred for 30 min. at room temperature.
This reagent, preformed DTPA-OBtdiester, was directly injected into the free-amine GLP-1/Glb-resin and stirred overnight. The resin was washed with DMSO, THF, 5% DIEA 5% water in THF (5 min), THF, and DCM. The compound was cleaved from the resin as described above and purified by HPLC. The purified peptide was dissolved in 0.1 M ammonium acetate buffer pH 8.0, 3.0 eq. Eu(III)Cl3 was added and the reaction was stirred at room temperature overnight. The Eu-labelled peptide was separated using Solid-Phase Extraction (SPE) and lyophilized to yield an amorphous white powder. The final compound was characterized by HPLC (TEAA buffer pH 6.0), ESI-MS and/or FT-ICR.
Solid-phase Synthesis of non-labelled GLP-1/Glb (11)
An aliquot of the dry protected GLP-1 resin 7 (10 μmol, from previous step) was placed in a 2 mL syringe reactor. The resin was treated with similar reactant sequence as for compounds 10: PEGO, [Pro-Gly]3 repeats, Gly, PEGO, and compound 6.
Solid-phase Synthesis of Eu-GLP-1 (12)
An aliquot of the dry protected GLP-1 resin 7 (10 μmol, from previous step) was placed in a 2 mL syringe reactor. The resin was washed with DMF and DCM then treated with Pd(0) to remove the Aloc protecting group and washed with DCM and DMSO. Attachment of DTPA of was performed using preformed HOBt activation (as in 10, Scheme 3). The compound was cleaved from the resin and purified by HPLC. The purified peptide was dissolved in 0.1 M ammonium acetate buffer pH 8.0, 3.0 eq., Eu(III)Cl3 was added and the reaction was stirred at room temperature overnight. The Eu-labelled peptide was separated using Solid Phase Extraction (SPE) and lyophilized to yield an amorphous white powder.
Scheme 3.Synthesis of Eu-GLP-1/Glb 10 (i) Fmoc/tBu solid phase synthesis, ε-amine of Lys26 was protected with Aloc. (ii) Pd(0) tetrakis Ph3P, N,N-dimethyl barbituric acid, DCM, 30 min., room temp. (iii) Fmoc-PEGO, DIC, DCM, room temp. (iv) Fmoc/tBu solid phase synthesis, Pro-Gly repeats, Lys(Aloc), and PEGO. (v) Compound 6, HBTU/DIEA, room temp, overnight. (vi) (a) (ii) Pd(0) tetrakis Ph3P, N,N-dimethyl barbituric acid, DCM, 30 min, room temp. (b) DTPA HOBt ester, DMSO, overnight (vi) TFA, scavenges, 4 hrs, room temp. (vi) Eu(III)Cl3, pH 8.0, overnight, room temp.
Quality Control and Purification
The purity of products was checked by analytical PR-HPLC using a Waters Alliance 2695 Separation Model with a Waters 2487 dual wavelength detector (220 and 280 nm) on a reverse phase column (Waters Symmetry C18, 4.6 × 75 mm, 3.5 μm). Peptides were eluted with a linear gradient of aqueous CH3CN/0.1% CF3CO2H at a flow rate of 1.0 mL/min. Purification of ligands was achieved on a Waters 600 HPLC using a reverse phase column (Vydac C18, 15–20 μm, 22 X 250 mm). Peptides were eluted with a linear gradient of CH3CN/0.1% CF3CO2H at a flow rate of 5.0 mL/min. Separation was monitored at 230 and 280 nm. Size exclusion chromatography was performed on a borosilicate glass column (2.6 × 250 mm, Sigma, St. Louis, MO) filled with medium sized Sephadex G-25 or G-10. The compounds were eluted with an isocratic flow of 1.0 M aqueous acetic acid. Pure compounds were dissolved in DI water or DMSO at approximately 1-5 mM. Accurate concentrations were determined by HPLC at 280 nm. A solution of D-Trp in water or DMSO was co-injected as an internal standard. Structures were characterized by ESI (Finnigan, Thermoquest LCQ ion trap) or MALDI-TOF (Bruker Reflex-III, α-cyanocinnamic acid as a matrix). For internal calibration a mixture of standard peptides was used with an average resolution of 8,000–9,000. High resolution mass measurements were carried out on a FT-ICR IonSpec 4.7T instrument.
Eu(III)-based Ligand Binding Assays
Analysis of Cellular Receptor Number
Receptor number expression analysis was performed using high throughput saturation binding analyses, with Eu-labelled ligands.[4,33,35–37] Binding assays were performed in 96-well plates. Cells were plated in Perkin Elmer or black walled CoStar 96-well plates at a density of ~70,000 cells/well and were allowed to grow for 3 days. On the day of the experiment, media were aspirated from all wells prior to the addition of the ligands to be tested. Ligands were diluted in binding media (RPMI, 1 mM 1,10-phenanthroline, 200mg/L bacitracin, 0.5mg/L leupeptin, 0.2% BSA) and samples were tested in quadruplicate, unless otherwise noted. Cells were incubated for 1 hour in the presence varying concentrations of Eu-labelled ligands. Non-specific binding was evaluated at each concentration by measuring binding in the presence of 1 μM unlabelled GLP-1/Glb11 ligand. Following the incubation periods, cells were washed three times with binding media; the media were carefully replaced manually using a vacuum manifold: it is important to make certain that cells are not removed from the wells during the washing manoeuvre. To release Eu(III) from the DTPA-cage, and provide the antenna to boost the Eu(III) signal, enhancement solution (Perkin Elmer; 1244-105) was added directly to the wells (105 μL/well) after the wash, and the plates were incubated for at least 30 min. at 37°C prior to reading.
For Figure 3, cells were incubated in the presence of 15 nM Eu-GLP-1/Glb 10and either 4.1 fM or 1.6 nM unlabelled GLP-1 or Glb, without analysis of non-specific binding.
Analysis of Apparent Binding Affinity
To enhance assay signal to noise, saturation binding was carried out with a greater number of cells (receptors) using 6 well plates.βTC3 cells were seeded in six well plates and allowed to grow to ~90% confluence. On the day of the experiment, media were aspirated and cells were rinsed two times with wash buffer (RPMI, 0.2% BSA and 25 mM HEPES). Ligands were diluted in binding buffer (RPMI, 0.2% BSA, 25 mM HEPES, 200 mg/L bacitracin and 0.5 mg/L leupeptin) then added to cells and incubated for 1hr at 37°C. Non-specific binding was determined by competition with 300 nM cold GLP-1 300 nM Glb or GLP-1/Glb 11 at each concentration tested. Cells were washed 2times with wash buffer. After washing off unbound labelled ligand, cells were collected by scraping and centrifugation of the suspension. The resulting pellet was suspended in “enhancement solution” (ES). By minimizing ES volume, the number of cells (receptors) per sample volume was increased, and contribution of non-specific signal from ligand bound to the well was eliminated. Tubes with enhancement solution/cells were incubated at 37°C for 1hr. The solution was centrifuged to pellet cellular debris. 100 μl of the supernatant was added to wells of a 96 well Perkin Elmer B&W Isoplate. Each sample was replicated six times.
Insulin Assays
INS 832/3 cells, a robust model of β-cell glucose stimulated insulin secretion [38,39] were seeded at a density of 500,000 cells per well in a 24 well plate and allowed to proliferate for 72 hours. Prior to the experiment the cells were quickly rinsed with HBSS containing 1 mM glucose and were rinsed a second time for 2 hours in the same. Following the 2 hour rinse, the medium in each well was replaced with medium containing the appropriate concentration of glucose and secretagouges for each experimental condition. Following a 2 hour incubation period the medium from each well was collected and assayed using a high range rat insulin ELISA kit (ALPCO Diagnostics, Salem, NH). The values (ng/ml) were then normalized to the average level of insulin secretion in 1 mM glucose (basal secretion) for each experiment and are expressed as a percentage response relative to insulin secretion at 1 mM glucose.
Data Analysis
For both assays, bound ligand was evaluated in 96wellplatesread on a Wallac VICTOR3 instrument using the standard Eu(III) Time resolved fluorescence (TRL) measurement for Europium [4] (340 nm excitation, 400 μsec delay, and emission collection for 400 μsec at 615 nm). Data were analyzed with Sigma Plot Software (unless noted) using a 3 parameter sigmoid non-linear regression analysis which provided apparent Kd values.
Microscopic Imaging
Microscopic imaging provides rapid information regarding binding, and ligand/receptor distributions.[14,40] βTC3 and INS-1F cells were grown on 25 mm #1 coverslips housed in wells of a six well plate. A single coverslip was placed in a chamber held at 37°C while mounted on the stage of an inverted Olympus IX70 microscope equipped with a 40×1.4 NA ultrafluor objective, and a 300 W Xe lamp as the excitation source. Cy5 fluorescence was excited using a 20 nm band pass centered at 640 nm and emitted light collected through a 30 nm wide pass filter centered at 680 nm and focused onto a CCD camera (Photometrics CH-350). Cells were incubated with the ligand for 2 minutes, then the media containing the free ligand was replaced with ligand free media. Images were acquired 2 and 3 minutes thereafter.
Supplementary Material
Acknowledgements
The work was supported by an Innovation and Basic Research grants from the Juvenile Diabetes Research Foundation and by the NIH NHLBI (Heart Lung and Blood): Systems and Integrative Physiology training grant 5T32GM0084.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author
References
- 1.Lin M, Lubag A, Mcguire MJ, Seliounine SY, Tsyganov EN, Antich PP, Dean A, Brown KC, Sun X. Frontiers in Bioscience. 2008;13:4558–4575. doi: 10.2741/3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneider S. Diabetes, Obesity & Metabolism. 2008;10(S.4):109–118. doi: 10.1111/j.1463-1326.2008.00944.x. [DOI] [PubMed] [Google Scholar]
- 3.Souza F, Freeby M, Hultman K, Simpson N, Herron A, Witkowsky P, Liu E, Maffei A, Harris PE. Current Medicinal Chemistry. 2006;13:2761–2773. doi: 10.2174/092986706778521940. [DOI] [PubMed] [Google Scholar]
- 4.Josan JS, de Silva C, Yoo B, Lynch RM, Pagel MD, Vagner J, Hruby VJ. Fluorescent and Lanthanide Labeling for Ligand Screens, Assays, and Imaging. Humana Press; Totowa, NJ: 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiessling LL, Gestwicki JE, Strong LE. Current Opinion in Chemical Biology. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 6.Mammen M, Choi S-K, Whitesides GM. Angewandte Chemie International Edition. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 7.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Bioorganic & Medicinal Chemistry Letters. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 8.Vagner J, Xu L, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ. Angewandte Chemie. 2008;120:1709–1712. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma SD, Granberry ME, Jiang J, Leong SP, Hadley ME, Hruby VJ. Bioconjugate Chemistry. 1994;5:591–601. doi: 10.1021/bc00030a015. [DOI] [PubMed] [Google Scholar]
- 10.Caplan MR, Rosca EV. Annals of Biomedical Engineering. 2005;33:1113–1124. doi: 10.1007/s10439-005-5779-1. [DOI] [PubMed] [Google Scholar]
- 11.V Rosca E, Stukel JM, Gillies RJ, Vagner J, Caplan MR. Biomacromolecules. 2007;8:3830–5. doi: 10.1021/bm700791a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shewmake TA, Solis FJ, Gillies RJ, Caplan MR. Biomacromolecules. 2008;9:3057–3064. doi: 10.1021/bm800529b. [DOI] [PubMed] [Google Scholar]
- 13.Xu L, Josan JS, Vagner J, Caplan MR, Hruby VJ, Mash EA, Lynch RM, Morse DL, Gillies RJ. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21295–21300. doi: 10.1073/pnas.1211762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Josan JS, Handl HL, Sankaranarayanan R, Xu L, Lynch RM, Vagner J, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chemistry. 2011;22:1270–8. doi: 10.1021/bc1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monguchi Y, Vagner J, Handl HL, Jana U, Begay LJ, Hruby VJ, Gillies RJ, Mash EA. Tetrahedron Letters. 2005;46:7589– 7592. [Google Scholar]
- 16.Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chemistry. 2006;17:1545– 1550. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. Journal of the American Chemical Society. 2007;129:1312– 1320. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash E. a, Gillies RJ, Hruby VJ. Bioconjugate Chemistry. 2007;18:1101–9. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gillies RJ, Hruby VJ. International Journal of Peptide Research and Therapeutics. 2008;14:293–300. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brabez N, Lynch RM, Xu L, Gillies RJ, Chassaing G, Lavielle S, Hruby VJ. Journal of Medicinal Chemistry. 2011;54:7375–84. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kutchma A, Quayum N, Jensen J. Nucleic Acids Research. 2007;35:D674–679. doi: 10.1093/nar/gkl990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quayum N, Kutchma A, Sarkar SA, Juhl K, Gradwohl G, Mellitzer G, Hutton JC, Jensen J. Experimental Diabetes Research. 2008;2008:1–9. doi: 10.1155/2008/312060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cline GW, Zhao X, Jakowski AB, Soeller WC, Treadway JL. Biochemical and Biophysical Research Communications. 2011;412:413–418. doi: 10.1016/j.bbrc.2011.07.077. [DOI] [PubMed] [Google Scholar]
- 24.Schneider S, Ueberberg S, Korobeynikov A, Schechinger W, Schwanstecher C, Schwanstecher M, Klein HH, Schirrmacher E. Regulatory Peptides. 2007;139:122–127. doi: 10.1016/j.regpep.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Sweet IR, Cook DL, Lernmark Å, Greenbaum CJ, Wallen AR, Marcum ES, Stekhova SA, Krohn KA. Biochemical and Biophysical Research Communications. 2004;314:976–983. doi: 10.1016/j.bbrc.2003.12.182. [DOI] [PubMed] [Google Scholar]
- 26.Wild D, Wicki A, Mansi R, Béhé M, Keil B, Bernhardt P, Christofori G, Ell PJ, Mäcke HR. Journal of Nuclear Medicine. 2010;51:1059–1067. doi: 10.2967/jnumed.110.074914. [DOI] [PubMed] [Google Scholar]
- 27.Baggio LL, Huang Q, Brown TJ, Drucker DJ. Diabetes. 2004;53:2492–2500. doi: 10.2337/diabetes.53.9.2492. [DOI] [PubMed] [Google Scholar]
- 28.Combettes MMJ. Current Opinion in Pharmacology. 2006;6:598–605. doi: 10.1016/j.coph.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Hansen KB, Vilsbøll T, Knop FK. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 2010;3:155–163. [PMC free article] [PubMed] [Google Scholar]
- 30.Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thøgersen H, Wilken M, Agersø H. Journal of Medicinal Chemistry. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 31.Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thøgersen H, Wilken M, Johansen NL. Journal of Medicinal Chemistry. 2007;50:6126–6132. doi: 10.1021/jm070861j. [DOI] [PubMed] [Google Scholar]
- 32.Maffei A, Liu Z, Witkowski P, Moschella F, Del Pozzo G, Liu E, Herold K, Winchester RJ, Hardy MA, Harris PE. Endocrinology. 2004;145:4513–4521. doi: 10.1210/en.2004-0691. [DOI] [PubMed] [Google Scholar]
- 33.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Analytical Biochemistry. 2005;343:299–307. doi: 10.1016/j.ab.2005.05.040. [DOI] [PubMed] [Google Scholar]
- 34.Krchňák V, Vágner J, Šafář P, Lebl M. Collection of Czechoslovak Chemical Communications. 1988;53:2542–2548. [Google Scholar]
- 35.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Analytical Biochemistry. 2004;330:242–250. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 36.Handl HL, Gillies RJ. Life Sciences. 2005;77:361–371. doi: 10.1016/j.lfs.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 37.De Silva CR, Vagner J, Lynch R, Gillies RJ, Hruby VJ. Analytical Biochemistry. 2010;398:15–23. doi: 10.1016/j.ab.2009.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Diabetes. 2000;49:424–430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- 39.Ronnebaum SM, Jensen MV, Hohmeier HE, Burgess SC, Zhou Y-P, Qian S, MacNeil D, Howard A, Thornberry N, Ilkayeva O, et al. The Journal of Biological Chemistry. 2008;283:28909–28917. doi: 10.1074/jbc.M804665200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou M, Nakatani E, Gronenberg LS, Tokimoto T, Wirth MJ, Hruby VJ, Roberts A, Lynch RM, Ghosh I. Bioconjugate Chemistry. 2007;18:323–332. doi: 10.1021/bc0601929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.