

Abstract
Previous reports have associated GRHL2 with tumor progression. However, the biological role of GRHL2 in human colorectal cancer (CRC) has not been explored. We examined the expression of GRHL2 in 75 CRC samples, as well as the paired non-tumor tissues, by immunohistochemistry, qRT-PCR, and western blot analysis. The association between GRHL2 expression and various clinicopathological parameters including Ki-67, a marker of proliferative activity, was also evaluated. We performed lentivirus-mediated shRNA transfection to knock down GRHL2 gene expression in HT29 and HCT116 CRC cells. Cell proliferation was examined by the CCK-8 (Cell Counting Kit-8) assay, colony formation, and cell cycle assay in vitro. Tumorigenesis in vivo was assessed using a mouse xenograft model. Moreover, we transiently silenced ZEB1 expression in GRHL2-knockdown CRC cells using specific shRNA, and then examined the effects on GRHL2 and E-cadherin expression, as well as cell proliferation. Herein, we demonstrated that enhanced GRHL2 expression was detected in CRC, and correlated with higher levels of Ki-67 staining, larger tumor size, and advanced clinical stage. Knocking down GRHL2 in HT29 and HCT116 CRC cells significantly inhibited cell proliferation by decreasing the number of cells in S phase and increasing that in the G0/G1 phaseof the cell cycle. This resulted in inhibition of tumorigenesis in vivo, as well as increased expression of ZEB1. Furthermore, transient ZEB1 knockdown dramatically enhanced cell proliferation and increased GRHL2 and E-cadherin expression. Collectively, our study has identified ZEB1 as a target of GRHL2 and suggested a reciprocal GRHL2-ZEB1 repressive relationship, providing a novel mechanism through which proliferation may be modulated in CRC cells.
Keywords: colorectal cancer, E-cadherin, GRHL2, progression, proliferation, ZEB1, tumorigenesis
Introduction
Colorectal cancer (CRC) is a major cause of cancer morbidity and mortality.1 Although the mortality associated with CRC has been steadily declining over the past few decades, partly due to earlier detection by screening, CRC remains to be a major public health concern. CRC development is a multistep process mediated by complex cascades of molecular events.2 Over the past three decades, molecular genetic studies have revealed some critical mutations underlying the pathogenesis of CRC. A number of oncogenes and tumor-suppressor genes are mutated in CRC, and a larger collection of genes that are mutated in subsets of CRC have begun to be defined.3
Grainyhead like 2 (GRHL2), an epithelium-specific mammalian homolog of Drosophila Grainyhead, functions in vivo to regulate epithelial differentiation in the otic vesicle, the gut endoderm, and the surface ectoderm.4,5 This protein expressed in epithelial structures acts as a transcriptional activator, regulating epidermal development.6,7 GRHL2 is associated with age-related hearing impairment,8 neural tube closure,9 epidermal integrity,10 wound healing,11 and the regulation of many physiological functions of human airway epithelium.12 In the context of cancer, GRHL2 has been reported to function as a tumor suppressor.13,14 However, increasing evidence indicates that GRHL2 may have an oncogenic role in cancer development.15-18 These controversial results suggest that the role of GRHL2 is possibly tumor-specific and highly dependent on its targets in different cancer cells.19 However, the expression of GRHL2 in CRC is unknown and its role in CRC progression is also elusive.
One of the major target genes of GRHL2 is E-cadherin repressor ZEB1 (zinc finger E-box binding homeobox 1; also known as δEF1).19 GRHL2 has been shown to repress ZEB1 expression and interact directly with the ZEB1 promoter.14 ZEB1, a dual zinc-finger, DNA-binding transcription factor,20 is a direct transcriptional repressor of E-cadherin and it has the most consistent inverse correlation with E-cadherin across different types of carcinomas.21,22 E-cadherin, encoded by the CDH1 gene on chromosome 16q22, is a signature member of the cadherin family and constitutes a key component of adherens junctions.23
Herein, we have showed that GRHL2 acted as an oncogene as it was upregulated in about 61% of CRCs and associated with tumor progression due to higher levels of cell proliferation. Knocking down GRHL2 inhibited G1/S cell cycle progression in CRC cells, decreased cell proliferation and impaired tumorigenesis in a nude mouse xenograft model. We further demonstrated that knocking down GRHL2 inhibited cell proliferation via upregulating ZEB1 and downregulating E-cadherin. The exploration of such a mechanism would provide promising therapeutic targets for CRC treatment.
Results
GRHL2 expression was upregulated in human CRC
To determine the role of GRHL2 in CRC development, we evaluated the expression of GRHL2 in 75 clinical specimens. We compared the endogenous GRHL2 expression in human CRC with that in adjacent non-tumor tissue by immunohistochemistry (IHC). As shown in Figure 1, IHC staining indicated that GRHL2 was predominantly present in the nuclei of CRC cells and normal cells in the paired normal colorectal mucosa. Among 75 cases, positive GRHL2 expression was observed in 61.3% (46 / 75) of CRC tissues compared with 44.0% (33 / 75) of the paired adjacent non-tumor tissue (P < 0.05, Fig. 1A–F; Table 1). Meanwhile, in the tumors paired with these normal tissues, GRHL2 expression was completely positive. Moreover, importantly, the GRHL2 score in cancer tissue was greater than that in the normal paired surface epithelium. To validate the IHC staining results, we performed western blot in 6 random cases of primary CRC tissues (T) and paired adjacent noncancerous tissues (N). We found that GRHL2 protein was significantly upregulated in tumor tissues compared with matched adjacent normal tissues (Fig. 1G). Moreover, the average GRHL2 mRNA expression in CRC tissues was markedly higher than that in the paired adjacent non-tumor tissues (P < 0.001, Fig. 1H). Further, RT-PCR was performed to assess GRHL2 gene transcripts in different CRC cell lines (Fig. 1I). GRHL2 expression was varied in seven different CRC cell lines, five of which showed a high expression. Altogether, these data indicated that GRHL2 was overexpressed in CRC, and its overexpression may contribute to the development of human CRC.

Figure 1. GRHL2 was overexpressed in CRC samples and associated with tumor progression. Immunohistochemical (IHC) staining of formalin-fixed, paraffin-embedded CRC tissues and paired non-tumor tissues was performed. Representative images illustrative of the different staining patterns are presented in (A–F). (A) Negative GRHL2 staining in no-tumor normal mucosa. (B) Nuclear staining of GRHL2 in no-tumor epithelium adjacent to CRC. (C) Negative staining of GRHL2 in CRC. (D) Weak nuclear intensity staining of GRHL2 in CRC. (E) Moderate nuclear staining of GRHL2 in CRC. (F) CRC sample showing strong nuclear staining for GRHL2. (G) Western blot analysis demonstrated GRHL2 expression was higher in six CRC specimens (T1–T6) compared with adjacent non-tumor specimens (N1–N6) relative to the loading control GAPDH. (H) Box plot depicting GRHL2 levels as assessed by qRT-PCR in the normal mucosa (NM) and our series of 75 CRC samples classified by according tumor stage (stage I+II, n = 30; stage III+ IV, n = 45). *indicates P < 0.05, *** indicates P < 0.001. (I) Semi-qRT-PCR analysis demonstrated GRHL2 expression was varied in seven different CRC cell lines.
Table 1. Expression of GRHL2 in colorectal cancer tissues and matched non-tumor mucosa.
| Total | GRHL2 (−) | GRHL2 (+) | P value | |
|---|---|---|---|---|
| Tumor specimens | 75 | 29 (38.7%) | 46 (61.3%) | 0.034* |
| Non-tumor mucosa | 75 | 42 (56.0%) | 33 (44.0%) |
Statistically significant.
Expression of GRHL2 positively correlates with cancer progression and proliferation of CRC
We then performed a correlation analysis to assess the relationship between GRHL2 expression and clinicopathological variables by classifying patients into two groups: GRHL2 negative or GRHL2 positive. As shown in Table 2, expression of GRHL2 was not related to gender (P = 0.824), age (P = 0.858), tumor histology (P = 0.906), or tumor site (P = 0.723), but positively associated with tumor size (P = 0.029) and advanced TNM stage (P = 0.033). Furthermore, we assessed GRHL2 mRNA expression in CRC tissues by qRT-PCR. The average GRHL2 mRNA level in stage III/IV was significantly higher than that in stage I/II (P < 0.05, Fig. 1H). Our clinical CRC samples were also tested for Ki-67, a nuclear marker of cell proliferation. IHC staining demonstrated that Ki-67 was present in the nuclei of CRC cells (Fig. 2A). Tumor samples with high levels of Ki-67 demonstrated higher GRHL2 mRNA expression levels (P < 0.01, Fig. 2B). Upregulation of GRHL2 was positively related to a high Ki-67 cell proliferative index (r = 0.269, P = 0.016, Table 2). Collectively, our results provided evidence that GRHL2 was associated with CRC progression and acted as a potential oncogene.
Table 2. Relationship between GRHL2 expression level and clinicopathological variables in colorectal cancer patients.
| Variables | Subgroup | GRHL2 expression | P value | ||
|---|---|---|---|---|---|
| Total | Negative | Positive | |||
| Gender | Male | 40 | 15 (37.5%) | 25 (62.5%) | 0.824 |
| Female | 35 | 14 (40.0%) | 21 (60.0%) | ||
| Age (years) | <60 | 32 | 12 (37.5%) | 20 (62.5%) | 0.858 |
| ≥60 | 43 | 17 (39.5%) | 26 (60.5%) | ||
| Histology | Tubular | 60 | 23 (38.3%) | 37 (61.7%) | 0.906 |
| Mucinous | 15 | 6 (40.0%) | 9 (60.0%) | ||
| Site | Rectum and sigmoid | 49 | 19 (38.8%) | 30 (61.2%) | 0.723 |
| Right colon | 18 | 6 (33.3%) | 12 (66.7%) | ||
| Left colon | 8 | 4 (50.0%) | 4 (50.0%) | ||
| Size | <3 cm | 25 | 14 (56.0%) | 11 (44.0%) | 0.029 |
| ≥3 cm | 50 | 15 (30.0%) | 35 (70.0%) | ||
| TNM stage | I+II | 30 | 16 (53.3%) | 14 (46.7%) | 0.033 |
| III+IV | 45 | 13 (28.9%) | 32 (71.1%) | ||
| Ki-67 | Low | 31 | 17 (54.8%) | 14 (45.2%) | 0.016 |
| High | 44 | 12 (27.3%) | 32 (72.7%) | ||
Statistically significant
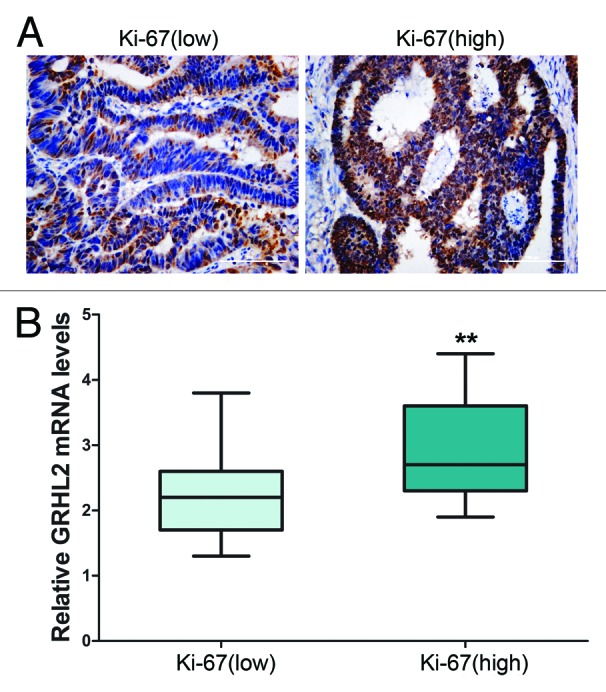
Figure 2. Overexpression of GRHL2 was associated with a high Ki-67 proliferative index. (A) Representative immunostaining of Ki-67. The sample with negative GRHL2 expression (left panel) showed a low proliferation index as indicated by the “Ki-67 (low)” label, whereas the sample with positive (right panel) had a high proliferation index as indicated by the “Ki-67 (high)” label. Representative photos of stained cells are shown with the original magnification of 200×. (B) Average GRHL2 mRNA expression in tumor tissues was stratified according to Ki-67 level. Cases with high Ki-67 levels also demonstrated higher GRHL2 mRNA expression (low Ki-67, n = 31; high Ki-67, n = 44). The levels of GRHL2 mRNA were examined using qRT-PCR by the 2−ΔCT method with GAPDH as the internal control. ** indicates P < 0.01.
Downregulation of GRHL2 inhibited cellular proliferation in vitro
Given that GRHL2 was positively associated with tumor growth in clinical samples, we sought to further investigate its influence on cell proliferation by knocking down GRHL2 in CRC cells and carrying out a series of functional assays. We utilized lentiviral shRNA to stably silence GRHL2. Efficient knockdown of GRHL2 was assessed by qRT-PCR (Fig. 3A) and western blot (Fig. 3B). The CCK-8 (Cell Counting Kit-8) assays indicated that knocking down GRHL2 in HT29 and HCT116 cells inhibited cell proliferation compared with control cells (Fig. 3C). GRHL2-knockdown produced less and smaller plate colonies (Fig. 3D). Importantly, flow cytometry demonstrated that the percentage of cells in S phase was significantly decreased in GRHL2-knockdown (GRHL2-KD) cells, and the population of cells in G0/G1 phase was increased (Fig. 3E). Taken together, these data suggested that GRHL2-knockdown inhibited cell proliferation by regulating the cell cycle in CRC cells.

Figure 3. Downregulation of GRHL2 induced a reduction in CRC cell proliferation in vitro. (A) Knockdown of GRHL2 expression using shRNA in HT29 and HCT116 CRC cells was achieved by lentiviral shRNA transfection and confirmed by qRT-PCR. Fold change was relative to parental cells. (B) Western blot analysis of GRHL2 expression in HT29 and HCT116 cells with or without GRHL2-knockdown (left panel). Densitometric analysis is expressed relative to the loading control, GAPDH (right panel). (C) Knockdown of GRHL2 in HT29 and HCT116 cells induced a remarkable reduction in cell proliferation, as determined by CCK-8 assays. (D) The ability of CRC cells to form colonies was analyzed in a clonogenic assay. GRHL2-knockdown cells produced less and smaller plate colonies than Scr-con cells. (E) Representative charts for cell-cycle distribution in GRHL2-KD and Scr-con cells. The percentage of cells in S phase was significantly decreased in GRHL2-KD cells compared with Scr-con cells, while the population of cells in G0/G1 phases was increased. Data are expressed as means ± SD from 3 separate experiments. *Relative to Scr-con control; #relative to Parental control. *P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.05, ##P < 0.01, ###P < 0.001.
Downregulation of GRHL2 suppressed tumor growth in vivo
To confirm the potential role of GRHL2 in cell growth in vivo, we established a BALB/c nude mouse xenograft model using the stable GRHL2-knockdown cell lines (GRHL2-KD). GRHL2 knockdown and scrambled shRNA control (Scr-con) cells were generated using HT29 and HCT116 CRC cells. As shown in Figure 4, HT29/GRHL2-KD and HCT116/GRHL2-KD cells grew more slowly than the scrambled control cells and formed smaller tumors (Fig. 4A and B). The tumor volumes and weights of the mice injected with HT29/GRHL2-KD or HCT116/GRHL2-KD cells were significantly lower than those injected with Scr-con cells (Fig. 4C and D). Additionally, western blot confirmed the downregulation of GRHL2 expression in tumors from GRHL2-KD mice (Fig. 4E). These results indicated that GRHL2 knockdown inhibited tumorigenesis of HT29 and HCT116 cells in vivo.

Figure 4. GRHL2-knockdown suppressed growth of CRC primary tumors in a mouse xenograft model. (A) Tumor growth was monitored in BALB/c mice implanted with Scr-con and GRHL2-KD cells. Tumor size was measured every 4 d and tumor volume was calculated as described in the Materials and Methods section. Tumors in the GRHL2-KD groups grew more slowly than those in the Scr-con groups. (B) Representative tumors from the mice of each group are shown. Tumors from the GRHL2-KD groups were smaller than those from the Scr-con groups. (C) As compared with Scr-con controls, the average tumor volumes from mice in the GRHL2-KD groups were markedly smaller. (D)The average weight of primary tumors originating from GRHL2-KD cells was lower than those from Scr-con cells. (E) Western blot was performed to confirm the downregulation of GRHL2 expression in tumors from GRHL2-KD mice. Samples were selected and paired randomly. Data were expressed as mean ± SD *indicates P < 0.05, **indicates P < 0.01.
GRHL2-knockdown inhibited proliferation by targeting ZEB1
The findings above further led us to explore the potential molecules with which GRHL2 interacted to regulate proliferation in CRC. Interestingly, we found that ZEB1 expression was inversely correlated with GRHL2 expression in CRC cell lines (Fig. 5A). To further investigate whether ZEB1 was involved in GRHL2-mediated tumor progression, ZEB1 expression was examined by qRT-PCR and western blot in GRHL2-KD cells. As shown in Figure 5B and C, ZEB1 expression was increased both at mRNA and protein levels in GRHL2-KD cells compared with Scr-con control cells. Accordingly, as detected by immunofluorescence, GRHL2 was downregulated in the nuclei of GRHL-KD cells, while ZEB1 accumulated in the nuclei of these cells (Fig. 5D). Taken together, this indicates that ZEB1 expression is regulated by GRHL2.

Figure 5. GRHL2-knockdown inhibited proliferation by targeting ZEB1 (A) Inverse expression pattern of GRHL2 and ZEB1 in human CRC cell lines, as demonstrated by western blot analysis. (B) Downregulation of GRHL2 resulted in increased ZEB1 mRNA expression, and decreased E-cadherin mRNA expression. Fold change was calculated relative to the mRNA expression of Scr-con cells. **P < 0.01. (C) GRHL2-knockdown increased ZEB1 and decreased E-cadherin protein expression, as determined by western blot analysis. (D) Representative images of immunofluorescence analysis using anti-GRHL2 and anti-ZEB1 antibodies are shown. Nuclei were counterstained with DAPI. Scale bar: 50 μm. (E) Transient knockdown of ZEB1 using sh-ZEB1 resulted in decreased ZEB1 mRNA expression, but increased GRHL2 and E-cadherin mRNA in GRHL2-KD HT29 and HCT116 cells. Fold change was calculated relative to the mRNA expression of Sh-con cells. **P < 0.01. (F) Sh-ZEB1 transfection decreased ZEB1 protein and increased GRHL2 and E-cadherin protein levels in GRHL2-KD cells, as determined by western blot analysis. (G) ZEB1 knockdown using Sh-ZEB1 enhanced the proliferative ability of GRHL2-KD cells to a similar level as observed in Scr-con cells. *Relative to Sh-con vs. Sh-ZEB1. *P < 0.05, **P < 0.01. #Relative to Scr-con vs. Sh-con. #P < 0.05, ##P < 0.01. Data were expressed as the means ± SD from 3 separate experiments.
Additionally, we asked whether downregulating ZEB1 in GRHL2-KD cells could lead to enhanced cell proliferation. To this end, ZEB1 expression was downregulated by transient transfection of sh-ZEB1 in GRHL2-KD cells. Efficient knockdown of ZEB1 in GRHL2-KD cells was verified by qRT-PCR (Fig. 5E) and western blot (Fig. 5F). As shown in Figure 5G, compared with GRHL2-KD cells transfected with the sh-Con negative control, the proliferative ability of GRHL2-KD cells transfected with sh-ZEB1 was significantly enhanced to levels similar to those observed in Scr-con cells. These findings demonstrated that ZEB1 was a target through which GRHL2 regulated cell proliferation in CRC cells.
Intriguingly, we observed that repression of ZEB1 induced a striking upregulation of GRHL2 mRNA (Fig. 5E) and protein (Fig. 5F) levels in GRHL2-KD cells. This indicated that ZEB1 might also regulate the expression of GRHL2. Thus, there may exist a double-negative GRHL2/ZEB1 transcriptional regulatory feedback loop in human CRC cells. Furthermore, since ZEB1 is a direct transcriptional repressor of E-cadherin, we detected E-cadherin expression in GRHL2-KD and sh-ZEB1 cells. In addition to the previously observed increase in ZEB1 following knock-down of GRHL2, E-cadherin expression was downregulated both at mRNA (Fig. 5B) and protein (Fig. 5C) levels. Further, transient silencing of ZEB1 in GRHL2-KD cells resulted in upregulation of E-cadherin expression (Fig. 5E and F). We also transfected the E-cadherin plasmid into HT29 and HCT116 GRHL2-KD cells to identify whether re-expression of E-cadherin could restore the proliferative ability of GRHL2-KD CRC cells. Indeed, as shown in Figure S1, transient expression of E-cadherin in GRHL2-KD cells enhanced their proliferative abilities.
Discussion
Our study has identified several novel findings: GRHL2 acts as an oncogene in CRC, and that GRHL2 functions to regulate cell proliferation both in vivo and in vitro. We showed that GRHL2-knockdown in CRC cells resulted in dramatic growth inhibition. Moreover, we provided molecular evidence that GRHL2 suppressed CRC cell proliferation by targeting ZEB1.
The balance between proliferation and programmed cell death is crucial to maintain tissue and organ integrity. Disturbance of this balance by disrupting the program that regulates cell cycle entry and death can result in the transformation of normal cells into tumor cells.24 However, to date, there is no information available describing a direct effect of GRHL2 on CRC proliferation and the underlying molecular mechanisms remain to be fully delineated. A previous study demonstrated that knockdown of GRHL2 resulted in a significant reduction in cell proliferation and led to congruent loss of telomerase activity in human oral squamous cell carcinoma cells.15 In this study, for the first time, we investigated the association between GRHL2 expression and CRC proliferation status and found that GRHL2 expression, which is upregulated in CRC, was positively correlated with CRC progression. Both in vitro and in vivo assays supported that downregulation of GRHL2 decreased the cell proliferation and tumorigenesis of CRC cells. Collectively, these results indicate that GRHL2 has an oncogenic role in CRC.
GRHL2 plays an important role in epidermal junctions, in part due to its activation of target genes including E-cadherin,14 by binding to a cis-regulatory region localized in intron 2 of the E-cadherin gene.6 GRHL2 determines the epithelial phenotype of breast cancers and tightly co-regulates with E-cadherin in cancer cells. Knockdown of GRHL2 in the human mammary epithelial cell line MCF10A leads to downregulation of E-cadherin.16 GRHL2 is downregulated during epithelial-mesenchymal transition with kinetics similar to the loss of E-cadherin.14 Functional inactivation of GRHL2 results in a remarkable reduction in cell proliferation of breast cancer.19 In the present study, we demonstrated that knocking down GRHL2 in CRC cells significantly reduced the expression of E-cadherin, a surrogate marker of epithelial differentiation.25 Despite the abundance of literature supporting an anti-proliferative role for E-cadherin, there is also evidence indicating that enhanced E-cadherin expression is associated with increased cell proliferation.26,27 In CRC, E-cadherin does not always show absent or reduced expression.28 In fact, there have been reports of increased E-cadherin expression in tumors from patients with sporadic colorectal carcinomas,29 and higher expression of E-cadherin occurring in metastatic tumors.29,30 High numbers of Ki-67-expressing cells were found in the central areas of the CRC primary tumors and metastases with expression of E-cadherin. Loss of the epithelial phenotype in tumor cells was shown to be accompanied by a loss of proliferative capacity.31 The proliferative stimulus was mediated by E-cadherin engagement and coordinated through Rac1 and p120-catenin.27 E-cadherin regulates several signaling pathways that are involved in the regulation of proliferation.32 Moreover, in our CRC cell models, we demonstrated that GRHL2-knockdown decreased expression of E-cadherin, and re-expression of E-cadherin restored the proliferative capacity of GRHL2-knockdown CRC cells.
E-cadherin expression is regulated at multiple levels, with carcinomas displaying genetic, epigenetic, transcriptional, and posttranslational alterations of this protein.33 The major transcriptional repressors that are recognized to inhibit E-cadherin expression include those in the Snail and ZEB families, which directly bind to the E-cadherin promoter and repress its transcription.34 Of these, ZEB1 has the most consistent inverse correlation with E-cadherin across different types of carcinomas.35,36 ZEB1 has been demonstrated to interact with either E-box elements (especially 5′-CAGGTG-3′, 5′-CATGTG-3′, or 5′-CACCTG-3′) or Z-box elements (especially 5′-CAGGTA-3′ or 5′-TACCTG-3′) in the proximal promoters of the CDH1 (E-cadherin) gene.37 Knockdown of ZEB1 reduced cell death in response to the EGFR inhibitor erlotinib in an E-cadherin-dependent manner.38 ZEB1 regulates the cell cycle and proliferation and increases the percentage of cells in G1 phase.39 GRHL2 is also a target of ZEB1.40 ZEB1 represses GRHL2 expression by binding two Z-box elements (at positions −129 and −106) and one E-box element (at position −76) of the GRHL2 promoter region.19 Mutual regulatory relationships have been defined for GRHL2 and ZEB1,14,19 and a functional and reciprocal relationship between ZEB1 and GRHL2 has been established.40 In our study, we demonstrated that ZEB1 was a downstream target of GRHL2 in the regulation of CRC cell proliferation. We presented evidence that suggested GRHL2 might repress ZEB1 and hence relieve ZEB1-mediated CDH1 transcriptional repression, thus restoring E-cadherin expression. On the contrary, we found GRHL2 was upregulated by transient knockdown of ZEB1. Thus, similar to the ZEB1-miR200 reciprocal regulatory relationship,41,42 our data suggests that there also exists a negative feedback loop between ZEB1 and GRHL2.
In conclusion, our study demonstrated that GRHL2 expression was upregulated in CRC and was associated with CRC progression. By targeting ZEB1, GRHL2-knockdown induced downregulation of E-cadherin and inhibited cell proliferation in CRC cells. Although the function of GRHL2 in cancer remains to be fully understood, our findings have identified a new molecular target of GRHL2 and a mechanism of action by which GRHL2 may contribute to CRC progression. Therefore, GRHL2 may potentially be an important novel target for CRC prevention and therapy.
Materials and Methods
Clinical specimens
Fresh CRC tissues were collected from January 2011 to December 2011. Each sample was matched with the adjacent non-tumor mucosa removed during the same surgery. Cancer tissues were cut into wedge shapes and non-tumor mucosa were cut at least 5 cm away from the tumor margin. Tissues were cut as soon as the surgical specimens were excised and then washed with saline. For PCR and western blot analysis, specimens were snap-frozen in liquid nitrogen, and then stored at −80 °C until use. For immunohistochemistry stains, samples were then fixed with formalin for two weeks and embedded with paraffin. All patients were diagnosed with histologically confirmed CRC and were classified according to the 7th edition of the TNM staging system.43 Informed consent for sample collection was obtained from all patients. This study was approved by the Ethical Committee of Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine.
Immunohistochemical staining
Immunohistochemistry (IHC) stains were performed using formalin-fixed, parafin-embedded tissue sections of tissue blocks as previous described.44 The antibodies used were the rabbit monoclonal anti-GRHL2 antibody (1:300, HPA004820, Sigma) and the mouse monoclonal anti-Ki-67 antibody (1:200, sc-23900, Santa Cruz Biotechnology). The slides were analyzed by standard light microscopy. Negative controls were stained with IgG as primary antibody. Nuclear immunostaining in tumor cells was considered positive staining. GRHL2 IHC staining was based on the proportion of cell staining (0 = 0%, 1 ≤ 25%, 2 = 25% to 50%, 3 = 51% to 75%, 4 ≥ 75% positive cells) and the staining intensity (0 = no staining, 1 = weak, 2 = moderate, 3 = strong). The scores for intensity and percentage were multiplied. An overall score of ≤ 3 was defined as negative, while a score > 3 was defined as positive. The percentage of tumor cells positive for Ki-67 was considered low or high when < 40% or > 40% of tumor cells, respectively, showed positive staining. IHC stains were scored by two independent researchers, blinded to the clinical characteristics of the patients.
Cell culture
HT29 and HCT116 cells were obtained from the American Type Culture Collection (ATCC) and maintained in McCoy’s 5A medium (Corning Cellgro®) supplemented with 10% fetal bovine serum (FBS, Corning Cellgro®). The human colorectal cancer cells SW116, LoVo, SW480, SW620, and Caco2 were preserved in our institute. These cells were grown in RPMI-1640 (Corning Cellgro®) medium containing 10% fetal bovine serum (FBS, Corning Cellgro®). All the cells were cultured at 37 °C in a humidified atmosphere of 5% CO2.
Generation of GRHL2 stable knockdown cells by lentiviral transduction
Plasmids with sh-GRHL2 (GRHL2-KD; sc-77606-V) or the scrambled shRNA control (Scr-con; sc-108080) encoded in lentiviral particles were purchased from Santa Cruz. Lentiviral transfection was performed according to the manufacturer’s instruction to establish stable GRHL2 knockdown in HT29 and HCT116 cells. For establishment of stably transfected cells, puromycin dihydrochloride (sc-108071, Santa Cruz, US, 5.0 ug/mL for HCT116; 3.0 ug/mL for HT29) selection was performed for 2 wk.
Transient transfection
Short hairpin RNA against human ZEB1 (Sh-ZEB1) and negative control shRNA (Sh-con) were purchased from GenePharma. Human E-cadherin-pcDNA3.0 plasmid, encoding E-cadherin protein, was gifted from Tao Du in our institute. HT29-GRHL2-KD and HCT116-GRHL2-KD cells were seeded in 6-well culture plates one day before transfection, and then were transfected transiently using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instruction. Forty-eight hours after transfection, cells were verified and used for analysis.
RT-PCR and quantitative real-time PCR
Total RNA was extracted from cells and tissues using Trizol (Invitrogen) according to the manufacturer’s protocol. Total RNA (0.5 μg) from each sample was used for first-strand cDNA synthesis using a reverse transcriptional kit (Promega). For RT-PCR, samples were analyzed on a 1.5% agarose gel. The qRT-PCR was performed using cDNA as a template and Universal PCR Master Mix (Applied Biosystems) on an Applied Biosystems 7900HT sequence detection system (Applied Biosystems). Primers used for qRT-PCR analysis were as follows: GRHL2, GGAAATCTAG CCCTGGGTTT G (forward) and TCAGGGAGGA ACGCACTGA (reverse); GAPDH, AAGGTGAAGG TCGGAGTCAA C (forward) and GGGGTCATTG ATGGCAACAA TA (reverse). The relative amount of mRNA was normalized using GAPDH as an endogenous control.
Western blot
Whole cellular and tissue proteins were extracted with RIPA lysis buffer (Solarbio) containing 0.2 mM phenylmethylsulfonyl fluoride (PMSF), according to standard methods. Western blot analysis was performed on protein extracts from cells and tissues. Primary antibodies used in this study were as follows: GRHL2 (1:300, HPA004820, Sigma), ZEB1 (1:500, sc-25388, Santa Cruz Biotechnology), E-cadherin (1:500, 3195, Cell Signaling Technology). Goat anti-mouse or goat anti-rabbit IgG conjugated to horseradish peroxidase (1:1000, HRP, Pierce) was used as the secondary antibody. Chemiluminescent signals were visualized using Super Signal West Pico Chemiluminescent Substrate (Pierce) and the signal intensity was analyzed using the Image Lab™ Software Version 4.0.1 (BIO-RAD). The experiments were performed in triplicate with GAPDH (1:10 000; Sigma-Aldrich) as an endogenous control.
Cell proliferation assays
Cell Counting Kit-8 (CCK-8; CK04, DOJINDO) was used to assess cell proliferation according to the manufacturer’s protocol. Cells were seeded onto 96-well plates at a density of 1.5 × 103 cells/well and cultured for 24, 48, 72, 96, and 120 h. Then, 10 μL of the CCK-8 solution was added to each well of the plate and incubated for another 3 h. The absorbance was measured at 450 nm with a microplate reader.
Immunofluorescence
Immunofluorescence was performed as previous described.45 Briefly, GRHL2-KD and Scr-con cells were incubated with the primary antibody overnight at 4 °C, followed by incubation with fluorescent secondary antibody for 2 h at room temperature. Primary antibodies used were: GRHL2 (1:100, HPA004820, Sigma-Aldrich), ZEB1 (1:100, sc-25388, Santa Cruz). Secondary antibodies used were Alexa Fluor® 555 conjugated anti-rabbit (1:1000, 4413) and Alexa Fluor® 488 conjugated anti-mouse (1:1000, 4408) from Cell Signaling Technology. After final washes with phosphate-buffered saline (PBS), the coverslips were mounted using an anti-fade mounting solution containing 4',6-diamidino-2-phenylindole (DAPI; P36935, Invitrogen). Images were captured using the AMG EVOS® FL Color Imaging System (Applied Biosystems).
Tumor formation in vivo animal model
HT29 and HCT116 cells with stable knock-down of GRHL2 (HT29-GRHL2-KD and HCT116-GRHL2-KD, respectively) or the scrambled empty vector (HT29-Scr-con and HCT116-Scr-con, respectively) were collected and suspended in 100 μL of PBS at a concentration of 1 × 107 cells/mL, then subcutaneously implanted into nude mice (male BALB/c nu/nu nude mice, 4-wk-old, 6 mice per group). The tumor size (V) was determined by measuring the length and width of the tumor and using the formula V = (width2 × length) / 2. Tumor size was measured every 4 d using calipers. Tumor tissues were resected after 32 d, and tumor masses were examined by western blot. All animal experiments were performed according to study protocols that comply with the institution’s guidelines and animal research laws.
Statistical analysis
IBM SPSS Statistical software (version 19.0) was utilized for statistical analysis. Correlations between GRHL2 expression in CRC tissues and clinicopathological parameters were analyzed using the Pearson Chi-square (χ2) test. Differences were compared using a two-tailed Student t test. All P values were determined from 2-tailed tests and differences with a P value < 0.05 were considered to be statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank Ms Goldie Lui for her kind help with reviewing the manuscript. We wish to thank Joey Jia, Zhonghua Zhao, Ying Qu, Bingqing Yu, Minmin Shi, and Tao Du for help, advice and reagents. We thank Yan Shen for help with performing the mouse xenografts experiment. This study was supported by grants from National Natural Science Foundation of China (NSFC, 30971488, 81300290, 81372187, and 81172521).
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 3.Ewing I, Hurley JJ, Josephides E, Millar A. The molecular genetics of colorectal cancer. Frontline Gastroenterol. 2014;5:26–30. doi: 10.1136/flgastro-2013-100329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auden A, Caddy J, Wilanowski T, Ting SB, Cunningham JM, Jane SM. Spatial and temporal expression of the Grainyhead-like transcription factor family during murine development. Gene Expr Patterns. 2006;6:964–70. doi: 10.1016/j.modgep.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Wilanowski T, Tuckfield A, Cerruti L, O’Connell S, Saint R, Parekh V, Tao J, Cunningham JM, Jane SM. A highly conserved novel family of mammalian developmental transcription factors related to Drosophila grainyhead. Mech Dev. 2002;114:37–50. doi: 10.1016/S0925-4773(02)00046-1. [DOI] [PubMed] [Google Scholar]
- 6.Werth M, Walentin K, Aue A, Schönheit J, Wuebken A, Pode-Shakked N, Vilianovitch L, Erdmann B, Dekel B, Bader M, et al. The transcription factor grainyhead-like 2 regulates the molecular composition of the epithelial apical junctional complex. Development. 2010;137:3835–45. doi: 10.1242/dev.055483. [DOI] [PubMed] [Google Scholar]
- 7.Varma S, Cao Y, Tagne J-B, Lakshminarayanan M, Li J, Friedman TB, Morell RJ, Warburton D, Kotton DN, Ramirez MI. The transcription factors Grainyhead-like 2 and NK2-homeobox 1 form a regulatory loop that coordinates lung epithelial cell morphogenesis and differentiation. J Biol Chem. 2012;287:37282–95. doi: 10.1074/jbc.M112.408401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Laer L, Van Eyken E, Fransen E, Huyghe JR, Topsakal V, Hendrickx J-J, Hannula S, Mäki-Torkko E, Jensen M, Demeester K, et al. The grainyhead like 2 gene (GRHL2), alias TFCP2L3, is associated with age-related hearing impairment. Hum Mol Genet. 2008;17:159–69. doi: 10.1093/hmg/ddm292. [DOI] [PubMed] [Google Scholar]
- 9.Pyrgaki C, Liu A, Niswander L. Grainyhead-like 2 regulates neural tube closure and adhesion molecule expression during neural fold fusion. Dev Biol. 2011;353:38–49. doi: 10.1016/j.ydbio.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senga K, Mostov KE, Mitaka T, Miyajima A, Tanimizu N. Grainyhead-like 2 regulates epithelial morphogenesis by establishing functional tight junctions through the organization of a molecular network among claudin3, claudin4, and Rab25. Mol Biol Cell. 2012;23:2845–55. doi: 10.1091/mbc.E12-02-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moussian B, Uv AE. An ancient control of epithelial barrier formation and wound healing. Bioessays. 2005;27:987–90. doi: 10.1002/bies.20308. [DOI] [PubMed] [Google Scholar]
- 12.Gao X, Vockley CM, Pauli F, Newberry KM, Xue Y, Randell SH, Reddy TE, Hogan BL. Evidence for multiple roles for grainyheadlike 2 in the establishment and maintenance of human mucociliary airway epithelium. Proc Natl Acad Sci U S A. 2013;110:9356–61. doi: 10.1073/pnas.1307589110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiang J, Fu X, Ran W, Chen X, Hang Z, Mao H, Wang Z. Expression and role of grainyhead-like 2 in gastric cancer. Med Oncol. 2013;30:714. doi: 10.1007/s12032-013-0714-5. [DOI] [PubMed] [Google Scholar]
- 14.Cieply B, Riley P, 4th, Pifer PM, Widmeyer J, Addison JB, Ivanov AV, Denvir J, Frisch SM. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 2012;72:2440–53. doi: 10.1158/0008-5472.CAN-11-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang X, Chen W, Kim RH, Kang MK, Park N-H. Regulation of the hTERT promoter activity by MSH2, the hnRNPs K and D, and GRHL2 in human oral squamous cell carcinoma cells. Oncogene. 2009;28:565–74. doi: 10.1038/onc.2008.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiang X, Deng Z, Zhuang X, Ju S, Mu J, Jiang H, Zhang L, Yan J, Miller D, Zhang HG. Correction: grhl2 determines the epithelial phenotype of breast cancers and promotes tumor progression. PLoS One. 2013;8:e50781. doi: 10.1371/annotation/aa633603-370b-4613-bf35-2e5f999cff38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Vasudevan P, Parekh V, Penev A, Cunningham JM. Bridging cancer biology with the clinic: relative expression of a GRHL2-mediated gene-set pair predicts breast cancer metastasis. PLoS One. 2013;8:e56195. doi: 10.1371/journal.pone.0056195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka Y, Kanai F, Tada M, Tateishi R, Sanada M, Nannya Y, Ohta M, Asaoka Y, Seto M, Shiina S, et al. Gain of GRHL2 is associated with early recurrence of hepatocellular carcinoma. J Hepatol. 2008;49:746–57. doi: 10.1016/j.jhep.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 19.Werner S, Frey S, Riethdorf S, Schulze C, Alawi M, Kling L, Vafaizadeh V, Sauter G, Terracciano L, Schumacher U, et al. Dual roles of the transcription factor grainyhead-like 2 (GRHL2) in breast cancer. J Biol Chem. 2013;288:22993–3008. doi: 10.1074/jbc.M113.456293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci. 2009;66:773–87. doi: 10.1007/s00018-008-8465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–66. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 22.Sánchez-Tilló E, Lázaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29:3490–500. doi: 10.1038/onc.2010.102. [DOI] [PubMed] [Google Scholar]
- 23.Benjamin JM, Kwiatkowski AV, Yang C, Korobova F, Pokutta S, Svitkina T, Weis WI, Nelson WJ. AlphaE-catenin regulates actin dynamics independently of cadherin-mediated cell-cell adhesion. J Cell Biol. 2010;189:339–52. doi: 10.1083/jcb.200910041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Paredes J, Figueiredo J, Albergaria A, Oliveira P, Carvalho J, Ribeiro AS, Caldeira J, Costa AM, Simões-Correia J, Oliveira MJ, et al. Epithelial E- and P-cadherins: role and clinical significance in cancer. Biochim Biophys Acta. 2012;1826:297–311. doi: 10.1016/j.bbcan.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez FJ, Lewis-Tuffin LJ, Anastasiadis PZ. E-cadherin’s dark side: possible role in tumor progression. Biochim Biophys Acta. 2012;1826:23–31. doi: 10.1016/j.bbcan.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu WF, Nelson CM, Pirone DM, Chen CS. E-cadherin engagement stimulates proliferation via Rac1. J Cell Biol. 2006;173:431–41. doi: 10.1083/jcb.200510087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang W, Hiscox S. beta-catenin-cell adhesion and beyond (review) Int J Oncol. 1997;11:635–41. doi: 10.3892/ijo.11.3.635. [review] [DOI] [PubMed] [Google Scholar]
- 29.El-Bahrawy MA, Poulsom R, Jeffery R, Talbot I, Alison MR. The expression of E-cadherin and catenins in sporadic colorectal carcinoma. Hum Pathol. 2001;32:1216–24. doi: 10.1053/hupa.2001.28948. [DOI] [PubMed] [Google Scholar]
- 30.Bukholm IK, Nesland JM, Børresen-Dale AL. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients [see comments] J Pathol. 2000;190:15–9. doi: 10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 31.Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A. 2001;98:10356–61. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207–35. doi: 10.1146/annurev.cellbio.19.011102.111135. [DOI] [PubMed] [Google Scholar]
- 33.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 34.Peinado H, Portillo F, Cano A. Transcriptional regulation of cadherins during development and carcinogenesis. Int J Dev Biol. 2004;48:365–75. doi: 10.1387/ijdb.041794hp. [DOI] [PubMed] [Google Scholar]
- 35.Sánchez-Tilló E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci U S A. 2011;108:19204–9. doi: 10.1073/pnas.1108977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sánchez-Tilló E, Siles L, de Barrios O, Cuatrecasas M, Vaquero EC, Castells A, Postigo A. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am J Cancer Res. 2011;1:897–912. [PMC free article] [PubMed] [Google Scholar]
- 37.Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, Jung A, Kirchner T, Brabletz T. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology. 2006;131:830–40. doi: 10.1053/j.gastro.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 38.Haddad Y, Choi W, McConkey DJ. Delta-crystallin enhancer binding factor 1 controls the epithelial to mesenchymal transition phenotype and resistance to the epidermal growth factor receptor inhibitor erlotinib in human head and neck squamous cell carcinoma lines. Clin Cancer Res. 2009;15:532–42. doi: 10.1158/1078-0432.CCR-08-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Postigo AA. Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J. 2003;22:2443–52. doi: 10.1093/emboj/cdg225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cieply B, Farris J, Denvir J, Ford HL, Frisch SM. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013;73:6299–309. doi: 10.1158/0008-5472.CAN-12-4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop–a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11:670–7. doi: 10.1038/embor.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–9. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471–4. doi: 10.1245/s10434-010-0985-4. [DOI] [PubMed] [Google Scholar]
- 44.Huang A, Zhou H, Zhao H, Quan Y, Feng B, Zheng M. High expression level of TMPRSS4 predicts adverse outcomes of colorectal cancer patients. Med Oncol. 2013;30:712. doi: 10.1007/s12032-013-0712-7. [DOI] [PubMed] [Google Scholar]
- 45.Dong TT, Zhou HM, Wang LL, Feng B, Lv B, Zheng MH. Salinomycin selectively targets ‘CD133+’ cell subpopulations and decreases malignant traits in colorectal cancer lines. Ann Surg Oncol. 2011;18:1797–804. doi: 10.1245/s10434-011-1561-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.