

Abstract
Microtubules are essential cytoskeletal components with a central role in mitosis and have been particularly useful as a cancer chemotherapy target. We synthesized a small molecule derivative of a symmetrical 1,3-phenyl bis-thiourea, (1,1'-[1,3-phenylene]bis[3-(3,5-dimethylphenyl)thiourea], named “41J”), and identified a potent effect of the compound on cancer cell survival. 41J is cytotoxic to multiple cancer cell lines at nanomolar concentrations. Cell death occurred by apoptosis and was preceded by mitotic arrest in prometaphase. Prometaphase arrest induced by 41J treatment was accompanied by dissociation of cyclin B1 levels from the apparent mitotic stage and by major spindle abnormalities. Polymerization of purified tubulin in vitro was directly inhibited by 41J, suggesting that the compound works by directly interfering with microtubule function. Compound 41J arrested the growth of glioblastoma multiforme xenografts in nude mice at doses that were well-tolerated, demonstrating a relatively specific antitumor effect. Importantly, 41J overcame drug resistance due to β-tubulin mutation and P-glycoprotein overexpression. Compound 41J may serve as a useful new lead compound for anticancer therapy development.
Keywords: chemotherapy, microtubule, mitosis, drug resistance, xenograft
Introduction
Tubulin-binding agents (TBAs) are established chemotherapeutic drugs that have been used successfully for the treatment of numerous adult and pediatric malignancies, since they were initially discovered.1,2 These compounds interfere with the cytoskeleton by binding to distinct sites along the hollow tube structure of microtubules. Assembled by α- and β-tubulin heterodimer constituents, the microtubule network performs critical functions in many cellular processes, including the maintenance of cell shape, motility, intracellular transport, and cell division.3 Specifically, during the mitotic stage of prometaphase, sister chromatids become attached at the kinetochore by spindle microtubules that radiate from the microtubule-organizing center at the centrosome. This process occurs during the spindle-assembly checkpoint until all kinetochores are attached, which permits irreversible progression into anaphase by the separation of sister chromatids.
When tumor cells are treated with TBAs, mitosis is halted in prometaphase to cause cell cycle arrest. This prolonged cell cycle arrest leads to apoptosis through an unknown mechanism that likely involves mitotic slippage and catastrophe.4 At high concentrations, microtubule-stabilizing agents bind and prevent the depolymerization of tubulin, while microtubule-destabilizing agents inhibit tubulin polymerization. At clinically relevant concentrations, however, both stabilizing and destabilizing agents suppress dynamic instability to prevent microtubule shortening and growth, and effectively block the transition from metaphase to anaphase. This accepted model has been recently reevaluated, in the context of patient therapy for cancer, since TBAs are paradoxically effective in malignancies when few cells are thought to be undergoing cell division.5 Thus, it has been proposed that TBAs, in addition to their antimitotic effects, interfere with vesicle trafficking and other cellular functions as additional basis for their cytotoxicity.
Classical examples of microtubule-stabilizing and destabilizing agents are the taxanes and Vinca alkaloids, respectively. Both are widely used, derived from natural products, and continue to be delivered in combinatorial treatment strategies, even with the development of newer targeted therapies. Nonetheless, the drawbacks of TBAs significantly limit their usage and efficacy in the clinical setting. These obstacles include complex synthesis, difficult route of administration (i.v.), low bioavailability, neural and systemic toxicity, and drug resistance.6 For TBAs, examples of drug resistance may be intrinsic or acquired and encompass multiple molecular mechanisms. Acquired drug resistance in patients may arise from upregulated expression of the multidrug resistance (MDR1) gene to promote drug efflux, overexpression of non-targeted microtubule isoforms, and mutations in the targeted microtubule isoforms that prevent drug binding. Overall, drugs that can circumvent clinically relevant modes of resistance and can address other disadvantages of TBAs are greatly needed to improve this important chemotherapeutic strategy.
We performed a search for known compounds that could be used as anticancer therapy, and focused on non-peptidic cyclophilin inhibitors as a possible strategy.7,8 With the goal of targeting subsets of cyclophilins, we synthesized various derivatives of the lead compound 41 (which has been shown to bind to cyclophilin A), and performed preliminary cytotoxic screens for several compounds. One compound, named 41J, was discovered to be cytotoxic to cells at nanomolar concentrations; however, subsequent testing revealed that it lacks the high affinity for cellular cyclophilins previously demonstrated for the parent compound 41. 41J is cytotoxic, causing multiple cancer and TBA-resistant cell lines to die via apoptosis. Furthermore, 41J treatment caused a robust cell cycle arrest, which was accompanied by the upregulation of mitotic transcripts. Moreover, compound 41J dramatically increased mitotic transit time and accelerated mitotic entry. To elucidate the mechanism of drug action, we performed in vitro tubulin polymerization assays and found that 41J is a direct inhibitor of microtubule growth. Lastly, compound 41J significantly arrested the growth of glioblastoma xenografts in an in vivo model. Thus, our findings demonstrate the discovery of a novel microtubule-destabilizing agent that has a simple chemical synthetic procedure, and which may serve as a useful lead compound for the discovery of novel anticancer therapeutics.
Results
Following a search for cyclophilin inhibitors, we synthesized compound 41 (due to its reported high affinity for cyclophilin A) and an unbiased set of derivatives of this structure to target the compound for other cyclophilin family proteins.7 Testing of compound 41J revealed that it was uniquely highly effective at inhibiting cell viability (Table 1).
Table 1. Cytotoxicity of 41J in tumor and drug-resistant cell lines.
| 41J | Vincristine | Colchicine | Paclitaxel | |||||
|---|---|---|---|---|---|---|---|---|
| IC50a | RRb | IC50 | RR | IC50 | RR | IC50 | RR | |
| Jurkat | 161 ± 7.3 | 1.9 ± 0.4 | 10.5 ± 0.9 | 1.4 ± 0.2 | ||||
| HeLa | 440 ± 36.2 | 37.0 ± 28.4 | 13.7 ± 5.0 | 51.2 ± 2.0 | ||||
| U251 | 825 ± 568 | 1.7 ± 0.8 | 23.3 ± 3.4 | 16.9 ± 13.6 | ||||
| HT29 | 1132 ± 519 | 9.4 ± 0.4 | 13.1 ± 4.4 | 4.2 ± 0.2 | ||||
| T24 | 1231 ± 392 | 128 ± 99.4 | 22.2 ± 2.3 | 65.0 ± 24.0 | ||||
| 1A9 | 269 ± 70.1 | 2.3 ± 0.4 | 12.3 ± 1.3 | 2.6 ± 0.3 | ||||
| PTX10 | 1412 ± 255 | 5.2 | 7.5 ± 1.8 | 3.2 | 20.8 ± 2.2 | 1.7 | 77.6 ± 6.1 | 29.3 |
| PTX22 | 981 ± 224 | 3.6 | 6.7 ± 0.8 | 2.9 | 18.6 ± 2.4 | 1.5 | 56.7 ± 5.7 | 21.5 |
| CCRF-CEM | 377 ± 18.8 | 2.0 ± 0.2 | 8.9 ± 0.1 | 1.8 ± 0.01 | ||||
| CEM/VCR R | 1349 ± 24 | 3.6 | 2861 ± 552 | 1446 | 527 ± 65.0 | 59.1 | 928 ± 214 | 518 |
| AuxB1 | 664 ± 458 | 133 ± 83.4 | 214 ± 2.5 | 143 ± 77.1 | ||||
| CHRC5 | 2640 ± 551 | 4.0 | 1035 ± 797 | 7.8 | 1824 ± 167 | 8.5 | 2701 ± 2264 | 18.9 |
aIC50, average concentration (nM) required to inhibit half of maximal cell viability (n = 3, mean IC50 ± SEM). bRR, relative resistance is the IC50 of the resistant cell line divided by the IC50 of the parental cell line.
Compound 41J is cytotoxic to cancer cells in vitro
To define the activity of this newly identified compound, we quantified the cytotoxic activity of 41J in a range of cell lines (Table 1; Fig. 1A). Following 48 h incubation with the compound or control drugs, we determined the percent of viable cells using the resazurin assay. Of the cell lines tested, we found a range of average concentrations for 50% inhibition of cell viability (IC50) values from 161 ± 7.3 nM in Jurkat cells to 1231 ± 392 nM in T24 cells. 41J was approximately 20 times more effective than the parent compound 41 (data not shown).

Figure 1. 41J is cytotoxic to cancer cell lines and inhibits proliferation after removal. (A) Jurkat cells were treated with 41J, colchicine (COL), vincristine (VCR), or paclitaxel (PTX) for 48 h, and cell viability was measured by the resazurin assay. Fluorescence intensity from control, DMSO-treated cells was set at 100%. Nonlinear least-sqaures regression was performed to determine the half-maximal concentration (IC50) for each drug. (B and C) Jurkat (B), U251, and T98G cells were treated with DMSO or concentrations of 41J for 72 h followed by annexin V-Cy5 and propidium iodide staining. (D) T98G cells were treated with DMSO or 500 nM 41J (12 or 24 h), and stained with Hoechst 33342. Cells arrested in prometaphase (arrowheads) and apoptotic nuclei (arrows) were visible at both time points. (E) U251 cells were treated with DMSO, 500 nM 41J, or 2.5 μM 17-AAG for 48 h. Culture media was removed, cells were washed with PBS and replated. Proliferation was measured by flow cytometric counting every 24 h.
To explain the diminished viability of 41J-treated cells, we next aimed to determine if cell death was an outcome of treatment. Therefore, we performed annexin V and propidium iodide (PI) staining of cells to distinguish between apoptotic and necrotic forms of cell death, and Hoechst 33342 staining to visualize whether apoptotic nuclei could be seen. At all concentrations tested, a substantial population that was annexin V-positive and PI-negative appeared, consistent with induction of apoptosis by 41J treatment. At higher concentrations, the percentage of cells that were positive for both annexin V and PI increased, due to increased amounts of cell death, which could either represent necrosis or late stages of apoptosis (Fig. 1B). Conversely, the live cell population (annexin V-negative, PI-negative) decreased with increasing 41J concentrations (Fig. 1B and C). These results were recapitulated in glioblastoma cell lines, U251 and T98G (data not shown), although concentrations for 100% death of T98G cells were not achievable. In addition, nuclear morphology of 41J-treated T98G cells stained with Hoechst 33342 revealed bright, densely staining areas consistent with cellular apoptosis (Fig. 1D).
Although a small fraction of treated cells did not appear to die, based on microscopic appearance and normal light scattering upon flow cytometry, cultures of treated cells did not readily grow back. We wondered whether 41J treatment might impair proliferation in addition to causing cell death. This type of response to chemotherapeutic drugs has been previously demonstrated following treatment with the HSP90 inhibitor 17-AAG.9 We exposed U251 cells to 500 nM 41J or 2.5 µM 17-AAG for 48 h, and then removed any residual drug by washing and re-plating the remaining live cells in complete medium. There was a clear impairment of proliferation that extended for at least 6 d following this treatment (Fig. 1E), indicating a potential impact of the compound on proliferation.
Compound 41J induces cell cycle arrest in mitosis
To identify a possible molecular mechanism for the effects of 41J in cell killing, we performed a gene expression analysis using the Illumina HumanHT-12 v4 BeadChip platform on U251 cells treated for 12 h with 41J (2 μM) or DMSO (Table S1). Notable among the ten most upregulated probes were those corresponding to proteins involved in mitosis, including cyclin B1, the Aurora-A activator Bora, CDC28 regulatory subunit, centromere protein CENP-A, NIMA-related kinase 2 (NEK2), and protein kinase Aurora-A. Among the most downregulated probes were four different tubulin genes (TUBB3, 4A, 2A, and 2B). Consistent with this enrichment of mitotic genes, microscopic examination of Hoechst 33342 stained cells demonstrated, in addition to apoptotic nuclei, numerous cells that appeared to be arrested in prometaphase (Fig. 1D). Treatment of U251 cells with 600 nM 41J, (below the IC50 for U251 of 825 nM) caused a potent arrest at the tetraploid (4N) stage, as judged by PI staining (Fig. 2A). Likewise, we observed a dose-dependent arrest in G2/M with treatment of T98G glioblastoma cells (Fig. 2B). This effect was observed as early as 6 h post-treatment and became more prominent by 24 h (Fig. 2C).

Figure 2. 41J causes G2/M arrest. Cells were harvested by trypsinization, permeabilized with 0.1% Triton X-100, and stained with propidium iodide. (A and B) U251 (A) and T98G (B) cells were treated for 24 h with DMSO or 41J at the indicated concentrations. (C) U251 cells were treated for 6, 12, or 24 h with DMSO, 500 nM 41J, 165 nM nocodazole (NOC), or 8 nM paclitaxel (PTX). Cell cycle populations were determined by software analysis.
Entry into mitosis is guided by a rise in cyclin B1 levels beginning in S phase with peak levels in prometaphase. The activation of the anaphase-promoting complex (APC/C) at the transition from metaphase to anaphase leads to the rapid degradation of cyclin B1.10 Because cyclin B1/CDK1 are critical controllers of mitotic entry and exit, we hypothesized that the expression levels of cyclin B1 protein would be increased, consistent with transcript levels, in 41J-treated cells. We quantified cyclin B1 expression by immunofluorescence in treated cells that were arrested at prometaphase. Interestingly, following treatment with 41J at 500 nM or 2 µM, we discovered that arrested cells express a wide range of cyclin B1 levels. While DMSO-treated cells showed high cyclin B1 levels in prometaphase, 41J-treated cells that had prometaphase chromatin showed variable expression of cyclin B1 (Fig. 3A and B). The absence of P-H3Ser10 expression may indicate a spontaneous exit from mitosis in a subset of the arrested 41J-treated cells. Thus, cyclin B1 protein expression becomes decoupled from its normal mitotic levels due to prometaphase arrest by 41J treatment.

Figure 3. 41J causes variable cyclin B1 expression, mitotic arrest, and accelerates mitotic entry. (A and B) T98G cells were treated with DMSO, 500 nM or 2 μM 41J for 12 h. Immunofluorescence staining was conducted for cyclin B1 and phospho-histone H3 Ser-10 (P-H3). Cyclin B1 expression was quantified for DMSO-treated cells in all stages of mitosis and 41J-treated cells arrested in prometaphase using ImageJ software. Cells positive (arrowhead) and negative for cyclin B1 (arrow) are indicated for DMSO and 2 μM 41J treatment. (C and D) T98G cells were treated with DMSO or 500 nM 41J for 35 h and observed by time-lapse microscopy. Duration of mitosis (C) and time of mitotic entry (D) were recorded for cells treated with DMSO (n = 75) or 41J (n = 76).
41J accelerates mitotic entry and prolongs mitosis
Live cell imaging allowed the visualization of the temporal and phenotypic nature of the mitotic arrest induced by 41J. Time-lapse imaging was performed for T98G cells treated with 500 nM of compound 41J, and individual cells were monitored from the time of mitotic entry (based on loss of attachment to the plate) to the time of mitotic exit. We discovered that 41J dramatically extended the time in mitosis compared with DMSO-treated cells (DMSO: 1.09 ± 0.27 h; 41J: 10.64 ± 0.45 h) (Fig. 3C). In addition, cells treated with 41J entered mitosis sooner than cells treated with diluent alone (P = 0.0225) (Fig. 3D). Taken together, our results demonstrate that 41J causes severe mitotic dysregulation.
Compound 41J causes spindle abnormalities and inhibits tubulin polymerization
41J-treated U251 cells stained for α-tubulin demonstrated spindle abnormalities (Fig. 4A), resembling repeated observations of cells arrested in mitosis by exposure to vincristine or paclitaxel.11,12 Similar to paclitaxel treatment, 41J caused the formation of multiple microtubule asters, surrounded by uncongressed prometaphase chromosomes. Both centrosomes colocalized to asters; however they were unseparated in contrast to the normal, bipolar prometaphase spindle (Fig. 4A). Notably, these defective spindles were observed at 41J concentrations 8-fold lower than the cell viability IC50, suggesting potent arrest achieved below the threshold of cell death induction. The uncongressed chromosomes, as labeled by centromere staining, did not colocalize with microtubule ends (Fig. 4B). Collectively, these observations suggest that the drug-induced mitotic arrest is controlled by a slow search-and-capture process, resulting in immobile chromosomes, and leading to a failure to turn off the spindle assembly checkpoint.

Figure 4. 41J causes spindle abnormalities and inhibits the rate of in vitro tubulin polymerization. (A) U251 cells were treated for 10 h with DMSO (interphase, prometaphase, anaphase), 50 nM paclitaxel (PTX), 50 nM vincristine (VCR), or 100 nM 41J. Cells were stained for α-tubulin and centrin. (B) T98G cells were treated for 8 h with DMSO (interphase, metaphase), 330 nM nocodazole (NOC), or 500 nM 41J. Cells were stained for α-tubulin and centromeres. (C) The in vitro tubulin polymerization assay was conducted using purified tubulin with 15% glycerol. Controls were DMSO, 3 μM PTX, and 3 μM VCR. The results are representative of three independent experiments.
Perturbed microtubule dynamics could stem from direct or indirect interaction between 41J and tubulin. To distinguish between these two possibilities, we performed an in vitro tubulin polymerization assay using fluorescent reporter readout in real-time with purified tubulin (Fig. 4C). Compared with DMSO alone, 41J potently inhibited the growth rate of microtubules in a dose-dependent manner. Even the lowest concentration of 41J tested (3 µM), with a molar ratio of 41J to tubulin at 1:12, inhibited polymerization.
Compound 41J inhibits the growth of xenografts
While compound 41J demonstrated reproducible toxicity in vitro, whether 41J exhibited the ability to kill tumor cells in vivo was unknown. To establish xenografts, we injected 1 × 106 GBM43 cells into the flanks of nude mice. Upon reaching a tumor volume of 400 mm3 on day 10, we initiated daily intraperitoneal injections of 120 mg/kg of 41J in 50 µL DMSO until an endpoint volume of 2000 mm3 was achieved for the control, vehicle-injected mice. We observed the mice on each day and saw no signs of drug toxicity. GBM43 tumors were significantly smaller in the 41J-treated group compared with the control group (P < 0.05) (Fig. 5). We conclude that 41J was able to inhibit the growth of tumors in mice at levels achievable in vivo. Of note, the pharmacokinetic characteristics of this compound are unknown, and we have not conducted an extensive test of various dosing schedules. It is very likely that more optimized conditions of compound administration or medicinal chemistry enhancements to 41J may yield even better results in longer term assays.
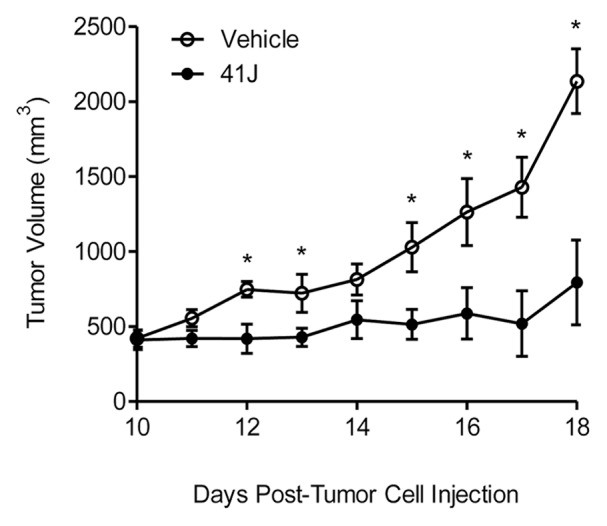
Figure 5. 41J prevents growth of glioblastoma xenografts. Human glioblastoma multiforme (GBM) 43 cells (1 × 106) were injected into the flanks of nude mice and upon reaching a tumor volume of 400 mm3, daily intraperitoneal injections of vehicle (DMSO) or 120 mg/kg of 41J were administered. Five animals were used per treatment group. Qualitatively similar results were obtained in two independent assays.
Compound 41J is able to overcome microtubule drug resistance
A common problem of clinically used tubulin-binding agents is inherent or acquired resistance in tumors, rendering them ineffective as therapeutic agents. New microtubule compounds would be potentially more valuable if they would be effective at killing resistant cells. We tested 41J in three models of resistance. Derived from 1A9 human ovarian carcinoma cells, PTX10 and PTX22 are resistant to paclitaxel, most likely due to β-tubulin mutations in the paclitaxel binding site.13 CEM/VCR R cells are resistant to vincristine, mostly through the upregulation of proteins associated with microtubules or cytoskeleton filaments.14,15 In the third model, CHRC5 cells are multi-drug resistant to several compounds, including colchicine, and have been shown to overexpress P-glycoprotein.16,17 Based on the relative resistance (RR, defined as IC50 of the resistant cell line divided by that of the parental cell line), we found that 41J was able to overcome resistance in each of these three models (Table 1; Fig. S1). For PTX10 and PTX22, the resistance factor of 41J was 6- to 7-fold lower than that of paclitaxel. For CEM/VCR R, the resistance factor of 41J was 400-fold lower than that of vincristine. For CHRC5, the resistance factor of 41J was half the resistance factor of colchicine. We conclude that 41J may not be subject to the typical modes of resistance induced by the conventionally used microtubule-specific chemotherapy drugs, and could provide a useful alternative mode of cancer cell killing.
Discussion
In this study, we report the discovery of a novel, synthetic, microtubule polymerization inhibitor. 41J kills cancer cells at nanomolar concentrations by inducing apoptosis. Similar to known tubulin-binding agents (TBAs), 41J causes potent mitotic arrest in prometaphase that is accompanied by upregulation of cyclin B1 and increased transcription of other mitotic regulators. Visualization of the spindle during mitotic arrest showed multiple asters comprised of shortened microtubule fibers, surrounded by uncongressed chromosomes. These cellular observations correspond with a dose-dependent decrease in the rate of in vitro tubulin polymerization. Furthermore, in vivo data reveal that 41J inhibits the growth of xenografts, and maintains cytotoxic activity in drug-resistant cell lines.
We have demonstrated through several different assays that 41J is cytotoxic to numerous cancer cell lines and induces apoptosis (Table 1; Fig. 1B and D). As observed by time-lapse microscopy, cell death occurred in response to prolonged mitotic arrest of approximately 11 h (Fig. 3C). We postulate that 41J-treated cells die as a consequence of mitotic catastrophe.18 This mechanism adapts to mitotic failure by driving the cell to an irreversible fate, namely necrosis, apoptosis or senescence, and has been well-described as a consequence of TBA treatment.19 Indeed, we found that 41J treatment may also result in a non-proliferative state (Fig. 1E), consistent with the occurrence of mitotic catastrophe.
Previous studies have shown that the likelihood of cell death following mitotic arrest varies greatly within and between cell lines due to TBA treatment.20-22 Cell death signals have been proposed to compete with cyclin B expression, whereby signaling leads to death during mitosis while cyclin B proteolysis leads to mitotic exit. Indeed, paclitaxel-treated cells accelerate cyclin B decay before exiting mitosis.23 Our staining of 41J-treated cells arrested in prometaphase demonstrate variable cyclin B1 levels, which likely represent snapshots of cells in different phases of cyclin B1 decay (Fig. 3A and B). Two examples we highlight are: (1) early arrest (cyclin B1high and P-H3high) and (2) late arrest, nearing mitotic exit (cyclin B1low and P-H3low). Whether cyclin B1low cells undergo 41J-induced cell death is an area for future investigation. Moreover, the identification of the apoptotic events initiating cell death should be examined.24,25
Our gene expression analysis found that seven of the ten most upregulated genes by 41J treatment are regulators of mitosis (Table S1). In addition to cyclin B1, upregulation of CKS2 was detected and is known, with CKS1, to be required for the G2-M transition by stimulating expression of cyclin B1, cyclin A, and Cdk1.26 Other transcripts included Bora, which activates protein kinase Aurora-A,27 and Nek2, a mitotic regulator of centrosome separation.28 Along with Aurora-A, Bora and Nek2 function in the polo-like kinase 1 (Plk1) pathway. Plk1 has a central role in centrosome separation for mitosis. Both centromere protein A (CENP-A) and KIF14, a mitotic kinesin, are expressed at elevated levels during mitosis as well.29,30 Among the most downregulated genes were the tubulin isotypes, αIVa, βIIa/IIb, and βIII. Published microarray profiling also observed downregulation of tubulin isotypes due to polymerization inhibition.31 Since it is known that exposure to microtubule inhibitors elicits tubulin heterodimer accumulation and autoregulation depressing tubulin mRNA,32 we can speculate that 41J also increases the heterodimer pool in cells. This would explain the decline in tubulin isotype gene expression.
Visualization of prometaphase arrest by 41J treatment revealed major spindle abnormalities, notably multiple microtubule asters, lack of chromosome congression, and unseparated centrosomes (Fig. 4A and B). Aster formation due to paclitaxel has been proposed to result from stabilization of microtubule dynamics and thus, prevention of spindle components from accumulating at centrosomes. This may cause loss of cohesion at centrosomes and the release of microtubules which become redistributed as asters.33 This process may explain the spindle abnormalities seen in the presence of 41J.
Normal mitosis involves the initiation of centrosome separation during prophase,34 but we observed 41J-treated cells with centrosomes still in close proximity after separation should have occurred (Fig. 4A). Because centrosome movement, and hence bipolar spindle formation, involves Plk1 signaling, it is possible that this pathway is inhibited by 41J. Alternatively, 41J could prevent centrosome separation through destabilization of tubulin polymerization, since microtubule motor proteins including kinesin-5 motor Eg5 and dynein are implicated in this phenomenon.35 While it has been shown that microtubule stabilizers can impair centrosome separation,36 it remains to be determined how this influences treatment responses like cell death.
We discovered that the spindle abnormalities and prometaphase arrest produced by 41J are likely caused by inhibition of microtubule assembly (Fig. 4C). There are several possible explanations for this decrease in polymerization growth rate: 41J may bind to soluble tubulin, to the microtubule ends, or along the assembled microtubule polymer, or could incorporate into the lattice.19 If the in vitro microtubule polymerization assay had revealed an effective molar ratio of 41J to tubulin of 1:1, this would have suggested that 41J binds to soluble tubulin in a stoichiometric interaction. However, since the effective ratio of 41J to tubulin was significantly less than 1:1, we speculate that 41J is able to bind to assembled microtubules either by incorporation, binding to the ends, or along the helix. Future investigations should define the exact binding interaction and characterize intracellular tubulin assembly kinetics, because most TBAs stabilize polymerization to suppress dynamic instability in cells. As well, it would be interesting to see whether 41J can depolymerize intact microtubules, as other compounds have been found to perturb dynamics without depolymerizing fibers.37
The most useful newly discovered TBAs would be those able to overcome drug resistance. In our study, we found that 41J killed vincristine-resistant cells and almost completely overcame paclitaxel resistance (Table 1; Fig. S1). Since CEM/VCR R cells have mutations in cytoskeletal proteins,15 we suspect that 41J perturbs microtubule-associated proteins or other cytoskeletal components, such as actin. Regarding a distinct model, because PTX10 and PTX22 cells have mutations in β-tubulin at the paclitaxel binding site decreasing affinity of drug binding,13 41J may associate with a unique pharmacophore on microtubules. Our gene expression data showed potent downregulation of βIII-tubulin by 41J treatment, suggesting that 41J may be useful in cases where resistance is attributable to βIII-tubulin upregulation.38,39
The discovery of improved TBAs will help expand this valuable class of anticancer agents. Taken together, these data have uncovered a new lead compound that may be useful for further development and investigation. While it is clear that 41J causes mitotic arrest, inhibits tubulin polymerization, and leads to cell death, the exact mechanistic details of this process warrant further investigation. Discovery of this novel small molecule represents an opportunity to explore a potential new class of TBAs, the basic function of the microtubule cytoskeleton, and new ways in which anticancer chemotherapy can be optimized.
Materials and Methods
Synthesis
Compound 41J was prepared following a general method as described.7 Thus, 1,3-diisothiocyanatobenzene (1 equiv) was stirred overnight with 3,5-dimethylaniline (2 equiv) in dimethylacetamide (DMA) at ambient temperature (Fig. 6). Addition of water gave a white precipitate that was filtered and washed with water and dichloromethane. The crude was purified by recrystallization with dry pure acetone in over 80% yield. The product was characterized by 1H-NMR, IR, and MS spectroscopy. 41J was dissolved in DMSO.
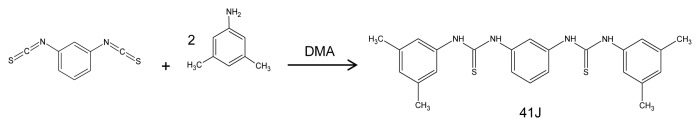
Figure 6. Synthesis reaction for compound 41J.
Antibodies and reagents
The primary antibodies used were rabbit polyclonal centrin (MC1) (kind gift from Jeffrey Salisbury, Mayo Clinic), human polyclonal anti-centromeric antibody (Antibodies Inc.), mouse monoclonal anti-cyclin B1 (BD Biosciences), rabbit polyclonal anti-phospho-histone H3 (Ser10) (Upstate), and mouse anti-α-tubulin (Sigma). Secondary antibodies used were from Molecular Probes. The drugs used in this study were paclitaxel, vincristine, colchicine, nocodazole, and verapamil (Sigma), as well as 17-AAG (Enzo Life Sciences). Colchicine and verapamil were dissolved in water, while the other drugs were dissolved in DMSO. Hoechst 33342 (Invitrogen) was dissolved in water.
Cell culture
U251, T98G, and HeLa cells were cultured in high glucose DMEM (ATCC). HT29 and T24 cells (kind gift from Scott Kaufmann, Mayo Clinic) were cultured in McCoy’s 5A medium. CCRF-CEM, CEM/VCR R (kind gift from Maria Kavallaris, University of New South Wales), and Jurkat cell lines were cultured in RPMI-1640. The Chinese hamster ovary (CHO) cell lines, AuxB1 and CHRC5 (kind gift from Scott Kaufmann), were cultured in MEM-α medium. The 1A9 parental cell line and drug-resistant cell lines, PTX10 and PTX22 (kind gifts from Antonio Tito Fojo and Marianne Poruchynsky, National Cancer Institute) were cultured in RPMI-1640. In further detail, PTX10 and PTX22 cells were cultured in RPMI-1640 until established, 15 ng/mL paclitaxel and 5 μg/mL verapamil was added, and removed at least 5–7 d before an experiment. All culture media was supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 mM l-glutamine, and incubated at 37 °C with 5% CO2.
Cytotoxicity assay
Cells were plated and incubated for 12 to 24 h. Drug treatment was initiated by media replacement with DMSO or drug-containing media. After 48 h of incubation, resazurin (Invitrogen) was added at a tenth of the total volume, and incubated at 37 °C until color development occurred. The emission at 572 nm was measured, percent fluorescence of DMSO-treated cells was calculated, and dose response curves were plotted based on nonlinear regression analysis and least squares fit. Reported values represent the mean IC50 from three independent experiments in triplicate.
Annexin V and propidium iodide assay
Cells were harvested, washed with 1× PBS, and stained with annexin V-Cy5 (BD Biosciences) and propidium iodide (Sigma) in 1× binding buffer (10 mM HEPES, pH 7.4; 140 mM NaCl; 2.5 mM CaCl2).
Cell cycle analysis
Cells were harvested, washed with 1× PBS, and stained in cold buffer containing 0.1% Triton X-100 and 50 µg/mL propidium iodide (Sigma) in 0.1% sodium citrate. Cell cycle analysis was performed using FlowJo software.
Gene expression analysis
U251 cells were treated with DMSO or 41J (2 μM) for 12 h in triplicate. Total RNA was isolated from U251 cell pellets using the RNeasy Plus Mini Kit (QIAGEN). cDNA was synthesized by reverse transcription with priming from the poly-A tail of mRNA. Next, amplification with an in vitro transcription step was used to generate biotin-labeled cRNA. This amplified pool was then assembled onto the Illumina BeadChip platform (HumanHT-12 v4). Visualization of the array was performed by staining with streptavidin-Cy3, followed by hybridization to the array. Expression values were calculated using Illumina GenomeStudio® software, and analysis was performed by the Gene Expression Core at Mayo Clinic. Fold changes between DMSO and 41J treatment were calculated based on the mean of three technical replicates, and the two-sample t test was performed to determine the P value.
Time-lapse microscopy
T98G cells were plated on glass bottom wells 16 h before treatment. DMSO or compound-containing media was added at the initiation of the time-lapse experiment. Images of the cells were captured every 7 min for approximately 36 h.
Immunofluorescence
Fixation procedures were as follows: cyclin B1, 3% paraformaldehyde in 1× PBS for 15 min at room temperature (RT); phospho-histone H3 Ser-10, 1% formaldehyde for 5 min at RT. Cells stained for cyclin B1 and phospho-histone H3 Ser-10 were permeabilized in 0.2% Triton X-100 in PBS for 12 min at RT. For α-tubulin and centrin staining, microtubules were stabilized by 5 min incubation in PHEM buffer (25 mM HEPES, 10 mM EGTA, 60 mM PIPES, 2 mM MgCl2, [pH 6.9] containing 0.5% Triton X-100), fixed and permeabilized using chilled 100% methanol for 10 min at −20 °C, and rehydrated in PBS for 5 min. Standard washing (1× PBS), blocking (3% low fat milk in PBS), and antibody incubation times were used.
Microtubule polymerization
In vitro tubulin polymerization was performed using a fluorescence reporter assay (Cytoskeleton).40 Assay conditions were 2 mg/mL porcine brain tubulin in 80 mM PIPES (pH 6.9), 2 mM MgCl2, 0.5 mM EGTA, 1 mM GTP, and 15% glycerol at 37 °C. Fluorescence intensity was measured each minute by excitation at 355 nm and emission at 460 nm. Compound concentrations were chosen based on the molar ratio of 41J to tubulin, as follows: 3 μM (1:12), 9 μM (1:4), 17 μM (1:2), and 35 μM (1:1).
Animal studies
Human glioblastoma multiforme cells (GBM43) were previously isolated from patient tumors,41 and 1 × 106 cells/animal were injected into the flanks of 8-wk-old, female, athymic nude mice (Foxn1nu, Harlan). Five mice were injected per treatment group. At tumor volumes of 400 mm3, daily intraperitoneal injections of 3 mg 41J in 50 μL DMSO (single doses of 120 mg/kg) were initiated until tumors reached 2000 mm3. Tumors were measured using digital calipers, and volumes were calculated by the formula width2 × length / 2.
Statistical analyses
Data analyses were conducted using GraphPad Prism software. All error bars presented represent mean ± standard error. The Student t test was conducted to determine statistical significance (P < 0.05).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Antonio Tito Fojo, Justin Gundelach, Scott Kaufmann, Maria Kavallaris, Liviu Malureanu, Marianne Poruchynsky, Ann Reed, Jeffrey Salisbury, Jan van Deursen, Ying Li from the Mayo Clinic Gene Expression Core Facility, and the Mayo Clinic Flow Cytometry/Optical Morphology Resource for technical advice and assistance. This work was supported by a grant from the University of Minnesota, Mayo Clinic Partnership and from the National Institutes of Health, R01NS077555-01.
Glossary
Abbreviations:
- TBA
tubulin-binding agent
- COL
colchicine
- PTX
paclitaxel
- NOC
nocodazole
- VCR
vincristine
- RR
relative resistance
- IC50
half-maximal inhibitory concentration
References
- 1.Johnson IS, Armstrong JG, Gorman M, Burnett JP., Jr. The Vinca Alkaloids: A New Class of Oncolytic Agents. Cancer Res. 1963;23:1390–427. [PubMed] [Google Scholar]
- 2.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971;93:2325–7. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 3.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 4.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–92. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 5.Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol. 2011;8:244–50. doi: 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- 6.Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y-Q, Belyakov S, Choi C, Limburg D, Thomas BE, IV, Vaal M, Wei L, Wilkinson DE, Holmes A, Fuller M, et al. Synthesis and biological evaluation of non-peptidic cyclophilin ligands. J Med Chem. 2003;46:1112–5. doi: 10.1021/jm020409u. [DOI] [PubMed] [Google Scholar]
- 8.Lee J, Kim SS. Current implications of cyclophilins in human cancers. J Exp Clin Cancer Res. 2010;29:97. doi: 10.1186/1756-9966-29-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Restall IJ, Lorimer IAJ. Induction of premature senescence by hsp90 inhibition in small cell lung cancer. PLoS One. 2010;5:e11076. doi: 10.1371/journal.pone.0011076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–7. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Kenny AE, Brito DA, Rieder CL. Cells satisfy the mitotic checkpoint in Taxol, and do so faster in concentrations that stabilize syntelic attachments. J Cell Biol. 2009;186:675–84. doi: 10.1083/jcb.200906150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordan MA, Thrower D, Wilson L. Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res. 1991;51:2212–22. [PubMed] [Google Scholar]
- 13.Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, Poruchynsky MS. Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem. 1997;272:17118–25. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 14.Haber M, Norris MD, Kavallaris M, Bell DR, Davey RA, White L, Stewart BW. Atypical multidrug resistance in a therapy-induced drug-resistant human leukemia cell line (LALW-2): resistance to Vinca alkaloids independent of P-glycoprotein. Cancer Res. 1989;49:5281–7. [PubMed] [Google Scholar]
- 15.Kavallaris M, Tait AS, Walsh BJ, He L, Horwitz SB, Norris MD, Haber M. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res. 2001;61:5803–9. [PubMed] [Google Scholar]
- 16.Abraham EH, Prat AG, Gerweck L, Seneveratne T, Arceci RJ, Kramer R, Guidotti G, Cantiello HF. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proc Natl Acad Sci U S A. 1993;90:312–6. doi: 10.1073/pnas.90.1.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling V, Thompson LH. Reduced permeability in CHO cells as a mechanism of resistance to colchicine. J Cell Physiol. 1974;83:103–16. doi: 10.1002/jcp.1040830114. [DOI] [PubMed] [Google Scholar]
- 18.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–92. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 19.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 20.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–22. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res. 2008;68:3269–76. doi: 10.1158/0008-5472.CAN-07-6699. [DOI] [PubMed] [Google Scholar]
- 22.Brito DA, Rieder CL. The ability to survive mitosis in the presence of microtubule poisons differs significantly between human nontransformed (RPE-1) and cancer (U2OS, HeLa) cells. Cell Motil Cytoskeleton. 2009;66:437–47. doi: 10.1002/cm.20316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z, Kenny AE, Brito DA, Rieder CL. Cells satisfy the mitotic checkpoint in Taxol, and do so faster in concentrations that stabilize syntelic attachments. J Cell Biol. 2009;186:675–84. doi: 10.1083/jcb.200906150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terrano DT, Upreti M, Chambers TC. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol. 2010;30:640–56. doi: 10.1128/MCB.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–20. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinsson-Ahlzén H-S, Liberal V, Grünenfelder B, Chaves SR, Spruck CH, Reed SI. Cyclin-dependent kinase-associated proteins Cks1 and Cks2 are essential during early embryogenesis and for cell cycle progression in somatic cells. Mol Cell Biol. 2008;28:5698–709. doi: 10.1128/MCB.01833-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutterer A, Berdnik D, Wirtz-Peitz F, Zigman M, Schleiffer A, Knoblich JA. Mitotic activation of the kinase Aurora-A requires its binding partner Bora. Dev Cell. 2006;11:147–57. doi: 10.1016/j.devcel.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Fry AM, Meraldi P, Nigg EA. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998;17:470–81. doi: 10.1093/emboj/17.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shelby RD, Vafa O, Sullivan KF. Assembly of CENP-A into centromeric chromatin requires a cooperative array of nucleosomal DNA contact sites. J Cell Biol. 1997;136:501–13. doi: 10.1083/jcb.136.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carleton M, Mao M, Biery M, Warrener P, Kim S, Buser C, Marshall CG, Fernandes C, Annis J, Linsley PS. RNA interference-mediated silencing of mitotic kinesin KIF14 disrupts cell cycle progression and induces cytokinesis failure. Mol Cell Biol. 2006;26:3853–63. doi: 10.1128/MCB.26.10.3853-3863.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yokoi A, Kuromitsu J, Kawai T, Nagasu T, Sugi NH, Yoshimatsu K, Yoshino H, Owa T. Profiling novel sulfonamide antitumor agents with cell-based phenotypic screens and array-based gene expression analysis. Mol Cancer Ther. 2002;1:275–86. [PubMed] [Google Scholar]
- 32.Cleveland DW, Lopata MA, Sherline P, Kirschner MW. Unpolymerized tubulin modulates the level of tubulin mRNAs. Cell. 1981;25:537–46. doi: 10.1016/0092-8674(81)90072-6. [DOI] [PubMed] [Google Scholar]
- 33.Hornick JE, Bader JR, Tribble EK, Trimble K, Breunig JS, Halpin ES, Vaughan KT, Hinchcliffe EH. Live-cell analysis of mitotic spindle formation in taxol-treated cells. Cell Motil Cytoskeleton. 2008;65:595–613. doi: 10.1002/cm.20283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piehl M, Tulu US, Wadsworth P, Cassimeris L. Centrosome maturation: measurement of microtubule nucleation throughout the cell cycle by using GFP-tagged EB1. Proc Natl Acad Sci U S A. 2004;101:1584–8. doi: 10.1073/pnas.0308205100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanenbaum ME, Medema RH. Mechanisms of centrosome separation and bipolar spindle assembly. Dev Cell. 2010;19:797–806. doi: 10.1016/j.devcel.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 36.Rohena CC, Peng J, Johnson TA, Crews P, Mooberry SL. Chemically diverse microtubule stabilizing agents initiate distinct mitotic defects and dysregulated expression of key mitotic kinases. Biochem Pharmacol. 2013;85:1104–14. doi: 10.1016/j.bcp.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamath K, Okouneva T, Larson G, Panda D, Wilson L, Jordan MA. 2-Methoxyestradiol suppresses microtubule dynamics and arrests mitosis without depolymerizing microtubules. Mol Cancer Ther. 2006;5:2225–33. doi: 10.1158/1535-7163.MCT-06-0113. [DOI] [PubMed] [Google Scholar]
- 38.Gan PP, Pasquier E, Kavallaris M. Class III beta-tubulin mediates sensitivity to chemotherapeutic drugs in non small cell lung cancer. Cancer Res. 2007;67:9356–63. doi: 10.1158/0008-5472.CAN-07-0509. [DOI] [PubMed] [Google Scholar]
- 39.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10:194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 40.Bonne D, Heuséle C, Simon C, Pantaloni D. 4′,6-Diamidino-2-phenylindole, a fluorescent probe for tubulin and microtubules. J Biol Chem. 1985;260:2819–25. [PubMed] [Google Scholar]
- 41.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A, Pandita A, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–71. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.