

Abstract
The bone marrow (BM) is one of the organs that is sensitive to acute exposure of ionizing radiation (IR); however, the mechanism of its high sensitivity to IR remains to be elucidated. BM is differentiated into dendritic cells (DC) with granulocyte macrophage-colony stimulating factor (GM-CSF). Using this in vitro model, we studied whether radiosensitivity is distinctly regulated in undifferentiated and differentiated BM. We discovered that levels of DNA damage repair (DDR) proteins are extremely low in BM, and they are markedly increased upon differentiation to DC. Efficiency of both homologous recombination (HR)- and non-homologous end joining (NHEJ)-mediated repair of DNA double strand breaks (DSBs) is much lower in BM compared with that of DC. Consistent with this, immunofluorescent γH2AX is highly detected in BM after IR. These results indicate that increased radiosensitivity of BM is at least due to low expression of the DNA repair machinery.
Keywords: DNA damage, ionizing radiation, DNA double strand breaks, homologous recombination repair, non-homologous end joining
Introduction
The first report of bone marrow (BM) damage by ionizing radiation (IR) was reported more than 80 y ago.1 Failure of development of hematopoietic lineage has been extensively studied when BM is exposed to IR, however, the mechanism underlying this high sensitivity remains to be elucidated. IR-induced DNA damage results in the activation of biological responses collectively, namely the DNA damage response (DDR). The DDR represents a network of signaling pathways that regulates a response within the cells to DNA damage. Numerous proteins have been implicated as a component of the DDR pathway: (1) DNA damage-sensing proteins, such as Ku70/80 and MRN complex (Mre11, Rad50, Nbs1), and signaling proteins, including ATM, ATR and DNA-PKcs; (2) transducer proteins that relay and amplify the DNA damage signal, establishing a local self-feeding loop that leads to accumulation of upstream DDR proteins, which including γH2AX, MRN complex (Mre11, Rad50, Nbs1); (3) effector proteins that determine biological outcomes, such as cell cycle checkpoint, DNA repair, transcriptional regulation, apoptosis, and senescence, which including Chk1, Chk2, Rad51, and p53.2 Thus, cellular phenotypes caused by DNA stress are determined by an orchestrated regulation and function of the signal cascade controlled by these multiple proteins.
DC were first recognized by Steinman and Cohn in 1973 as a “novel cell type in peripheral lymphoid organs of mice”.3 DC are central modulators in both innate and adaptive immune responses and are specialized for presentation of antigens to T cells.4 From transfer studies in rodents and bone marrow transplantation in humans, it was demonstrated in vivo that DC originated in the bone marrow.5 GM-CSF is the major growth factor required for DC development from bone marrow precursors, as well as for DC differentiation.6-8 Thus, primary BM has often been used to investigate a mechanism of cell differentiation.
In the present studies, we investigated increased radiosensitivity and the mechanism of DNA repair in un-differentiated BM and differentiated DC induced by GM-CSF treatment.
Results
Differential expression of the DDR proteins in BM and GM-CSF-induced DC
HR- and NHEJ-mediated DNA repair pathways have been implicated to repair the DNA lesion when cells are exposed to genotoxic insult. Many different proteins are involved in these pathways, such as ATM/ATR/DNA-PKcs. We began to characterize whether DNA repair pathways could be differentially regulated when BM is differentiated to DC upon treatment with GM-CSF. We examined first the levels of a panel of the DDR proteins and their mRNA by western blot and RT-PCR, respectively (Fig. 1). BM was isolated from B129Sv/J mice (male, age 10 wk), followed by treatment with GM-CSF (20 ng/mL) to induce differentiation to DC, as described previously.7,9 Protein and mRNA samples were obtained after 3, 6, and 9 d. Surprisingly, some of the DDR proteins examined are not expressed, or are expressed at only extremely low levels, in BM, including DNA-PKcs, ATM, ATR, Rad50, NBS1, BRAT1, Chk1, Chk2, and p53 (Fig. 1A). Ku70 and DNA ligase IV were expressed in BM (D0). After GM-CSF treatment, Chk1 and Chk2 were weakly induced on day 3 (D3), and DNA-PKcs, ATM, NBS1, BRAT1, and p53 became detectable on day 6 (D6) through day 9 (D9). Levels of ATR and Rad50 were highly expressed on day 6 and decreased on day 9. We next attempted to study whether induced expression of these proteins is transcriptionally regulated. mRNA was extracted at D3, D6, and D9, and transcripts of these proteins were analyzed by RT-PCR (Fig. 1B). Interestingly, even though protein expression of DNA-PKcs, ATM, ATR, Rad50, NBS1, BRAT1, Chk1, Chk2, and p53 were not detected in BM at D3, their mRNA was transcribed. mRNA of DNA-PKcs and DNA ligase IV decreased on D9, and p53 mRNA increased from D3 to D6. mRNA of Ku70 and Chk1 slightly decreased in D9. mRNA of ATR was high at D3, and decreased from D6 to D9. These results demonstrate that protein expression of significant numbers of the DDR proteins is posttranscriptionally regulated in the process of the DC differentiation.
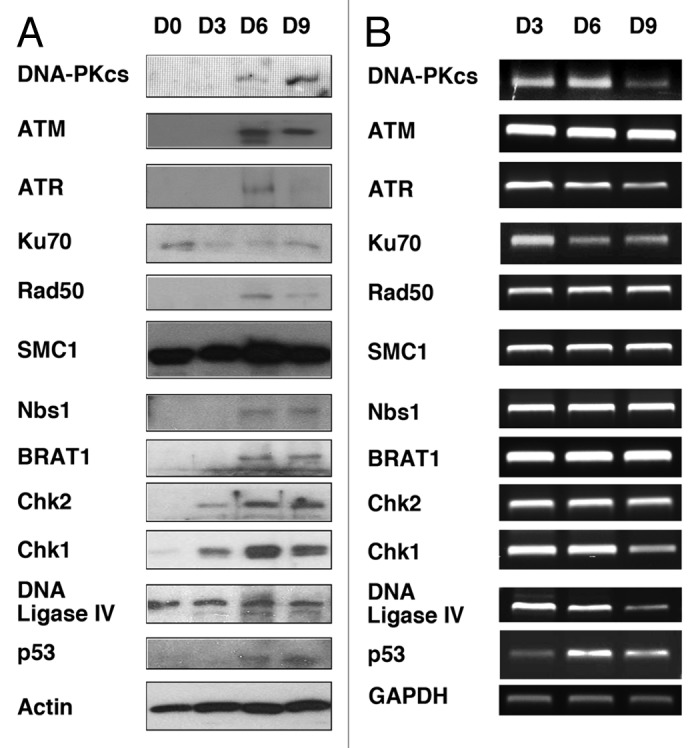
Figure 1. Levels of expression of the DDR proteins in BM treated or untreated with GM-CSF. BM obtained from B129Sv/J mice (day 0/D0) were immediately treated with GM-CSF (20 ng/mL) for 3, 6, and 9 d (D3, D6, and D9, respectively). (A) Indicated proteins were immunoblotted with BM cell lysates obtained at D0, D3, D6, and D9. Actin serves as a control. (B) RT-PCR analysis of mRNA of the indicated proteins. Total RNA was extracted from BM of D3, D6, and D9. PCR primers are shown in Table S1. GAPDH serves as a control.
Homologous recombination (HR)- and non-homologous end joining (NHEJ)-mediated repair are impaired in BM
Given that expression of ATM, ATR, and DNA-PKcs is induced when BM is differentiated into DC with GM-CSF treatment, we reasoned that HR- and NHEJ- DNA repair processes are impaired in BM, compared with those in matured DC. This hypothesis was confirmed by using artificial chromosome substrates to detect HR- or NHEJ-mediated DNA repair. GFP-Pem1 containing DNA DSBs substrates to quantify the efficiency of both HR- and NHEJ-mediated repair have been established previously.10 BM treated with GM-CSF for 1, 3, or 4 d was transfected with the plasmid DNA of either HR or NHEJ substrates together with I-SceI expression vector that introduces DSBs into these substrate DNA. Efficiency of repair of the HR- and NHEJ-substrates was determined by the ratio of emitting green fluorescence to red fluorescence signals after 24 h (samples are from D2, D4, and D5). We found that efficiency of both HR- and NHEJ-mediated repair is much lower in D2 and D4 BM, compared with that of D5 (Fig. 2). We assume that low efficiency of DNA repair activity in un-differentiated BM is due to much lower levels of the DDR proteins. These results are consistent with the immunoblot data shown in Figure 1, demonstrating that ATM, ATR, and DNA-PKcs, key molecules for HR- and NHEJ-mediated repair become detectable only after differentiation is induced by GM-CSF treatment.
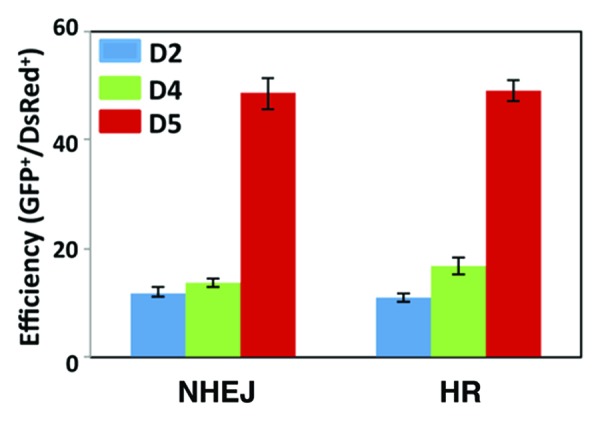
Figure 2. Efficiency of HR- and NHEJ-mediated repair of DSBs in BM. BM cells cultured in GM-CSF-containing media (20 ng/mL) at day 1, day 3, and day 5 were transfected with HR- or NHEJ-substrates plasmids together with I-SceI expression vector and pDsRed2-N1 plasmid. Fluorescent signals were analyzed as described in Methods to quantify NHEJ or HR events during DC development by flow cytometry at 24 h after transfection. The DsRed-positive cells were used to validate transfection efficiency, and the ratio of GFP/DsRed
BM is not extensively radiosensitive compared with DC
The orchestrated regulation of the DDR proteins is essential in order to induce DNA damage checkpoint. Since many of the DDR proteins are undetectable in BM and they increase as the cells differentiate into DCs, it is assumed that radiosensitivity of BM is higher than DC due to low expression of these proteins. BM at D2 and D5 was treated with 2, 5, or 10 Gy of IR and apoptosis was determined by FACS analysis of Annexin V staining after 24 h (Fig. 3). BM at D3 showed constitutive levels of apoptosis (8.2%), which was further increased by IR treatment (total of early-to-late apoptosis is 12.0% [2 Gy], 13.9% [5 Gy], 17.8% [10 Gy], respectively). When BM at D5 was irradiated, induction of apoptosis after 24 h was lower than BM at D3 (8.2% [2 Gy], 10.2% [5 Gy], 10.6% [10 Gy]). Of note, induced levels of apoptosis were not remarkably high even though expression of the DDR proteins is extremely low in BM at D2 (Fig. 1), suggesting that unidentified DNA repair mechanism(s) functions in the early stage of BM (see below).
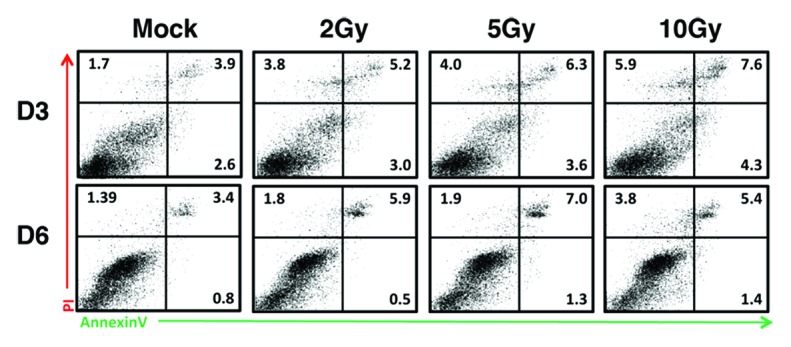
Figure 3. BM(D2) is more radiosensitive than BM(D5). BM at D2 and D5 were untreated (mock) or treated with ionizing radiation (2, 5, and 10 Gy). After 24 h (D3 and D6, respectively), apoptosis of these cells were quantified with FITC-conjugated Annexin V (as early apoptotic marker) and PI (as marker for late apoptotic and necrosis). Representative results of two independent experiments are shown.
Repair of DSBs in BM
Immunofluorescence analysis of γH2AX has been implicated in determining the appearance and completion of repair of DSBs when cells are exposed to IR.10,11 We irradiated asynchronous BM populations (at D2 and D6) with physiological doses of IR (4 Gy) and applied immunofluorescence analysis to study DSBs repair by enumerating γH2AX foci at defined time points after IR treatment (Fig. 4). In BM at D2, γH2AX rapidly increased in 0.5 h, sustained until 2 h, and decreased to the basal level in 4 h (Fig. 4, upper panel). In contrast, in BM at D6, γH2AX foci similarly increased in 0.5 h; however, numbers of foci were less than those of BM at D2 (Fig. 4, lower panel). Then, γH2AX signals decreased to the almost basal level in 2 h, indicating that repair of DSBs is more effective in BM at D6 than at D2.

Figure 4. Immunocytochemical analysis of γH2AX in BM after IR treatment. BM at D2 or D6 after GM-CSF treatment were irradiated (4 Gy), followed by immunostaining analysis of γH2AX (green). DAPI was used for nuclear staining (blue). Cells were fixed at 0.5, 2, and 4 h post-IR.
These results indicate that DNA repair machinery is not completely compromised in DC at D2, and DSBs are more rapidly repaired in differentiated DC than in BM.
Discussion
Homologous recombination (HR) and non-homologous end joining (NHEJ) represent two conceptually different pathways for repairing DNA double-strand breaks (DSBs), the lesion at the heart of many physiological and pathophysiological processes in mammalian cells. NHEJ repairs DSBs without requiring sequence homology and involves the core components DNA-PKcs/Ku70/Ku80 and DNA ligase IV/XRCC4/XLF.12,13 Recent studies have also illustrated that ATM is involved in the initiation of the HR-mediated DNA repair by phosphorylating cellular substrates such as NBS1 and BRCA1.14 It was surprising that many components of the DNA repair system were not detectable at early phase BM before DC differentiation is induced by GM-CSF. Interestingly, transcripts of these proteins exist during differentiation. These results demonstrate that expression of these DDR proteins is posttranscriptionally regulated in the process of the BM-to-DC differentiation. We did not observe retardation of the cell growth of BM in the presence of GM-CSF, showing that low expression of those proteins does not affect cell proliferation significantly. However, cell growth of BM obtained from ATM-deficient mice is much lower than that of wild type BM, indicating ATM’s essential roles in GM-CSF-mediated cell growth (submitted).
On the basis of these observations, we assumed that HR- and NHEJ-mediated DNA repair pathways do not function normally when BM is exposed to genotoxic stress because of a low expression of these DDR proteins in undifferentiated BM.
As shown in Figures 2 and 3, although efficiency of both NHEJ and HR DNA repair in early stage BM is lower than in late stage BM (BMDC), we found that rate of IR-induced apoptosis in early stage BM is not extensively higher than in DC (Fig. 3). Importantly, IR-induced γH2AX staining confirmed that repair of DSBs is more effective in BM at D6 than at D2 (Fig. 4). γH2AX have been implicated in existence and localization of DSBs, and we showed here that immunofluorescence signals of γH2AX eventually decreased in early stage BM, suggesting at least two possibilities: (1) a potential γH2AX phosphatase(s) is similarly activated in both BM and DC, even though DSBs are not fully repaired in BM; (2) other uncharacterized DNA repair pathway(s) is activated in early stage BM. Of note: Even though levels of major DDR kinases are not detectable in early stage BM, phosphorylation of H2AX is observed, suggesting that an unknown kinase(s) is activated in response to DNA damage and that the DDR pathway is not completely compromised. There is another less-characterized repair mechanism, called alternative non-homologous end joining (Alt-NHEJ).15,16 Alt-NHEJ is also referred to as microhomology-mediated end joining (MMEJ). This process depends on the Mre11/NBS1/Rad50 complex12-15,17 and is regulated by ATM.18 Because levels of any of these proteins are extremely low in BM, it is unlikely that Alt-NHEJ repairs DSBs in BM.
Bone marrow (BM) damage by ionizing radiation (IR) has been investigated for more than 80 y.1 Failure of development of hematopoietic lineage has been extensively studied when BM is exposed to IR. We have demonstrated that levels of many of the DDR proteins are extremely low in BM, those including DNA-PKcs, ATM, ATR, Rad50, Nbs1, BRAT1, Chk1, Chk2, and p53. Since mRNA of most of these proteins is comparably expressed in both BM and DC, these results illustrate that post-transcriptional mechanism determines the levels of these proteins. Thus, our studies point out that not only gene expression array but also proteome analysis are essential to understand BM’s radiosensitivity.
The mechanism underlying BM’s high sensitivity to IR is largely unknown. Considering that our studies of apoptosis in response to IR are determined by the isolated BM, it is possible that extensive BM damage caused by IR in vivo might be regulated by microenvironment, such as presence of BM stromal cells.
Materials and Methods
Bone marrow preparation and DC development
Mice were housed in animal facilities of Roswell Park Cancer Institute under SPF (specific pathogen free) conditions. Bone marrow cells were isolated from femora and tibiae of 10 wk old male B129Sv/J mice (Jackson Lab) through flushing method using a syringe. Single cells of BM were treated with 20 ng/mL murine GM-CSF (PeproTech) to develop DCs, as described previously.7,9
Immunoblot studies of the expression of the DDR proteins
BM were harvested at D0 (day 0), D3 (day 3), D6 (day 6), and D9 (day 9) after GM-CSF treatment, and cell lysates were prepared in ice-cold lysis buffer (50 mM TRIS-HCl [pH 7.6],150 mM NaCl, 1 mM EDTA [pH 8.0], 20 mM NaF, 1 mM Na3VO4, 1% NP40, 0.5 mM dithiothreitol) in the presence of protease-inhibitor mix (leupeptin, aprotinin, and PMSF, 10 mg/mL, respectively). Total cell lysate (20 μg/sample) was loaded and separated by 6% SDS-polyacrylamide gels and transferred to PVDF membranes (Immobilon-P). Membraines were blocked in 5% non-fat dried milk in Tris-buffered saline/0.1% Tween-20 and incubated with primary antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology). Primary antibodies used in this study were anti-ATM (GeneScript), anti-SMC1, anti-Nbs1, anti-Chk1/2, anti-Rad50, anti-Ku70, anti-DNA-PKcs, anti-DNA ligase IV (Santa Cruz Biotechnology), anti-BRAT1 (Abcam), and anti-p53, anti-ATR (Cell Signaling). Anti-actin antibody (Santa Cruz Biotechnology) was used to validate protein amounts.
RT-PCR analysis
Total RNA was isolated from D3, D6, and D9 BM using TRIzol Reagent (Invitrogen), according to the manufacturer’s instructions. First-strand cDNA was synthesized from 2 μg total RNA using moloney murine leukemia virus (MMLV) reverse transcriptase. PCR was conducted using specific primer sets for DNA damage response genes (Table S1).
Immunohistochemistry
BM at D2 and D6 were irradiated with 4 Gy in a 137Cs irradiator (MARK2, JL Stephen and Associate). Immediately after irradiation, BM was seeded in cell culture plate for indicated times. Suspended BM was placed onto a glass slide by cyto-centrifugation (Cyto-Tek, Fisher Scientific) after fixed in 2% paraformaldehyde. After blocking with PBS-TT (PBS containing 0.5%Tween-20 and 0.1% Triton X-100) containing 5% bovine serum albumin (BSA, Santa Cruz), cells were incubated with anti-γ-H2AX (Novus Biologicals, CO) in PBS-TT containing 1% BSA for 2h followed by three washed with PBS-TT. Cells were stained with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen) for 1 h. Slides were mounted with VECTASHIELD mounting medium containing DAPI (Vector Laboratories) followed by sealing with nail polish. Fluorescent images were captured using Nikon TE2000-E inverted microscope equipped with a Roper CoolSnap HQ CCD camera (Melville).
Flow cytometry analysis for apoptosis after IR
Suspended BM at D2 and D5 were irradiated with 2, 5, or 10 Gy and cultured in fresh media for 24 h. Cells were stained with FITC conjugated anti-AnnexinV and propidium iodide (PI) according to manufacturer’s instructions (BD Biosciences) and analyzed by flow cytometer (FACSCalibur, BD). The green (AnnexinV) and red (PI) fluorescence from each cell were separated and quantified using Cell Quest pro software (BD).
Analysis of the efficiency of HR- and NHEJ-mediated DSBs repair
BMs (2 × 106 cells/mL) were isolated from B129Sv/J mice as described. These cells were cultured in a 24 well cell culture plate for 2 to 5 d with RPMI media supplemented with 10% fetal bovine serum containing GM-CSF (20 ng/mL). To analyze the efficiency of HR or NHEJ repair, reporter plasmids were cotransfected with I-SceI endonuclease expression vector and a control plasmid vector, DsRed-expressing plasmid (pDsRed2-N1), for 24h as described previously.10 Transfection was performed with Lipofectamin LTX (Invitrogen) by following manufacturer’s instructions.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Ouchi laboratory for helpful discussion. HR and NHEJ substrate plasmids were kindly provided by Dr Vera Gorbunova (University of Rochester, Rochester, NY). This work is supported by grants R01CA79892, R01CA90631, and 5P30CA16056 from National Institutes of Health, and Breast Cancer Research Grant from Susan Komen Foundation.
Author Contributions
E.-Y. S. and T.O. planned and generated all the data. E.-Y. S. and T.O. discussed the results and wrote the paper.
References
- 1.Warren SL, Whipple GH. Roentgen Ray Intoxication: Ii. A Study of the Sequence of Clinical, Anatomical, and Histological Changes Following a Unit Dose of X-Rays. J Exp Med. 1922;35:203–11. doi: 10.1084/jem.35.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 3.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–62. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 5.Katz SI, Tamaki K, Sachs DH. Epidermal Langerhans cells are derived from cells originating in bone marrow. Nature. 1979;282:324–6. doi: 10.1038/282324a0. [DOI] [PubMed] [Google Scholar]
- 6.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-alpha cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360:258–61. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 7.Inaba K, Steinman RM, Pack MW, Aya H, Inaba M, Sudo T, Wolpe S, Schuler G. Identification of proliferating dendritic cell precursors in mouse blood. J Exp Med. 1992;175:1157–67. doi: 10.1084/jem.175.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witmer-Pack MD, Olivier W, Valinsky J, Schuler G, Steinman RM. Granulocyte/macrophage colony-stimulating factor is essential for the viability and function of cultured murine epidermal Langerhans cells. J Exp Med. 1987;166:1484–98. doi: 10.1084/jem.166.5.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lutz MB, Kukutsch N, Ogilvie AL, Rössner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/S0022-1759(98)00204-X. [DOI] [PubMed] [Google Scholar]
- 10.Mao Z, Seluanov A, Jiang Y, Gorbunova V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc Natl Acad Sci U S A. 2007;104:13068–73. doi: 10.1073/pnas.0702410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JH. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–2. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- 12.Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131:223–5. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 13.van Gent DC, van der Burg M. Non-homologous end-joining, a sticky affair. Oncogene. 2007;26:7731–40. doi: 10.1038/sj.onc.1210871. [DOI] [PubMed] [Google Scholar]
- 14.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 15.Boulton SJ, Jackson SP. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996;15:5093–103. [PMC free article] [PubMed] [Google Scholar]
- 16.Liang F, Jasin M. Ku80-deficient cells exhibit excess degradation of extrachromosomal DNA. J Biol Chem. 1996;271:14405–11. doi: 10.1074/jbc.271.24.14405. [DOI] [PubMed] [Google Scholar]
- 17.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–27. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 18.Riballo E, Kühne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–24. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.