

Abstract
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors in the US. The majority (~85%) of GISTs possess gain-of-function mutations in KIT or PDGFRA, causing constitutive activation of the kinase receptor. GIST management has been transformed by the identification of tumor driver mutations leading to unprecedented disease control of advanced GIST with the introduction of imatinib mesylate (IM). Despite IM’s efficacy, most patients experience primary and/or secondary resistance within 2 y of treatment. Additional therapies and methods to optimize screening of novel approaches in preclinical studies are warranted. Clinically, treatment efficacy is typically assessed using Response Evaluation Criteria In Solid Tumors (RECIST) guidelines or Choi criteria. Both require a period of time on therapy before changes indicative of response can be observed. In addition, neither informs directly about cell death. We evaluated the use of molecular imaging technology in an animal model using near-infrared (NIR) imaging probes together with three-dimensional fluorescence molecular tomography (FMT) for assessing therapeutic response and ultimately optimizing our understanding of the biologic effects of these agents. We determined the potential of NIR probes (PSVueTM794 and cell-penetrating KcapQ647) for detecting distinct markers of apoptosis and compare this to tumor size measured by MRI in response to IM treatment in GIST-T1 xenografts. Our studies revealed statistically significant increases in apoptosis due to IM treatment using both probes as early as 24 h post IM treatment which was confirmed by IHC. Molecular imaging will allow for faster and more effective screening of novel therapies in preclinical GIST models.
Keywords: GIST, imatinib mesylate, apoptosis, molecular imaging
Introduction
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract, afflicting an estimated 3000 to 6000 new patients each year.1 It typically presents between the fifth and sixth decade of life with abdominal pain, symptoms of anemia or acute bleeding. Management of the disease has been transformed by the identification of activating mutations in the tyrosine kinase receptors, KIT and PDGFRA, leading to the unprecedented disease control of advanced GIST with the introduction of the kinase inhibitors imatinib mesylate (IM), sunitinib malate, and regorafenib. Response to treatment has been correlated with the site of the mutation in KIT or PDGFRA, with those tumors carrying mutations in exons 11 and 12, respectively, having the best response to therapy.2 The success of IM in GIST has been tempered by the fact that treatment in the advanced disease setting only increases the median time to tumor progression by approximately 2 y. Reasons identified for this limited therapeutic efficacy include decreased effectiveness of IM for KIT mutations outside of exon 11 and in tumors lacking mutations in KIT and PDGFRA. Furthermore, GISTs that progress are likely to have secondary mutations in KIT or PGDFRA and/or epigenetic changes that act together to create a more aggressive tumor. Sunitinib therapy offers a median of an additional 5 mo delay to tumor progression3 and is currently used as the second-line therapy. Regorafenib, FDA approved in February 2013, is now the third-line drug of choice with a median progression free survival (PFS) of 4.8 mo.4,5 However, it is clear that additional therapies are still needed and methods to optimize screening of novel approaches are warranted.
In clinical trials, efficacy of therapies is measured utilizing RECIST criteria, a measure of change in tumor size over time using clinical imaging technologies (CT, MRI). RECIST, which looks at tumor size only, has been critiqued as not correlating with clinical improvement, metabolic response by [18F] fluorodeoxyglucose-positron emission tomography (FDG-PET), or pathologic response. For example, patients with IM-treated tumors that remain stable in size have similar survival to those with documented tumor response. FDG-PET metabolic response has predicted RECIST response. The Choi criteria defines treatment response as a >10% decrease in tumor size or a >15% decrease in tumor density of tumor lesions assessed on contrast-enhanced CT (CT) scans. Response by Choi criteria has been shown to correlate better with FDG-PET than standard RECIST response.6
A better understanding of imaging as it correlates with biologic markers of response is needed; imaging that informs researchers and clinicians more directly about cell death may be superior to current techniques. IM has been previously shown to cause apoptosis in vitro in IM-sensitive cell lines.7-10 Therefore, in this study we sought to evaluate the use of molecular imaging technology using near-infrared (NIR) imaging probes in combination with three-dimensional fluorescence molecular tomography (FMT) for assessing therapeutic response and ultimately improving our understanding of the biologic effects of these agents. Here, using GIST-T1 xenografts, we evaluate the potential of two NIR probes detecting two distinct markers of early apoptosis and compare this to tumor size measured by magnetic resonance imaging (MRI) in response to IM treatment.
Results
To develop methods for assessing novel therapeutic agents or combinations of agents to effectively treat GIST, we sought to evaluate whether we could successfully visualize treatment-related apoptosis in GIST xenografts using a treatment known to be effective both clinically and in this model. To do this, we utilized three NIR probes: IntegriSense680, PSVueTM794, and KcapQ647, in combination with FMT imaging. In preliminary pilot studies, we found that IntegriSense680, which detects the tumor-associated integrin αvβ3, localized strongly and specifically to the GIST xenograft tumors and could therefore be used to determine the three dimensional region of interest (ROI) in the FMT data sets. This was necessary because the signal from the apoptosis sensing probes was not as strong, and did not localize as specifically to the tumors as the IntegriSense680 probe. The time for optimal imaging (maximum signal in the tumor as compared with the whole-body background) for the IntegriSense680 probe was determined to be 96 h post-injection. We then performed additional preliminary studies using the PSVueTM794 and KcapQ647 NIR probes, in separate studies, to determine optimal time points for imaging following probe injection by acquiring longitudinal images. To do this, 8 mice with GIST-T1 cell xenografts (4 vehicle and 4 IM-treated) were given retro-orbital injections of either PSVueTM794 or KcapQ647 in combination with IntegriSense680. FMT Images were acquired at 0.5, 1, 3, 6, 12, 18, and 24 h post-probe injection. With both probes, all IM-treated GISTs had significantly more probe signal than vehicle-treated tumors at both 24 h post-IM treatment (Fig. 1) as well as 1 wk post-treatment, with the optimal imaging times of 18 h post-injection for PSVueTM794 and 1 h post-injection for KcapQ647. Following these pilot studies, a larger cohort of mice (n = 14) bearing GIST-T1 xenografts were randomized into two treatment groups: vehicle and IM. These mice were injected with all three probes (IntegriSense680, PSVueTM794, KcapQ647) at the previously determined optimal imaging times, and were treated with vehicle or IM (50 mg/kg daily). Twenty-four hours post treatment, all mice were imaged in the 645, 680, and 750 nm channels to determine individual probe retention in the tumor. This imaging schedule was repeated in these mice one week post treatment. To improve the power to detect a difference between vehicle and IM treated animals, we combined this data with data from both aforementioned studies. As outlined earlier, the GEE methodology was used to model treatment effect and time on apoptotic activity, adjusted for potential batch effects in the experiment. A statistically significant increase in the rate of apoptotic activity due to IM treatment (compared with control) was observed (P value = 0.002) using data from the KcapQ647 probe. However, there was neither a significant change in apoptotic activity with time (P value = 0.69) nor treatment effect with time (P value = 0.41). Similarly, a statistically significant increase in the rate of apoptotic activity due to IM treatment (compared with control) was observed (P value = 1.41e−04) using data from the PSVueTM794 probe. Again, there was neither a significant change in apoptotic activity with time (P value = 0.11) nor treatment effect with time (P value = 0.21). Figure 2A and B display the growth curves for the IM treated and vehicle treated control groups based on data obtained using the KcapQ647 and PSVueTM794 probes, respectively. It is evident that IM treatment increases apoptotic activity (as measured by both probes) compared with vehicle-treated control at each time point (days 1 and 7) (Fig. 2C and D). Vehicle treatment on day seven shows elevated PSVueTM794 retention compared with that on day one (Fig. 2C). This is most likely explained by the mechanism of the probe. PSVueTM794 probe works by targeting phosphatidylserines exposed on the cell surface of apoptotic cells; however, this probe cannot distinguish between apoptotic and necrotic cell membranes in vivo. KcapQ647, a caspase-activatable probe, allows for better discernment between apoptosis and necrosis. Therefore, the increased PSVueTM794 signal in vehicle treated tumors after 1 wk is most likely attributable to necrosis occurring in these tumors as they grow larger. As measured by the KcapQ647 probe, treatment with IM shows decreased signal on day 7 compared with that on day 1, while the PSVueTM794 probe shows an increase in signal, again most likely a combination of both apoptosis and necrosis detected by PSVueTM794 in these tumors, with no significant change over time (Fig. 2C and D).

Figure 1. Detection of GIST with IntegriSense680 and treatment-related apoptosis with KcapQ647. Representative images of FMT imaging of GIST xenografts treated with vehicle (top panel) or with IM (50 mg/kg) for 24 h (bottom panel). Images on the left are 2D reflectance images, in the middle, the 3D FMT reconstruction of the αvβ3 signal used to delineate the region of interest (ROI) and on the right is the FMT signal from the cell-penetrating KcapQ647 probe.

Figure 2. Quantification of IM-related apoptosis using NIR probes with FMT imaging. Change in apoptotic activity measured by the PSVueTM794 (A) and KcapQ647 (B) probes between day 1 and day 7 of treatment with IM or vehicle control. Each measurement on apoptotic activity was normalized to its corresponding tumor volume measurement obtained using MRI and expressed as a ratio (on the log-scale). A longitudinal model based on the generalized estimating equations approach was used to model the effect of treatment and time on apoptotic activity, adjusted for potential batch effects in the experiment. Statistically significant increases in the rate of apoptotic activity due to IM treatment (compared with control) was observed (P value = 1.41e−04 and 0.002, respectively) using data from the PSVueTM794 and KcapQ647 probes. Box-and-whisker plots of the apoptotic activity measured by the PSVueTM794 (C) and KcapQ647 (D) probes on the logarithmic scale for various treatment-time combinations. In each plot, box height represents the inter-quartile range (IQR) where the upper and lower ends indicate the third and first quartiles, respectively. The solid black horizontal line inside the box represents the median value while the whiskers (the two solid horizontal lines at either end, connected by dotted lines) extend to the most extreme data points which are no more than 1.5 times the IQR from the box in each direction. Using this criterion, points that lie beyond these whiskers are considered to be outliers (open circles).
Immediately following FMT imaging, we performed MRI scans to measure tumor volume. Due to the often flat and irregular morphology of these GIST-T1 xenografts (Fig. 3A, white arrow), we previously have found MRI tumor volume measurements to be more precise than standard caliper measurements, and our rapid MRI scanning protocol permits an efficient and cost effective method for obtaining these data. Not surprisingly, 24 h after IM treatment no tumor shrinkage was observed in the IM-treated cohort relative to their vehicle-treated counterparts (Fig. 3B, middle plot). These findings highlight the advantage of using the NIR probes in combination with FMT imaging to detect apoptosis at earlier time points, not possible with MRI, caliper measurements, or any method that only measures tumor volume. At one week post-treatment, a trend toward treatment-related shrinkage was observed by MRI (Fig. 3B, third plot; Fig. 3C).

Figure 3. Treatment-associated tumor shrinkage is not evident 24 h post IM treatment. (A) MR image showing GIST-T1 xenograft. (B) Box-and-whisker plots of tumor volume (mm3) measured 7 d prior to treatment (first plot), 1 d post-IM treatment (middle plot), and 7 d post-IM treatment by MRI (last plot). (C) Comparison of tumor volume change from 1 d post-treatment to 1 wk post-treatment between IM and vehicle-treated groups. P value is 0.094 using the Mann–Whitney test comparing the two differences IM day 7 − IM day 1 and vehicle day 7 − vehicle day 1. In all box plots, box height represents the inter-quartile range (IQR) where the upper and lower ends indicate the third and first quartiles, respectively. The solid black horizontal line inside the box represents the median value while the whiskers (the two solid horizontal lines at either end, connected by dotted lines) extend to the most extreme data points which are no more than 1.5 times the IQR from the box in each direction. Using this criterion, points that lie beyond these whiskers are considered outliers.
Finally, to validate these in vivo findings, immunohistochemical (IHC) analysis was performed ex vivo on tumors harvested from the mice upon conclusion of the one week post-treatment imaging. Tissue sections from all mice (n = 22) were stained for markers of apoptosis, cleaved Lamin A and cleaved Caspase-3. Histological examination demonstrated significant levels of both apoptotic markers in the mice treated with IM compared with their vehicle treated counterparts (Fig. 4).
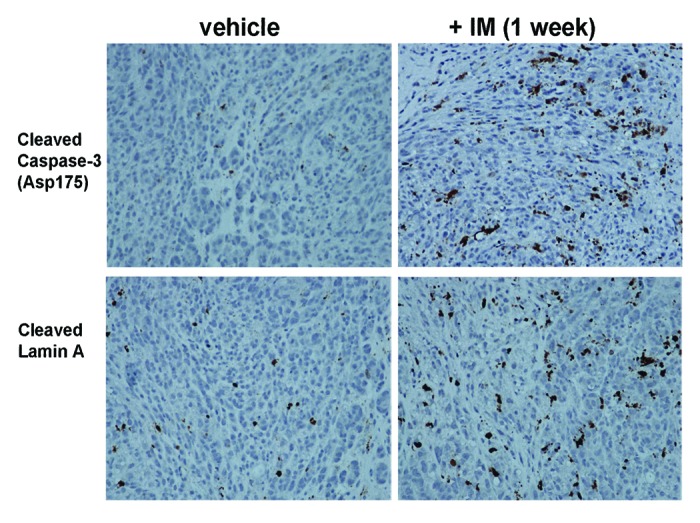
Figure 4. IM treatment causes higher levels of apoptotic markers, cleaved caspase-3 and cleaved Lamin A, in GIST xenografts. Immunohistochemistry of GIST-T1 xenografts, treated with vehicle (left column) or with IM (right column) for one week, for cleaved caspase-3 (top row) and cleaved Lamin A (bottom row).
Discussion
Since the FDA approval of IM in 2002, the treatment of GIST has radically improved. Advanced GIST, resistant to standard chemotherapy and radiation, can now be controlled initially with IM in the majority of cases. However, these effects are often short-lived, with IM demonstrating a median time to progression of approximately 2 y. More recently, sunitinib and regorafenib have been approved for second- and third-line options, though both have PFS of less than 6 mo. In addition, there are currently greater than 15 investigational agents being evaluated for the management of GIST, including a number of small molecule inhibitors of Hsp90 and IGF1R.11 With this wealth of agents currently being investigated as well as the fact that researchers are actively seeking out novel targets in GIST, it is becoming increasingly obvious that preclinical testing of these agents and/or novel combinations of agents will be essential in discovering optimal treatment regimens for GIST patients that have failed on the first three lines of treatment.
Much of the effort, to date, involving preclinical evaluation of therapeutics in vivo for GIST has consisted of imaging using small animal FDG-PET.12-16 Cullinane and colleagues (2005) demonstrated, in a xenograft model using FDC-P1 cell lines expressing either an IM-sensitive or -resistant KIT mutation, that IM led to a rapid reduction in FDG uptake on PET as early as 4 h post-treatment in the IM-sensitive model.15 In 2010, Revheim et al.14 showed decreased tumor-to-liver uptake ratios in GIST xenografts 1 d and 8 d post-treatment with both IM and sunitinib using a clinical PET/CT. Additionally, Pantaleo et al. (2010) preclinically evaluated two additional small molecule inhibitors, nilotinib and everolimus, in a GIST xenograft model, as single agents or in combination with IM, using FDG-PET imaging.13 This study demonstrated that all three single agents led to decreased glucose uptake at 4 and 13 d post-treatment, with the combination of IM and everolimus being superior to all other treatment arms. Most recently, Floris and colleagues demonstrated the successful combination of the PI3K inhibitor, GDC-0951, and IM in GIST xenografts using micro-PET imaging.16 An alternative approach has recently been reported using a fluorophore-conjugated anti-KIT antibody imaged by fluorescence laparoscopy to detect and visualize cecal GISTs in KIT K641E+/− transgenic mice.17 This approach yielded an accuracy of 92% in tumor detection and the authors speculate that this method has future potential in disease response as well as staging, evaluating tumor margin status, and identifying peritoneal metastases. It should be noted however, that the use of FDG PET or the labeled anti-KIT antibody do not assess apoptosis of cells.
Our study is a proof of concept demonstrating that in vivo molecularly targeted imaging can be successfully used to evaluate preclinical therapies non-invasively in GIST xenograft models. We successfully show that two NIR probes, PSVueTM794 and KcapQ647, can be used to quantify levels of apoptosis, as early as 24 h post-IM treatment. These probes have varying mechanisms of action with PSVueTM794 targeting phosphatidylserines exposed on the cell surface of both apoptotic and necrotic cells membranes, whereas the cell-penetrating probe, KcapQ647, possesses a cleavable domain (DEVD), a specific substrate for caspase-3, that therefore can better distinguish apoptotic from necrotic cells. KcapQ647 also has the advantage of a rapid and potentially translatable optimal imaging time (1 h post injection) compared with the other probes. Even with different mechanisms, both probes were able to detect IM-related apoptosis, which was subsequently confirmed by IHC staining of both cleaved Lamin A and cleaved caspase-3. This approach is superior to standard caliper tumor volume measurements, CT or MRI images, all of which are commonly used in the GIST preclinical setting as a readout of therapeutic efficacy, since this methodology allows for rapid detection of cell death in response to drug. In addition, we believe that this technology can be used preclinically as an alternative to PET imaging to avoid the use of radionuclides and to look at the effects of novel therapeutic agents or combination of agents at the molecular level. We hypothesize that using this non-invasive approach in preclinical models of GIST has potential in evaluating new agents, combination of agents or varying doses and dosing schedules and could allow us to assess the need to change treatment regimens or discontinue their development much earlier, when we see that there is no apoptosis induced by the investigational therapy. Future studies evaluating novel therapies are planned to test this hypothesis.
Methods
GIST xenografts and drug administration
All studies involving animals followed procedures approved by the Fox Chase Cancer Center Institutional Animal Care and Use Committee. GIST-T1 cells, a tumor cell line possessing a heterozygous mutation in KIT exon 11 kindly provided by Takahiro Taguchi,18 were washed and subsequently resuspended in phosphate-buffered saline (PBS) at a density of 3 × 106 cells/100 μL. One hundred microliters of cells in PBS were mixed thoroughly with 100 μL MatrigelTM Matrix (BD Biosciences) and the suspension was injected subcutaneously into the right flanks of athymic nude mice (strain NCRNU, Taconic Inc.). Mice were maintained on a standard diet (2018SX Teklad Global; Harlan Laboratories) and transitioned to a purified, alfalfa-free rodent chow (Teklad AIN76A; Harlan Laboratories) for a minimum of 48 h before imaging to minimize fluorescence in the gut, as well as drinking water ad libitium. Tumor volumes were measured twice weekly by MRI. When tumors reached approximately 300 mm3, mice were randomized into two groups. One group was administered IM orally at 50 mg/kg daily and the second group received only vehicle (sterile water).
Near-infrared fluorescent optical imaging agents
IntegriSense680 (PerkinElmer, Inc.), the αvβ3-targeted probe detecting tumor-associated integrin receptor expression was utilized to determine tumor regions of interest (ROI).19 PSVueTM794 (Molecular Targeted Technologies), used as a marker of apoptosis, binds to phosphatidylserine residues exposed on the cell surface of apoptotic cells and, in addition, binds to negatively-charged necrotic regions found in various tumors through its zinc (II)-dipicolylamine (Zn-DPA).20,21 A third probe, KcapQ647 (synthesized in the laboratory of Dr. David Piwnica-Worms), a cell-penetrating activatable probe was used to detect apoptosis-associated caspase activity.22-24
Magnetic resonance imaging
To accurately determine tumor volume, mice were imaged in a 7 Tesla Vertical bore MRI system with a Bruker DRX spectrometer and Paravision 3.0.1 software. A T2 weighted RARE (rapid acquisition of refocused echoes) pulse sequence provided rapid imaging times and good delineation of tumor margins. Twenty-six slices were acquired with MRI acquisition parameters: TR = 2300 msec, TE = 8.8 msec, rare factor = 8, effective TE = 36.8 msec, nex = 2, slice thickness = 1 mm, field of view = 2.56 mm, acquisition matrix = 256 × 256, and imaging time of 2.5 min. During imaging, animals were anesthetized with 2% isoflurane in O2. The total time to image one mouse, including animal preparation, was less than 10 min. Tumor volume was measured with the Paravision 3.0 software by manually outlining the tumor on each image slice where it appeared, and then summing the volumes from the individual slices. Because the subcutaneous tumors tended to have a relatively flat morphology, we found the MRI measurements to be superior to estimating tumor volume with calipers.
Fluorescence-molecular tomography (FMT)
In vivo FMT was performed with the VisEn FMT2500 Quantitative Tomography Imaging System (Perkin Elmer Inc. Waltham, MA). FMT imaging was performed according to the manufacturer’s standard procedures. Mice were anesthetized with isoflurane (2% in O2), and placed in the bi-planar imaging cassette supplied with the FMT2500. Mice were imaged in the 645 nm (KcapQ647), 680 nm (Integrisense680), and 750 nm (PSVueTM794) channels. The FMT imaging times were of approximately 15 min duration for each mouse. Transmission and fluorescence images were captured with a thermo-electrically cooled CCD camera. Three-dimensional volumetric FMT image sets were reconstructed to an isotropic spatial resolution of 1 mm3, and displayed with the TrueQuantTM software package supplied with the FMT2500. A small dose (0.5 nmoles, approximately ¼ the standard dose of 2 nmoles) of Integrisense680 (Perkin-Elmer) was injected 96 h prior to imaging to mark the tumor position. Prior work had indicated that in this model Integrisense680 imaged at this time provided very accurate delineation of the tumors, with very little background in the rest of the mouse. PSVueTM794 was injected 18 h and KcapQ647 1 h prior to imaging. All probes were injected retro-orbitally with an injection volume of 0.1 mL. Images were analyzed with the Truquant software supplied with the FMT imaging system.
Tissue preparation and immunohistochemical analysis
Upon conclusion of imaging, mice were euthanized by CO2 asphyxiation and tumors were harvested, fixed in 10% formalin overnight, and embedded in paraffin. GIST tumors were confirmed by staining with hematoxlyin and eosin (H&E) as well as CD117 (KIT, DAKO). Apoptosis was assessed using antibodies recognizing cleaved Lamin A and cleaved Caspase-3 (Cell Signaling Technology). Immunohistochemical staining was performed on 5 μm slides. After deparaffinization and rehydration, sections were subjected to heat-induced epitope retrieval by immersion in a 0.01 M citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked for 15 min in 3% hydrogen peroxide in methanol. Nonspecific binding was blocked by treatment with a blocking reagent (Protein Block Serum-Free, DAKO) for 30 min at room temperature. The slides were then incubated overnight with primary antibody at 4 °C in a humidified chamber. Immunodetection was performed by using the Sensitive™ Link-Label (Biotin-based) IHC Detection Systems.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by grants from the NIH K99 CA158061-01A1 (L.R.), R01 CA106588 (M.v.M. and A.K.G.), P50 CA094056 (D.P.-W.), R01 EY019587 (D.P.-W.), F32 EY20051 (J.R.J.), the NIH UL1 TR000001-02S1 (A.K.G.), and the NIH Core Grant CA 06-927. The authors would especially like to thank the GIST Cancer Research Fund for their continued support and to acknowledge support from the Fox Chase Cancer Center Keystone Initiative in Personalized Risk and Prevention. We would like to acknowledge the Laboratory Animal Facility and the Small Animal Imaging Facility at Fox Chase Cancer. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Trademarks and Patents
D.P.-W. is a named inventor on a relevant patent issued to Washington University.
References
- 1.Goettsch WG, Bos SD, Breekveldt-Postma N, Casparie M, Herings RM, Hogendoorn PC. Incidence of gastrointestinal stromal tumours is underestimated: results of a nation-wide study. Eur J Cancer. 2005;41:2868–72. doi: 10.1016/j.ejca.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, et al. EORTC Soft Tissue and Bone Sarcoma Group. Italian Sarcoma Group. Australasian GastroIntestinal Trials Group KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 3.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 4.FDA approves regorafenib (Stivarga) for GIST. Oncology (Williston Park) 2013;27:164. [PubMed] [Google Scholar]
- 5.Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H, et al. GRID study investigators Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, Chen LL, Podoloff DA, Benjamin RS. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–9. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 7.Henze J, Mühlenberg T, Simon S, Grabellus F, Rubin B, Taeger G, Schuler M, Treckmann J, Debiec-Rychter M, Taguchi T, et al. p53 modulation as a therapeutic strategy in gastrointestinal stromal tumors. PLoS One. 2012;7:e37776. doi: 10.1371/journal.pone.0037776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon PM, Fisher DE. Role for the proapoptotic factor BIM in mediating imatinib-induced apoptosis in a c-KIT-dependent gastrointestinal stromal tumor cell line. J Biol Chem. 2010;285:14109–14. doi: 10.1074/jbc.M109.078592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakatani H, Araki K, Jin T, Kobayashi M, Sugimoto T, Akimori T, Namikawa T, Okamoto K, Nakano T, Okabayashi T, et al. STI571 (Glivec) induces cell death in the gastrointestinal stromal tumor cell line, GIST-T1, via endoplasmic reticulum stress response. Int J Mol Med. 2006;17:893–7. [PubMed] [Google Scholar]
- 10.Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26:7560–8. doi: 10.1038/sj.onc.1210558. [DOI] [PubMed] [Google Scholar]
- 11.Vadakara J, von Mehren M. Gastrointestinal stromal tumors: management of metastatic disease and emerging therapies. Hematol Oncol Clin North Am. 2013;27:905–20. doi: 10.1016/j.hoc.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pantaleo MA, Landuzzi L, Nicoletti G, Nanni C, Boschi S, Piazzi G, Santini D, Di Battista M, Castellucci P, Lodi F, et al. Advances in preclinical therapeutics development using small animal imaging and molecular analyses: the gastrointestinal stromal tumors model. Clin Exp Med. 2009;9:199–205. doi: 10.1007/s10238-009-0033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pantaleo MA, Nicoletti G, Nanni C, Gnocchi C, Landuzzi L, Quarta C, Boschi S, Nannini M, Di Battista M, Castellucci P, et al. Preclinical evaluation of KIT/PDGFRA and mTOR inhibitors in gastrointestinal stromal tumors using small animal FDG PET. J Exp Clin Cancer Res. 2010;29:173. doi: 10.1186/1756-9966-29-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Revheim ME, Røe K, Bruland OS, Bach-Gansmo T, Skretting A, Seierstad T. Monitoring the effect of targeted therapies in a gastrointestinal stromal tumor xenograft using a clinical PET/CT. Mol Imaging Biol. 2011;13:1234–40. doi: 10.1007/s11307-010-0464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cullinane C, Dorow DS, Kansara M, Conus N, Binns D, Hicks RJ, Ashman LK, McArthur GA, Thomas DM. An in vivo tumor model exploiting metabolic response as a biomarker for targeted drug development. Cancer Res. 2005;65:9633–6. doi: 10.1158/0008-5472.CAN-05-2285. [DOI] [PubMed] [Google Scholar]
- 16.Floris G, Wozniak A, Sciot R, Li H, Friedman L, Van Looy T, Wellens J, Vermaelen P, Deroose CM, Fletcher JA, et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res. 2013;19:620–30. doi: 10.1158/1078-0432.CCR-12-2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metildi CA, Tang CM, Kaushal S, Leonard SY, Magistri P, Tran Cao HS, Hoffman RM, Bouvet M, Sicklick JK. In Vivo Fluorescence Imaging of Gastrointestinal Stromal Tumors Using Fluorophore-Conjugated Anti-KIT Antibody. Annals of surgical oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, Araki K, Akimaru K, Yuri K. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab Invest. 2002;82:663–5. doi: 10.1038/labinvest.3780461. [DOI] [PubMed] [Google Scholar]
- 19.Kossodo S, Pickarski M, Lin SA, Gleason A, Gaspar R, Buono C, Ho G, Blusztajn A, Cuneo G, Zhang J, et al. Dual in vivo quantification of integrin-targeted and protease-activated agents in cancer using fluorescence molecular tomography (FMT) Mol Imaging Biol. 2010;12:488–99. doi: 10.1007/s11307-009-0279-z. [DOI] [PubMed] [Google Scholar]
- 20.Smith BA, Akers WJ, Leevy WM, Lampkins AJ, Xiao S, Wolter W, Suckow MA, Achilefu S, Smith BD. Optical imaging of mammary and prostate tumors in living animals using a synthetic near infrared zinc(II)-dipicolylamine probe for anionic cell surfaces. J Am Chem Soc. 2010;132:67–9. doi: 10.1021/ja908467y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanshaw RG, Smith BD. New reagents for phosphatidylserine recognition and detection of apoptosis. Bioorg Med Chem. 2005;13:5035–42. doi: 10.1016/j.bmc.2005.04.071. [DOI] [PubMed] [Google Scholar]
- 22.Maxwell D, Chang Q, Zhang X, Barnett EM, Piwnica-Worms D. An improved cell-penetrating, caspase-activatable, near-infrared fluorescent peptide for apoptosis imaging. Bioconjug Chem. 2009;20:702–9. doi: 10.1021/bc800516n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson JR, Kocher B, Barnett EM, Marasa J, Piwnica-Worms D. Caspase-activated cell-penetrating peptides reveal temporal coupling between endosomal release and apoptosis in an RGC-5 cell model. Bioconjug Chem. 2012;23:1783–93. doi: 10.1021/bc300036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnett EM, Zhang X, Maxwell D, Chang Q, Piwnica-Worms D. Single-cell imaging of retinal ganglion cell apoptosis with a cell-penetrating, activatable peptide probe in an in vivo glaucoma model. Proc Natl Acad Sci U S A. 2009;106:9391–6. doi: 10.1073/pnas.0812884106. [DOI] [PMC free article] [PubMed] [Google Scholar]