

Abstract
The atrial G protein-regulated inwardly rectifying K+ (GIRK1 and GIRK4) heterotetrameric channels underlie the acetylcholine-induced K+ current responsible for vagal inhibition of heart rate and are activated by the G protein βγ subunits (Gβγ). We used a multistage protein-protein docking approach with data from published structures of GIRK1 and Gβγ to generate an experimentally testable interaction model of Gβγ docked onto the cytosolic domains of the GIRK1 homotetramer. The model suggested a mechanism by which Gβγ promotes the open state of a specific cytosolic gate in the channel, the G-loop gate. The predicted structure showed that the Gβ subunit interacts with the channel near the site of action for ethanol and stabilizes an intersubunit cleft formed by two loops (LM and DE) of adjacent channel subunits. Using a heterologous expression system, we disrupted the predicted GIRK1- and Gβγ-interacting residues by mutation of one protein and then rescued the regulatory activity by mutating reciprocal residues in the other protein. Disulfide crosslinking of channels and Gβγ subunits with cysteine mutations at the predicted interacting residues yielded activated channels. The mechanism of Gβγ-induced activation of GIRK4 was distinct from GIRK1 homotetramers. However, GIRK1-GIRK4 heteroterameric channels activated by Gβγ displayed responses indicating that the GIRK1 subunit dominated the response pattern. This work demonstrated that combining computational with experimental approaches is an effective method for elucidating interactions within protein complexes that otherwise might be challenging to decipher.
INTRODUCTION
Acetylcholine (ACh) released by the vagus nerve activates KACh potassium (K+) channels, which hyperpolarize the membrane potential of atrial cardiomyocytes and slow heart rate (1). These channels were the first to be identified as effectors of the βγ subunits of heterotrimeric G proteins (Gβγ) (2). Multiple Gβγ effectors have since been identified (reviewed in 3). Co-crystals of Gβγ with soluble proteins, such as phosducin (4) and G protein-coupled receptor kinase 2 (5), exist; the Gβγ interactions with these proteins serve largely to modulate membrane recruitment and orientation of the effector proteins relative to the membrane and other membrane-associated proteins. The mechanism by which Gβγ controls specific conformations and thus the functional activity of any of its membrane-associated downstream effectors, such as adenylyl cyclases, phospholipases, protein kinases, and Ca2+ or K+ channels, is not understood. KACh of heart atria is a heterotetramer of two G protein-regulated inwardly rectifying K+ (GIRK) channel subunits: GIRK1 and GIRK4 (also known as Kir3.1 and Kir3.4) (6–8). In the brain, all GIRK1–4 subunits can be detected (9), and they combine to form heterotetramers involved in the formation of inhibitory postsynaptic potentials, functioning in drug abuse and addiction, as well as in spatial memory and learning (reviewed in 10). GIRK channels also play important roles in hormone secretion and smooth muscle cell function (10). GIRK channels are gated not only by G proteins but also by alcohols (11, 12), and those containing the GIRK2 or GIRK4 subunits by Na+ ions (13). Any of these intracellular activators of GIRK channels also requires the presence of plasma membrane phosphatidylinositol bisphosphate (PIP2) to stimulate channel activity (14). Whereas most physiologic GIRK channels exist as heterotetramers, single point mutations in the pore helix (fig. S1A) enhance the activity of homomeric GIRK channels (referred to as GIRK*; where GIRK1* or G1* has the F137S mutation, and GIRK4* or G4* has the S143T) (15).
Structures of the heterotrimeric G protein complex and the Gβγ subunits alone were elucidated in the mid 1990s (16,17,18). Several atomic resolution structures of GIRK subunits are also available [GIRK1 (19,20) and GIRK2 (21,22)], but it has proven challenging to obtain structures of GIRK channels in complex with Gβγ and only recently has a cocrystal structure been published (while the present study was in its final stage of review) (49). These and other structures of Kir channels have revealed the presence of a cytosolic gate (G-loop gate) and two putative transmembrane gates, the inner helix and selectivity filter gates (fig. S1A). Although the structure of a fully activated GIRK tetramer with all gates in open conformations has not yet been determined, the intracellular G-loop gate of GIRK1 has been crystallized in the constricted or “closed” and the dilated or “open” conformations in a chimeric channel (19). This GIRK1 chimera consists of a mammalian GIRK1 channel for the intracellular domain and proximal one-third of the transmembrane domain and of a prokaryotic channel for the external two thirds of the transmembrane domain (fig. S1B). Open G-loop gate conformations have also been published for GIRK2 channels, in which an intracellular point mutation (R201A) was introduced that induced G-loop gate opening (22).
Given the difficulties in obtaining co-crystal structures of these membrane proteins, we pursued a computational docking strategy to develop an experimentally testable model of a GIRK channel in complex with Gβγ. We primarily focused on GIRK1 and used the published coordinates of the GIRK1 chimera (19) as the starting structure. We selected this GIRK1 chimera because, although cardiac KACh current results from GIRK1/GIRK4 heterotetramers (7), there are no known structures of GIRK4 channels and structures of GIRK1 are available. Furthermore, GIRK heterotetramers in other parts of the body are also almost universally combinations of GIRK1 with another GIRK isoform (reviewed in 10). The distal two-thirds of the transmembrane regions of the GIRK1 chimera are prokaryotic, but upon functional reconstitution this channel behaves as a bona fide inward rectifier (23). Importantly, the intracellular regions, where Gβγ binding is expected to occur, are mammalian in origin and the proximal transmembrane regions, which participate in PIP2 binding, are also of mammalian origin. Inclusion of the transmembrane region in the GIRK1 chimera offers advantages over other structures of the isolated intracellular cytoplasmic domains of the channel by obviating the need for an artificial linker to connect the N- and C- termini and by appropriately allowing the G-loop gate and surrounding residues to interact with the proximal transmembrane regions and other residues near the inner membrane leaflet.
Another advantage of the GIRK1 chimera structure is that it captures the same protein in two distinct conformations of its intracellular domain within the asymmetric unit of the crystal structure. Hence, the two conformations are seen in the exact same conditions without introduction of any mutations and thus likely represent energy minima in the conformational landscape of the intracellular domain. The two conformations have identical transmembrane segments, but differ in the cytosolic domains, including changes in the region that has been hypothesized to contribute to the Gβγ binding site by several studies and that contains the critical Leu333 residue that is necessary for Gβγ- and agonist-induced GIRK activation (33,34,39). Therefore, we used structures of the mammalian-derived intracellular domain of the GIRK1 chimera in the dilated “open” G-loop conformation (henceforth “open” channel structure) to hypothesize the Gβγ binding site in mammalian GIRK1 channels. For Gβγ, we selected the most complete structure with the fewest unresolved residues at the Gβ and Gγ termini (40). We then tested the resulting predicted interactions using experimental evidence from mammalian GIRK channels heterologously expressed in Xenopus oocytes.
RESULTS
A computational docking strategy predicts an energetically favorable complex between Gβγ and the GIRK1 cytosolic domain
For the computational docking approach, we used structures of the mammalian-derived intracellular domain of the GIRK1 chimera in the “open” G-loop conformation (mouse Kir3.1, PDB: 2QKS) and the Gβγ structure with the fewest unresolved residues at the Gβ and Gγ termini (bovine Gβ1 shares 100% amino acid identity with the human protein, Gβ1γ2, PDB: 2BCJ). We pursued a multistage strategy inspired by recent successes of protocols that combine several docking programs (25–28) (see Methods and Materials). Four computational stages identified the best energy scoring model presented in this study: (i) rigid body search, (ii) identification of regions of interest, (iii) refinement of docked structures through filtering by physiological criteria and hierarchical clustering, and (iv) selection of the best model (Fig. 1A). We used ZDock to perform rigid docking of Gβγ onto the channel (29,30). Because only the intracellular domain of the channel has been shown to interact with Gβγ, all channel residues except for the intracellular regions of two adjacent subunits were present but not permitted to participate in the binding. This reduced redundancy due to the channel’s 4-fold symmetry and excluded the transmembrane segments, which included all prokaryotic regions of the structure. This first step of rigid body search produced 54,000 poses that we retained (Fig. 1B).
Fig 1. Selection of a best-scoring computational model of Gβγ docked onto GIRK1.

(A) Summary of the docking protocol used to predict the GIRK1 channel-Gβγ binding mode. Individual steps are organized into phases of a generalized approach to multistage protein-protein docking as outlined by Vajda and Kozakov (25). (B) Cartoon representation of the channel structure. Two adjacent channel subunits are highlighted in cyan and magenta. Yellow dots represent the centers of mass of individual retained poses of Gβγ at the end of the rigid docking by ZDock phase (54,000 poses). (C) Centers of mass of poses retained after filtering by membrane distance restraints (~5000 poses). (D) Centers of mass of the 30 clusters of poses representing the centers of the 30 largest clusters obtained from Cluspro. The pose that was refined to yield the final model is highlighted in blue. Green highlights the pose representing the center of the largest cluster. (E) Scoring of RosettaDock refined models of the channel and Gβγ using a composite score. Scores are plotted versus interface root mean square deviation (RMSD) from the lowest-scoring model. There are 1000 points representing refined models for each of the 30 initial starting orientations obtained from clustering. Lower values represent more favorable scoring. Refined RosettaDock models from the starting orientation that yielded the final model are highlighted in blue. Refined models from the starting orientation representing the largest cluster are highlighted in green. Interface RMSD is calculated as follows: residues of Gβγ in the best scoring model containing at least 1 atom within 10Å of the channel are identified as reference. For each of the other poses, the positions of these same residues are identified within each of the other poses and RMSD is calculated compared to the reference.
We next filtered the results by rejecting any ZDock output model containing any atoms more than 8Å above the plane of the membrane or with the C-alpha atom of the C-terminal Gγ more than 30Å below the plane of the membrane (Fig. 1C). The first constraint reflects exclusion of the protein by the lipid bilayer and the second constraint reflects the expected prenylation (geranyl-geranylation) of the Gγ2 C-terminus, which would anchor it to the lipid bilayer (reviewed in 10).
Then, we subjected the top 2000 ZDock output poses remaining after filtering to hierarchical clustering by interface RMSD using Cluspro (27, 30) and retained 30 structures each representing the center pose of one of the 30 largest clusters (Fig. 1D). RosettaDock (30) refined these 30 structures through flexible sidechain docking. We produced 1000 poses for each of the 30 input structures and scored them using a composite energy score that combined the internal RosettaDock (Rscore), and the ZRank2.0 (Zscore) scoring functions (see Methods and Materials for details) (26,30). The best-scoring model was found within a steep energy well (Fig. 1D, E - blue) with poses of the largest cluster center localizing nearby (Fig. 1D, E, - green).
Analysis of the best-scoring model of a Gβγ-channel complex suggests a site of action for Gβγ-induced channel activation
Our best-scoring model of Gβγ and the “open” GIRK1 chimera structure predicts a binding mode with an ~1800 Å2, largely hydrophobic interaction surface with peripheral electrostatic interactions (Fig. 2A), which is typical of complexes of Gβγ with soluble proteins (4). The Gβγ binding interface on the channel predicted by our model agrees well with the results from an NMR study of Gβγ interactions with the cytosolic GIRK1 domain (32) (fig. S2). The two proteins interact through a large surface and is predicted to involve multiple interacting residues (Fig. 2B and 2C; fig. S3, left).
Fig 2. A computational docking strategy predicts an energetically favorable complex between Gβγ and channel.

(A) Surface representations of the channel (left) and Gβγ (right) are colored by residue hydrophobicity (44): Yellow is most hydrophobic, blue is least hydrophobic. Interface regions found in the best scoring model are outlined in magenta. (B) Cartoon illustration showing overall orientation of Gβγ docked to the channel. Gβ is yellow (ribbon), Gγ is orange (ribbon), two adjacent subunits of the homotetrameric channel are highlighted in cyan and magenta (cartoons). Interface residues of the channel subunits within 5Å of Gβ are depicted as spheres. (C) Close-up views depicting cartoon backbone and stick sidechain representations of interface residues (5Å cutoff) of the channel (left) and Gβγ (right). In C, red = acidic, blue = basic, green = polar, white = non-polar.
Inspection of this channel region in our best-scoring model of Gβγ docked to the “open” GIRK1 chimera structure showed Gβ residue Leu55 occupying the cleft between GIRK1 Leu333 and Phe243 (Fig. 3A). Previous experimental (33,34) and computational (24) studies have implicated the GIRK channel LM loop in Gβγ- and PIP2-mediated channel gating. Furthermore, the conserved GIRK1 LM loop residue Leu333 plays a critical role in stimulation of agonist-induced currents and stimulation of channel currents by Gβγ coexpression (34). Inspection of this region in the “closed” versus “open” conformations of the GIRK1 chimera crystal structure reveals a large upward movement of the LM loop in the open conformation (fig. S4A). In contrast, downward movement of the LM loop seen in the “closed” conformation brings the LM loop residue Leu333 to within van der Waals contact of the adjacent subunit's DE loop residue Phe243 and occludes the cleft that separates the two loops in the “open” conformation (fig. S4B). Both residues Phe243 and Leu333 are conserved in all human GIRK isoforms but are distinct from the residues found at these positions in the closely related, but G protein-insensitive, IRK channels (fig. S4C and D). These observations combined with previous evidence (36) for the involvement of Gβ Leu55 and other nearby Gβ residues in GIRK gating led us to hypothesize that Gβγ interactions stabilize the “raised” LM loop conformation seen in the “open” conformation of the GIRK1 structure and prevent the downward movement seen in the “closed” conformation. To test the interactions predicted from the computational docking procedure, we performed functional tests with the mammalian GIRK1* channel in the Xenopus laevis oocyte system (34).
Fig 3. Gβγ interactions predicted to open DE-LM cleft sterically or through electrostatic repulsion increase channel activity.

(A) Cartoon illustration of predicted functionally important residue interactions. Two adjacent subunits of the channel are highlighted in cyan and magenta. Gβ is yellow. Gβ Leu55 is seen inserting in a cleft between GIRK1 LM loop residue Leu333 and DE loop residue Phe243 of adjacent channel subunits. GIRK1 residues Glu334 and Glu335 are also seen near Gβ Leu55 and Lys89. Residue numbers are shaded as gray, hydrophobic; blue, positively charged; red, negatively charged. (B) Overlapped representative traces depicting currents at -80 mV measured by two-electrode voltage clamp in Xenopus laevis oocytes expressing either GIRK1* (G1*) or GIRK1* and Gβ1γ2. LK, Low potassium solution; HK, High potassium solution; Ba2+, High potassium solution containing 3 mM BaCl2. Ba2+is an inhibitor of GIRK channel activity. (C) The effect of side chain volume substitutions at G1* Phe243 on stimulation of G1* current by wild-type Gβγ. (D) The effect of side chain volume substitutions at G1*Leu333 on stimulation of G1* current by wild-type Gβγ. (E) The effect of side chain volume substitutions at Gβ Leu55 on stimulation of G1* channel current by Gβγ. † indicates estimated p-value < 0.05 by non-overlap of 90% confidence interval with that of Gβ(L55F)γ. (F) The effect of combining mutations that alter side chain volume in G1* and Gβγ. (Left) The effect of Gβ(L55C)γ mutation on G1* channels versus G1*L333W mutant channels. (Right) The ability of G1*(F243C) mutant channel to be activated by wild-type versus Gβ(L55W)γ. (G) The effect of neutralizing negatively charged LM loop channel residues. The effect of G1*E334Q and G1*E335Q on the ability of wild-type Gβγ (Left) or Gβ(L55E)γ (Right) to stimulate channel activity. In (C–G), each of the indicated channels or channel mutants was expressed alone or coexpressed with Gβγ and the fold change between the two groups (n=6–10 oocytes/group, repeated in two batches of oocytes) was calculated for each mutant. Fold-activation of control G1* channels by wild-type G βγ w as normalized t o 1 . Results are summarized in bar graphs (mean ± SEM). (** indicates estimated p-value < 0.01 by non-overlap of 95% confidence interval with that of wild-type; * indicates estimated p-value < 0.05 by non-overlap of 90% confidence interval with that of wild-type; see Materials and Methods for details).
Analysis of the input structure representing the center of the largest cluster showed an interaction surface similar to that observed in the best-scoring model, including preservation of the interaction of Gβ Leu55 with GIRK1 Leu333 and Phe243 and close proximity of Gβ Lys89 to GIRK1 Glu334 (figs. S5A–D, fig. S3, right). However, in the largest cluster model the Phe243 side chain points upwards (fig. S5D), which is consistent with the original crystal structure’s (fig. S4, A and B) compared to the final model’s Phe243 position (Fig. 2C and 3A), which is turned downwards and enlarges the DE-LM cleft. The pose of Gβγ in the largest cluster model is slightly rotated, compared to that in the final best-scoring model, such that its interaction surface includes additional residues, such as Trp99 and Ser334, which are closer to the center of the Gβγ propeller structure, and excludes other residues included in the best-scoring model’s interaction surface, such as Asn88 and Gly131.
PDB coordinates for both the best-scoring model and the largest cluster model are provided as PDB file S1 and PDB file S2, respectively. Both files contain chains A–D representing the cytosolic and proximal transmembrane regions of the channel (mammalian derived regions of the GIRK1 chimera structure) as well as chains E and F representing Gβ and Gγ respectively. The distal transmembrane regions, although present during the initial rigid docking with ZDock, were removed prior to clustering to save computational time. Hydrogens are added by the RosettaDock program. Thus, the best-scoring model structure contains all hydrogens, and the largest cluster model, which is the center pose of the largest cluster prior to RosettaDock refinement, contains only polar hydrogens.
Gβγ interactions predicted to open the DE-LM cleft sterically or through electrostatic repulsion increase channel activity
Consistent with previous studies (34), coexpression of bovine Gβ1γ2 with the human GIRK1* (GIRK1 F137S or G1*) channel in Xenopus oocytes stimulated channel currents (Fig. 3B). To isolate the effect on Gβγ-induced activation of channels, we focused on fold-change in current amplitude induced by Gβγ coexpression. Using the GIRK1* channel as our control, we tested a number of channel mutations, none of which independently activated currents above those of the GIRK1* alone (fig. S6). This indicated that lack of activation of a channel mutant by Gβγ coexpression was unlikely due to the mutation itself activating the channel and Gβγ then being unable to further activate the saturated current.
To test if the insertion of Gβ Leu55 into the intersubunit DE-LM cleft between the channel Phe243 and Leu333 residues was necessary for Gβγ-mediated channel activation, we individually changed the side chain volume of the three residues involved in the DE-LM cleft interaction: Phe243 of the channel’s DE loop, Leu333 of the channel’s LM loop, and Leu55 of Gβ. Decreasing the side chain volume of residue Phe243 reduced the stimulation of current by coexpressed Gβ1γ2 (Fig. 3C). Increasing the volume of residue Leu333 progressively enhanced the ability of Gβγ to stimulate channel activity (Fig. 3D). Similarly, reducing the volume of residue Gβ Leu55 decreased the ability of Gβγ to stimulate the channel (Fig. 3E). Although the Gβ(L55F)γ mutant was not as effective as wild-type Gβγ in stimulating channel activity, it was significantly more effective than the smallest side chain mutants. Introduction of an L333W mutation (a volume-increasing mutation) into the channel rescued the ability of the Gβ(L55C)γ mutant (a volume-decreasing mutation) to stimulate channel currents(Fig. 3F, left). Similarly, introduction of the L55W mutation in Gβ rescued induction of channel activity in the G1*(F243C) channel mutant (Fig. 3F, right). Thus, decreases in the side chain volume of residues in one protein were compensated by increases in side chain volume of the other protein. These experiments suggested that steric opening of the DE-LM cleft by Gβ Leu55 is critical to channel activation.
Adjacent to the GIRK1 LM loop residue Leu333 are two negatively charged residues Glu334 and Glu335 (Fig. 3A). We tested if we could maintain opening of the DE-LM cleft by electrostatic repulsion of the LM loop rather than steric opening of the cleft (Fig. 3G). The negatively charged mutant Gβ(L55E)γ robustly stimulated G1* currents (Fig. 3G, right). Neutralizing mutations of the negatively charged LM loop residues G1*(E334Q or E335Q) abrogated the stimulating ability of the Gβ(L55E)γ mutant (Fig. 3G, right) but did not significantly alter the effect of wild-type Gβγ (Fig. 3G, left). Combined, these results support the hypothesis that enlarging the DE-LM cleft through either steric effects or electrostatic repulsion of the channel's LM loop by Gβ(L55E)γ stimulates channel activity.
Disulfide cross-linking of Gβγ to the channel stimulates channel activity
To verify that Gβ Leu55 and GIRK1 Leu333 were in close proximity as predicted by our model, we tested whether the Gβ(L55C) and the G1*(L333C) mutant residues could be functionally cross-linked. We used 10 mM H2O2 as the cross-linking agent and assessed the effect of crosslinking on the whole-cell current generated in oocytes expressing either the Gβ *(L55C)γ or the G1*(L333C) with the corresponding control as coexpressed with each other. Treatment of oocytes expressing G1* channels or the G1*(L333C) mutant channels with 10 mM H2O2 inhibited the whole-cell current (Fig. 4A-top, B-top, C). H2O2 also reduced the current in oocytes coexpressing Gβγ with either the G1* or G1*(L333C) mutant, although to a smaller extent than was observed for the channels alone (Fig. 4A-middle, B-middle, C). When Gβ(L55C)γ was coexpressed with G1*, H2O2 produced a large reduction in current (Fig. 4A-bottom, C). In contrast, H2O2 activated G1*(L333C) when it was coexpressed with the Gβ(L55C)γ (Fig. 4B- bottom, C), indicating that these residues formed a functionally active disulfide link when exposed to H2O2 and thus increased channel activity.
Fig 4. Disulfide cross-linking of Gβγ to the channel stimulates channel activity.

(A–B) Representative two-electrode voltage clamp traces showing the effect of H2O2 on G1* and cysteine mutants [G1(L333C) or Gβ(L55C)γ] expressed in Xenopus oocytes. Abbreviations: LK, Low potassium Solution; HK, High potassium solution; Ba2+, High potassium solution containing 3 mM BaCl2. (C) Summary data showing % change in current due to H2O2 perfusion (mean ± SEM; n=5–9, repeated in at least two separate oocyte batches). All other groups were significantly less different than controls except the final which indicates the G1*L333C+Gβ(L55C)γ combination. (p<0.0001 by one way ANOVA and Dunnett’s post hoc test) (D) SDS PAGE analysis with Coomassie Brilliant Blue staining. Wild-type Gβγ or the L55C mutant was mixed with equal concentration of cysteine-less GIRK1 cytoplasmic domain (G1CP) containing the single cysteine mutation L333C [G1CP (L333C)] as indicated. Crosslinking was induced by treatment with increasing concentration of hydrogen peroxide up to 10 mM for 2.5 minutes. A representative image is shown from five individual experiments.
We verified the oocyte cross-linking results using purified proteins or protein fragments. We expressed and purified a cysteine-less version of the cytoplasmic GIRK1 fragment (amino acids 41–63 from the N-terminus directly linked to amino acids 190–371 from the C-terminus) (35) with the only cysteine resulting from the L333C mutation [G1CP(L333C)]. Similarly, we expressed and purified wild-type full-length Gβγ and the Gβ(L55C)γ mutant as previously described (37). By subjecting the combinations of purified proteins to reducing environments with varying concentrations of H2O2 (Fig. 4D), we determined that only the Gβ(L55C)γ could be crosslinked to G1CP(L333C) (Fig. 4D, band labeled G1CP+Gβ). These data further supported our computational model, indicating that the Gβ Leu55 residue is in close proximity to the GIRK1 Leu333.
A salt-bridge interaction that allows Gβγ to stabilize the open DE-LM cleft increases channel activity
Our model predicted that GIRK1 Glu334 and Gβ Lys89 form a salt bridge (Fig. 3A). We mutated these residues to reverse their respective charges and compared the resulting currents to G1* controls (Fig. 5A). The Gβ(K89E)γ mutant exhibited reduced ability to stimulate G1* (Fig. 5A, left). Simultaneous reversal of charge of the channel residue by coexpressing the G1*(E334K) mutant with the Gβ(K89E)γ mutant resulted in increased activation of the channel, (Fig. 5A, right). These results strongly support an electrostatic interaction between the two residues. Positioning of these residues in our model is consistent with their participation in a salt bridge that stabilizes the “raised” conformation of the LM loop found in the “open” state of the channel structure.
Fig 5. A salt-bridge interaction that allows Gβγ to stabilize the open DE-LM cleft increases channel activity.

(A) The effect of combining charge-reversing mutations at the predicted salt-bridge residues G1* Glu334 and Gβ Lys89. (Left) The ability of Gβ(K89E)γ versus wild-type Gβγ to stimulate G1* channels. (Right) The ability of Gβ(K89E)γ versus wild-type Gβγ to stimulate G1*E334K channels. (** indicates estimated p-value < 0.01 by non-overlap of 95% confidence interval with that of wild-type; * indicates estimated p-value < 0.05 by non-overlap of 90% confidence interval with that of wild-type; see methods section). (B) Overlapped representative traces depicting currents at −80 mV measured by two-electrode voltage clamp in Xenopus laevis oocytes. G1* ± Gβγ is co-expressed with mGluR2 receptors to monitor agonist-induced currents. (C) The effect of combining charge-reversing mutations at the predicted salt-bridge residues G1* Glu334 and Gβ Lys89 on basal and agonist-induced currents. (Left) The effect of co-expressed wild-type Gβγ versus Gβ(K89E)γ on basal and agonist induced activity of G1*. (Right) The effect of co-expressed wild-type Gβγ versus Gβ(K89E)γ on basal and agonist induced activity of G1*E334K. (Bar graphs are mean ± SEM of n=7–10, repeated in at least two separate oocyte batches; * indicates p<0.05, ** indicates p<0.001, *** indicates p<0.0001 for comparison of basal current to channel alone. † indicates p<0.05, †† indicates p<0.001, ††† indicates p<0.0001 for comparison of total maximal current to channel alone. Comparisons by one way ANOVA with Dunnet's post-hoc test).
We explored the relevance of these predicted interactions to agonist-induced GIRK currents. Coexpression of Gi-coupled receptors with GIRK channels in the Xenopus oocyte system enables monitoring of agonist-induced currents. Moreover, coexpression of exogenous Gβγ stimulates basal GIRK currents at the expense of agonist-induced currents, preserving the amplitude of total maximal current (34). We chose as a Gi-coupled receptor the metabotropic glutamate type 2 (mGluR2) (Fig. 5B) instead of the muscarinic type 2 (M2R) found in atrial cells to avoid ACh-mediated stimulation of the Gq-coupled M1 receptors endogenous to Xenopus oocytes, which can lead to PIP2 hydrolysis and current inhibition. The effect of charge reversal mutations of the salt-bridge pair residues G1* Glu334 and Gβ Lys89 on basal currents in the Xenopus oocytes also expressing mGluR2 (Fig. 5C) was the same as seen in the absence of receptor coexpression (Fig. 5A). Coexpression of Gβ(K89E)γ inefficiently stimulated G1* basal currents but robustly activated G1*(E334K) activity. Basal currents of both control and the mutant channel were effectively activated by Gβγ. The total currents in the presence of agonist also followed a similar pattern. The introduction of either Gβ(K89E) or G1*(E334K) mutations alone resulted in submaximal total currents compared to G1* with Gβγ. Coexpression of G1*(E334K) and Gβ(K89E)γ, however, resulted in maximally stimulated total currents. These data indicated that the GIRK1 Glu334 – Gβ Lys89 salt bridge plays an important role in mediating both basal and agonist-induced activation of GIRK1.
The GIRK1 response to Gβγ dominates over the GIRK4 response in the heteromeric channel
We next tested if the dependence on Gβ Leu55 side chain volume observed for G1* current stimulation (Fig. 3E) extended to agonist-activated G1* currents stimulated by coexpression of mGluR2 (Fig. 6A). The patterns for basal activity in the presence of receptor (Fig. 6A) were consistent with those already observed in its absence (Fig. 3A). Small side chain substitutions, such as Gβ(L55A)γ, were deficient in stimulating basal G1* currents compared to wild-type Gβγ; Large side chain mutations, such as Gβ(L55W)γ, performed comparably to wild-type Gβγ. Total currents produced by the addition of agonist were also consistent with this pattern (Fig. 6A).
Fig 6. The G1 response to Gβγ dominates over the G4 response in the heteromeric channel.
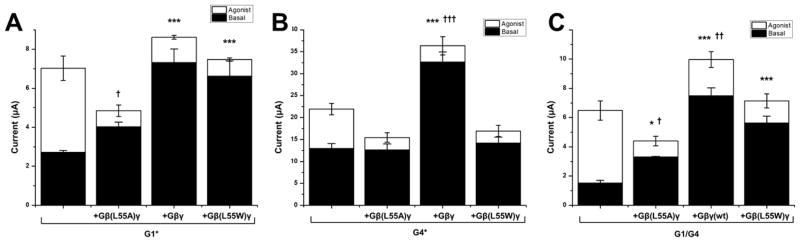
(A) The effect of side chain volume substitutions in coexpressed Gβγ at position Leu55 on basal and agonist-induced currents of G1*. (B) The effect of side chain volume substitutions in coexpressed Gβγ at position Leu55 on basal and agonist-induced currents of G4*. (C) The effect of side chain volume substitutions in coexpressed Gβγ at position Leu55 on basal and agonist-induced currents of wild-type heteromeric G1/G4 channels. (All bar graphs are mean ± SEM of n=6–8, repeated in at least two separate oocyte batches; * indicates p<0.05, ** indicates p<0.001, *** indicates p<0.0001 for comparison of basal current to channel alone. † indicates p<0.05, †† indicates p<0.001, ††† indicates p<0.0001 for comparison of total maximal current to channel alone. Comparisons by one way ANOVA with Dunnet's post-hoc test).
We extended our tests of dependence on Gβ Leu55 side chain volume to GIRK4* (mammalian GIRK4 S143T or G4*) channels and to the heteromeric wild-type GIRK1/GIRK4 (or G1/G4) channels (Fig. 6B and C, respectively). In contrast to G1*, neither the basal nor the total G4* currents showed the characteristic dependence on Gβ Leu55 side chain volume. Both the small and large side chain substitutions were deficient in stimulating either basal or agonist-induced G4* currents (Fig. 6B). This result suggests possible underlying structural differences between the DE-LM clefts of G1 and G4. However, a similar experiment with the wild-type G1/G4 channels revealed that the heteromeric channels exhibit a dependence on the Gβ Leu55 side chain volume that was similar to that of G1* (Fig. 6C). This result suggested that the dependence of the response of the wild-type heteromeric channels to the Gβ Leu55 side chain volume is dominated by the interactions with the GIRK1 channel subunit.
DISCUSSION
Using a multistage approach to protein-protein docking and utilizing a composite energy scoring function, we identified a model of Gβγ in complex with the intracellular portion of the GIRK channel structure. This model predicted a large surface of interaction and additional bioinformatic analysis further localized the site of action of Gβγ to the channel’s DE-LM cleft. Experimental tests for the functional importance of the protrusion of the Gβ residue Leu55 into the DE (Phe243) – LM (Leu333) cleft indicated that opening of this cleft is critical for the ability of Gβγ to stimulate GIRK1* and GIRK1/GIRK4 channel activity.
The GIRK1 chimera structure used here includes two conformations of the protein with dramatic changes in its LM loop, correlating with “opening” of its G-loop gate, between the two conformations. Although other structures exist that show “open” G-loop conformations (22), these structures are of the neuronally prevalent mouse GIRK2 channel that had a mutation (GIRK2 R201A) to induce opening of the G-loop gate and did not include multiple conformations with dramatic changes of the LM loop conformation, as seen in the GIRK1 chimera’s intracellular domain. Thus, it is not clear if the lack of LM loop conformational change is due to differences between GIRK1 and GIRK2 mechanisms of regulation by Gβγ or if it is due to the R201A mutation transducing conformational changes “downstream” of changes in the Gβγ binding site. Because proper backbone conformation of the binding site would be essential for successful docking, we chose to work with the GIRK1 chimera.
The purified GIRK1 chimera into planar lipid bilayers shows functional differences in response to Gβγ perfusion compared to mammalian GIRK1* channels heterologously expressed in cells (23). Whether these apparent functional differences can be reconciled when the entire signaling complex found in cells is reconstituted in planar lipid bilayers remains to be investigated. Regardless, in the present study we only used the mammalian-derived regions of the GIRK1 chimera that are identical in sequence to the mammalian GIRK1 channels in the computational docking analysis and to hypothesize the Gβγ binding site of the mammalian channels. Experimental testing was then carried out using the mammalian GIRK1* channel.
Here, we used the mammalian derived intracellular regions of the “open” structure of the GIRK1 chimera for our docking studies. Whether Gβγ binds to the “closed” structure of the intracellular domain or the closed state of the channel is a more challenging experimental question that is not yet addressed. In our structural modeling studies the channel is in an “open” or dilated conformation with regard to the G-loop, but the inner helix gate remains closed in this structure, meaning that the channel is non-conducting. In contrast to molecular dynamics simulations, which can allow for all-atom flexibility, the docking steps that we used at most allow flexibility of residue sidechains, while backbone conformations remain fixed. Thus, our computational docking methods only predict binding sites and “ignore” regions of the protein that are excluded from the search space because the docking does not attempt to predict conformational changes transduced in distant parts of the protein. Although this justifies our exclusion of the transmembrane regions of the GIRK1 chimera structure, it also implies that taken alone, the docking results address only the stabilization of a particular conformation of the intracellular domain and its G-loop gate and not necessarily to full channel activation. Evidence for the relevance of stabilization of this conformation of the intracellular domain to full channel activation comes not from the docked structure itself, but from the functional studies that demonstrated that residue mutations predicted to stabilize the “raised” LM loop conformation, equivalent to the “open” G-loop conformation, correlated with increased channel activation by Gβγ. The exact mechanism by which stabilization of the “open” conformation of the G-loop gate transduces changes in the other putative ion permeation gates in GIRK1 is not addressed here and remains an interesting question. Analogy to GIRK2 would suggest that PIP2 binding can help transmit the opening of the G-loop gate to the gates in the transmembrane segments (22).
In addition to this hypothesized role for PIP2 in GIRK1 channels, molecular dynamics simulations have suggested that PIP2 stabilizes the “open” conformation of the G-loop gate in the GIRK1 chimera. These simulations in the presence and absence of PIP2 revealed the rearrangement of intracellular structural elements that lead to stabilization of the “open” G-loop state (24). Briefly, in going from the “closed” conformation to the “open” conformation, the LM loop makes a large upward movement to interact closely with the N-terminus to form the βA-βM interaction. The CD loop, on the other hand, switches from close interactions with the N-terminus in the “closed” state to preferential interactions with the G-loop to stabilize its conformation in the “open” state. These movements are grossly visible in the structures of the two conformations of the G-loop gate (Fig. 7A) and the detailed changes in interactions are schematized in Figure 7B.
Fig 7. Various modulators stabilize the intracellular domain in the “open” G-loop conformation.

(A) Rearrangements of intracellular interactions grossly visible in comparison of the “closed” (left) and “open” (right) structures of the GIRK1 chimera. Some residues of the linker regions connecting the N- and C- termini to the transmembrane regions have been modeled in these structures. (B) Summary of the major results of Meng et al. (24) and their extension to include the DE loop and DE-LM loop cleft. Transitioning from the closed to open, the secondary structure elements switch their close interactions from adjacent elements on one side to the elements on the other side. PIP2 stabilizes the conformation by direct interactions with the CD loop and N-terminus. We propose that Gβγ works through a similar mechanism by stabilizing the same global conformation but by direct interactions with a different part of the channel. The proposed site of action at the DE-LM loop cleft is shared with the site of ethanol (EtOH) action. Pink and yellow highlight the alternating secondary structure elements involved in gating. The DE loop and the N-terminus would be from one subunit and all other elements would be from the other.
Results from our current study are fully consistent with this gating scheme and suggested that Gβγ interactions at the DE-LM cleft act to stabilize the “raised” conformation of the LM loop seen in the “open” state, allowing it to closely interact with the N-terminus to form the βA- βM interaction. Besides the critical steric effect of the Gβ Leu55 in opening the DE-LM cleft, the computed structure showed that Gβ Lys89 stabilizes the LM loop in the “raised” position through a salt bridge interaction with the Glu334 residue of GIRK1. Alcohols also interact with a hydrophobic pocket near the same cleft in GIRK channels to cause channel activation (38); whereas PIP2 acts near the CD loop and N-terminus to stabilize the G-loop gate to the open conformation (22,24) (Fig. 7B).
In our experiments, GIRK4* did not display the same dependence on Gβ Leu55 side chain volume as GIRK1*. Lack of crystallographic structures of GIRK4 precludes assessment of the putative differences in the DE-LM cleft between the two atrial channel subunits. On the basis of sequence similarity, GIRK4 is likely to be similar to GIRK2, which has a raised “LM” loop in the absence of Gβγ. We found that the heteromeric GIRK1/GIRK4 channel displayed a similar Gβ Leu55 side chain volume dependence as GIRK1*, suggesting that the need for opening the DE-LM cleft of GIRK1 dominates the heteromeric response.
The poses produced by RosettaDock were based on input structures representing centers of the 30 largest clusters of favorably scored ZDock output poses. The best-scoring model resided in a steep energy well (Fig. 1E, blue), consisting of poses produced from the input structure representing the center of one of the smaller clusters. This energy well appeared to connect to poses produced from the structure representing the center of the largest cluster (Fig. 1E, green). The existence of a large cluster of favorable conformations near the final model suggests the existence of a broad energy minimum near the final steep energy well (28, 47, 48), which increases the likelihood the protein will “find” the very stable (that is the deep energy well) conformation.
A larger search of conformational space around the center of the largest cluster may have allowed it to produce poses that also descended down the steep energy well leading to the final model. The input structure representing the center of the largest cluster included key features of the best-scoring structure: The interaction of Gβ Leu55 with GIRK1 Leu333 and Phe243 and close proximity of Gβ Lys89 to GIRK1 Glu334. However, the differences between the two models suggest that the distance between the DE-LM cleft is smaller in the largest cluster’s model and larger, more “open” in the final model. Gβγ in the largest cluster model is slightly rotated to include in its interaction surface residues, such as Trp99 or Ser334, on the “front” of the molecule closer to the center of the Gβγ propeller structure and excludes residues towards the “back” of the molecule, such as Asn88 and Gly131. Previously published mutagenesis data supports the importance of both of these relative front and back regions. Studies indicate that Trp99 (36), as well as residues Trp334, Thr86, Thr87, Gly130 (45), and the nearby Thr128 (46) all play a role in regulation of GIRK channels by Gβγ. Because the largest cluster model has a relatively low interface RMSD compared to the final model, it may be speculated to represent an alternate binding mode or represent an “encounter complex” resulting from favorable microcollisions prior to conformational descent down the final steep energy well (47, 48). Further investigation will require experimental probing of the binding dynamics.
Identification of the site of action of Gβγ by our computational model sets the stage for future experimental and computational studies. Gα-GDP binds Gβγ and terminates its ability to stimulate channel activity (2). The Gβ residues (such as Leu55, Lys89) identified to interact with the channel's LM loop also interact with the helical N-terminus of the Gα subunit in the GDP-bound inactive heterotrimer (16,17), hinting at the mechanism that may underlie the inhibitory role of Gα-GDP.
Our multistage docking strategy produced a compelling complex of the GIRK1 chimera with Gβγ, which allowed for experimental investigations which not only validated the predicted binding site in mammalian G1 channels, but allowed for localization of Gβγ’ s site of action within the binding site. Biochemical crosslinking of the two proteins in a functionally meaningful conformation offers a tool that could lead to successful experimental structural determination of the activated state of the GIRK1-Gβγ complex.
MATERIALS AND METHODS
Computational Docking
The structure selected to represent the GIRK channel was the “open” conformation (Chain A) of the crystal structure of a GIRK1 chimera (PDB code: 2QKS) consisting of mouse Kir3.1 (intracellular and proximal transmembrane regions) and the prokaryotic KirBac1.3 (middle and distal transmembrane regions) (19). A short loop linking the N-terminus to the first transmembrane domain is unresolved in the crystal structure. This region and any unresolved side chains were modeled using the loop modeling routine in Modeller (41). The first 40 N-terminal residues (Met1-Lys40) and the long, unique distal C-terminus of G1 (Ile372 to Thr501) are both missing from the crystal structure. However, previous experimental results suggest that activation of the channel or its binding to Gβγ can be preserved even with loss or alteration of large parts of these regions (34,39), thus we did not attempt to model these termini. Residues in the structure were renumbered to correspond to the equivalent human GIRK1 residue. All references to channel residue numbering are relative to GIRK1, unless otherwise stated. The bovine Gβγ structure used in our study was extracted from a co-crystal of Gαq, GRK2, and Gβγ (PDB code: 2BCJ) (40). This structure represents the most complete structure of Gβ1γ2 with the least residues missing from the termini (3 and 1 from the N- and C- termini respectively of Gγ2).
We used a multistage protein-protein docking approach as outlined by Vajda and Kozakov (25). The 6° sampling option of ZDock3.0.1 was used as the rigid body docking step to retain 54,000 poses (29,30). This was followed by application of membrane distance restraints where the plane of the membrane was defined by the C-alpha atoms of the channel's Leu75 residue in the interfacial helix that runs parallel to the inner membrane leaflet. ZDock output models containing any atoms more than 8Å above the plane of the membrane or with the C-alpha atom of the C-terminal Gγ more than 30Å below the plane of the membrane were excluded. The top 2000 ZDock output poses remaining after filtering were subject to hierarchical clustering by interface RMSD using ClusPro1.0 with default options and a 9.0Å clustering radius (28, 31). Structures of the centers of the 30 largest clusters were retained. Because of some redundancy due to the channel’s symmetry, some structures were rotated by 900 along the channel’s central axis to bring all structures to the same intersubunit interface. Next, refinement through flexible sidechain docking was performed using RosettaDock3.1 (30). 1000 models for each input structure were produced using the local refinement protocol of RosettaDock and scored using the internal scoring function (Rscore). Input side chain rotamers as well as extra chi1 rotamers and chi2 aromatic rotamers were included in the side chain search. All the poses produced by RosettaDock were rescored using the ZRank2.0 scoring function (Zscore) (26). Finally, a composite score combining the Rscore and Zscore was calculated to take into account the information coded in both scoring functions and to facilitate identification of models that are robust to both scores. Rscores and Zscores for all models were normalized on a scale from 0 (least favorable) to −1 (most favorable). Outlier models that were extremely unfavorably scored compared to the vast majority of other unfavorable models were excluded prior to normalization. The composite score for each model (Cscorei) was computed as the simple mean of its normalized Rscore and Zscore values:
where Xscore representing either Rscore or Zscore is normalized by:
Scoring with the Cscore allowed for identification of a single cluster as most favorable and pointed to a particular binding mode of the channel and Gβγ that was further analyzed and experimentally tested (Fig. 1D and E – in blue).
Mutagenesis
cDNAs of G1*, G4*, G1, G4, Gβ1, Gγ2, and mGluR2 were subcloned into an oocyte expression vector (pGEMHE). Desired mutations were introduced by the commercial Quickchange (Agilent Technologies) method. Briefly, complimentary primers containing and centered about the desired mutation were designed. PCR was carried out using the pfu polymerase and was allowed to cycle only 18 times to avoid errors. All mutants were confirmed by DNA sequencing (Genewiz).
Xenopus oocyte expression
All constructs were linearized using NheI restriction enzyme and in vitro transcribed using the “message machine” (Ambion) commercial kit. cRNA concentrations were quantified by optical density. Xenopus oocytes were surgically extracted, dissociated and defolliculated by collagenase treatment, and microinjected with 50 nl of a water solution containing the desired cRNAs. All constructs used in this study were injected to achieve 2 ng/oocyte. Oocytes were incubated for 2–4 days at 18 °C.
Electrophysiology
Whole-oocyte currents were measured by conventional two-electrode voltage clamp with a GeneClamp 500 amplifier (Axon Instruments). Agarose-cushion microelectrodes were used with resistances between 0.1 and 1.0 MΩ. Oocytes were held at 0 mv and currents were assessed by 800 ms ramps from −80 to +80 mv. Current at −80 mv was recorded. Barium-sensitive basal currents were defined as the difference between the steady-state currents while perfusing High potassium (ND96K or HK) solution or Barium solution. 10 μM Glutamate dissolved in HK solution was used to stimulate agonist-induced currents, defined as the difference in steady-state currents between perfusion of the Glutamate or the HK solution. HK solution contained: 91mM KCl, 1mM NaCl, 1mM MgCl2, 5mM KOH/HEPES, pH 7.4. Barium solution consisted of HK + 3mM BaCl2. 5–10 oocytes from the same batch were recorded for each group and the experiments were repeated in at least two batches.
Statistical Analysis
The quantity of interest in many of the experiments was the fold-change in current induced by co-expression of Gβγ. Thus, this fold-change represents the ratio of two unpaired means where each mean carries its individual degree of uncertainty. The mean current observed with Gβγ coexpression for a particular channel mutation is divided by the mean current observed for that channel expressed alone. Simply rescaling the individual observations of the numerator (channel+Gβγ) by the mean value of the denominator (channel alone) and then proceeding with a traditional comparison such as a t-test or ANOVA is not a valid solution because it ignores the uncertainty associated with the denominator's mean. A rigorous treatment to this problem is provided by Fieller's Theorem (42). Application of Fieller's theorem allows for rigorous construction of confidence intervals and calculation of standard error but it does not provide p-values for hypothesis testing. Non-overlap of confidence intervals does certainly confirm statistical significance, but it is a very conservative test and actual associated error rate would be much lower than the 0.05 implied by a single 95% confidence interval. Assuming an adequate sample, equal variance, and independence, the chance of finding non-overlapping 95% confidence intervals for the same mean purely due to chance (that is under the null hypothesis that the means are equal) can be calculated to be close to 0.0056 (43). Thus, we used Fieller's theorem to construct 95% confidence intervals for the fold-change values and we conservatively report p<0.01 when we find non-overlapping 95% confidence intervals for our data. For 90% confidence intervals, that value is calculated to be 0.02, which we conservatively report as p<0.05.
Other conventional statistical tests were employed in experiments reporting raw current values or within-oocyte % changes in current and these are clearly indicated in the appropriate figure legends.
Cross-linking of purified proteins
Gβγ and Gβ(L55C)γ were purified as previously described (37), followed by gel filtration with a Superdex-200 (S-200) column, equilibrated with 20 mM HEPES, 100 mM NaCl, 0.5 mM EDTA, 11 mM CHAPS, 1 mM DTT, and 10% Glycerol, pH 8.0. The cytoplasmic domain of GIRK1 (G1CP) was generated in a bacterial expression vector. All endogenous cysteine residues were mutated to serine by site-directed mutagenesis. A cysteine (L333C) was introduced at the position of Leu333 in the cysteine-less G1CP. This cysteine-less G1CP with L333C mutation [G1CP (cys-less, L333C)] was expressed and purified as previously described (19), followed by gel filtration with a S-200 column, equilibrated with 20 mM HEPES, 100 mM NaCl, 0.1 mM DTT, and 10% Glycerol, pH8.0. Gβ(L55C)γ (20 μM) was mixed with equal concentration of G1CP (cys-less, L333C) in 20 mM HEPES, 100 mM NaCl, 11 mM CHAPS, and 10% Glycerol, pH8.0. Crosslinking was induced by treatment with hydrogen peroxide up to 10 mM for 2.5 minutes. Gβ(L55C)γ alone or a mixture of G1CP (cys-less, L333C) with Gβγ was also tested with hydrogen peroxide treatment. Reactions were stopped by SDS gel loading buffer (without reducing agent) and analyzed immediately with SDS-PAGE. Protein bands were made visible by Coomassie Brilliant Blue staining.
Supplementary Material
Fig. S1. Permeation pathway, gates, and “closed” and “open” crystal forms of the intracellular G-loop gate of a GIRK1 chimera.
Fig. S2. Comparison of our best-scoring computational model with an NMR model of the footprint of Gβγ on GIRK1 residues.
Fig. S3. Gβ- and GIRK1-interacting residues in the best scoring and largest cluster models
Fig. S4. Critical elements of the “open” versus “closed” G-loop conformations of the channel structure
Fig. S5. Largest cluster model details
Fig. S6. Effect of channel mutations on mean currents relative to the G1*
PDB file S1. PDB coordinates of the best-scoring model
PDB file S2. PDB coordinates of the largest cluster model
Acknowledgments
We are grateful to members of the Logothetis lab and Drs. Clive Baumgarten, Jason Chen, Lou De Felice and Glen Kellogg for useful feedback throughout the length of this project and for useful comments on earlier drafts of the manuscript. We thank Zhe Zhang for producing the Gβ(L55C) mutant that was used for protein purification and Lia Baki for her input throughout the project and particularly with the crosslinking experiments. We thank Sophia Gruszecki and Heikki Vaananen for oocyte preparation and technical assistance, and Meng Cui and Xuanyu Meng for useful discussions on computational aspects of the work.
Funding: This work was partly supported by grants HL059949 and HL HL090882 from NIH to D. E. L. R.M. was supported by predoctoral NRSA fellowship F30HL097582 from NHLBI. M. Z. was supported by grant 13SDG16150007 from AHA.
Footnotes
Author contributions: R. M. designed and performed all of the computational and most of the electrophysiological experiments, analyzed the data and produced almost all figures, as part of his dissertation under the supervision of D. E. L. J. H. performed the experiments analyzed the data shown in Fig. 6. M. Z. produced and purified the cysteine-less G1 cytoplasmic protein construct that he used in the experiment of Fig. 4D. Ta. K. and To. K. were responsible for protein purification of wild-type Gβγ or the Gβ(L55C)γ protein from Sf9 cells that was used by M.Z. in Fig. 4D. R. M. and D. E. L. wrote the manuscript and all authors participated in revisions to its final form.
Competing interests: The authors have no competing financial interests to declare.
Data and materials availability: PDB coordinates for the best-scoring model and largest cluster model are provided in the Supplementary Materials.
REFERENCES AND NOTES
- 1.Trautwein W, Dudel J. Zum mechanismus der membranwirkung des acetylcholin an der herzmuskelfaser. Pflügers Archiv European Journal of Physiology. 1958;266:324–334. doi: 10.1007/BF00416781. [DOI] [PubMed] [Google Scholar]
- 2.Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 3.Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Insights into G Protein Structure, Function, and Regulation. Endocrine Reviews. 2003;24:765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- 4.Gaudet R, Bohm A, Sigler PB. Crystal structure at 2.4 angstroms resolution of the complex of transducin betagamma and its regulator, phosducin. Cell. 1996;87:577–588. doi: 10.1016/s0092-8674(00)81376-8. [DOI] [PubMed] [Google Scholar]
- 5.Tesmer JJG, Tesmer VM, Lodowski DT, Steinhagen H, Huber J. Structure of human G protein-coupled receptor kinase 2 in complex with the kinase inhibitor balanol. J Med Chem. 2010;53:1867–1870. doi: 10.1021/jm9017515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dascal N, Schreibmayer W, Lim NF, Wang W, Chavkin C, DiMagno L, Labarca C, Kieffler BL, Gaveriaux-Ruff C, Trollinger D. Atrial G protein-activated K+ channel: expression cloning and molecular properties. Proc Natl Acad Sci USA. 1993;90:10235–10239. doi: 10.1073/pnas.90.21.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krapivinsky G, Gordon EA, Wickman K, Vellimirovic B, Krapvinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K(+)-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 8.Kubo Y, Reuveny E, Slesinger PA, Jan YN, Jan LY. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature. 1993;364:802–806. doi: 10.1038/364802a0. [DOI] [PubMed] [Google Scholar]
- 9.Lesage F, Duprat F, Fink M, Guillemare E, Coppola T, Lazdunski M, Hugnot JP. Cloning provides evidence for a family of inward rectifier and G-protein coupled K+ channels in the brain. FEBS Lett. 1994;353:37–42. doi: 10.1016/0014-5793(94)01007-2. [DOI] [PubMed] [Google Scholar]
- 10.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 11.Lewohl JM, Wilson WR, Mayfield RD, Brozowski SJ, Morrisett RA, Harris RA. G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nature Neuroscience. 1999;2:1084–1090. doi: 10.1038/16012. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi T, Ikeda K, Kojima H, Niki H, Yano R, Yoshioka T, Kumanishi T. Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nature Neuroscience. 1999;2:1091– 1097. doi: 10.1038/16019. [DOI] [PubMed] [Google Scholar]
- 13.Sui JL, Chan KW, Logothetis DE. Na+ activation of the muscarinic K+ channel by a G-protein-independent mechanism. J Gen Physiol. 1996;108:381–391. doi: 10.1085/jgp.108.5.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sui JL, Petit-Jacques J, Logothetis DE. Activation of the atrial KACh channel by the betagamma subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc Natl Acad Sci USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, Logothetis DE. Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J Biol Chem. 1997;272:31553–31560. doi: 10.1074/jbc.272.50.31553. [DOI] [PubMed] [Google Scholar]
- 16.Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. The structure of the G protein heterotrimer Gi[alpha]1[beta]1[gamma]2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 17.Lambright DG, Sondek J, Bohm A, Skiba NP. The 2.0 Å crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 18.Sondek J, Bohm A, Lambright DG, Hamm HE, Sigler PB. Crystal structure of a G protein beta gamma dimer at 2.1A resolution. Nature. 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- 19.Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci. 2005;8:279–287. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- 21.Inanobe A, Matsuura T, Nakagawa A, Kurachi Y. Structural Diversity in the Cytoplasmic Region of G Protein-Gated Inward Rectifier K+ Channels. Channels. 2007;1:40–46. [PubMed] [Google Scholar]
- 22.Whorton MR, MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011;147:199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leal-Pinto E, Gómez-Llorente Y, Sundaram S, Tang Q-Y, Ivanova-Nikolova T, Mahajan R, Baki L, Zhang Z, Chavez J, Ubarretxena-Belandia I, Logothetis DE. Gating of a G protein-sensitive mammalian Kir3.1 prokaryotic Kir channel chimera in planar lipid bilayers. J Biol Chem. 2010;285:39790–39800. doi: 10.1074/jbc.M110.151373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng XY, Zhang HX, Logothetis DE, Cui M. The Molecular Mechanism by which PIP2 Opens the Intracellular G-Loop Gate of a Kir3.1 Channel. Biophysical Journal. 2012;102:2049–2059. doi: 10.1016/j.bpj.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vajda S, Kozakov D. Convergence and combination of methods in protein–protein docking. Current Opinion in Structural Biology. 2009;19:164–170. doi: 10.1016/j.sbi.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierce B, Weng Z. A combination of rescoring and refinement significantly improves protein docking performance. Proteins: Structure, Function, and Bioinformatics. 2008;72:993– 1004. doi: 10.1002/prot.21920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozakov D, Schueler-Furman O, Vajda S. Discrimination of near-native structures in protein-protein docking by testing the stability of local minima. Proteins. 2008;72:993–1004. doi: 10.1002/prot.21997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 29.Mintseris J, Pierce B, Wiehe K, Anderson R, Chen R, Weng Z. Integrating statistical pair potentials into protein complex prediction. Proteins: Structure, Function, and Bioinformatics. 2007;69:511–520. doi: 10.1002/prot.21502. [DOI] [PubMed] [Google Scholar]
- 30.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-Protein Docking with Simultaneous Optimization of Rigid-body Displacement and Side-chain Conformations. Journal of Molecular Biology. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 31.Kozakov D, Clodfelter KH, Vajda S, Camacho CJ. Optimal clustering for detecting near-native conformations in protein docking. Biophysical journal. 2005;89:867–875. doi: 10.1529/biophysj.104.058768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yokogawa M, Osawa M, Takeuchi K, Mase Y, Shimada I. NMR analyses of the Gbetagamma binding and conformational rearrangements of the cytoplasmic pore of G protein-activated inwardly rectifying potassium channel 1 (GIRK1) J Biol Chem. 2011;286:2215–2223. doi: 10.1074/jbc.M110.160754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finley M, Arrabit C, Fowler C, Suen KF, Slesinger PA. βL–βM loop in the C-terminal domain of G protein-activated inwardly rectifying K+ channels is important for Gβγ subunit activation. The Journal of Physiology. 2004;555:643–657. doi: 10.1113/jphysiol.2003.056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He C, Zhang H, Mirshahi T, Logothetis DE. Identification of a Potassium Channel Site That Interacts with G Protein βγ Subunits to Mediate Agonist-induced Signaling. Journal of Biological Chemistry. 1999;274:12517–12524. doi: 10.1074/jbc.274.18.12517. [DOI] [PubMed] [Google Scholar]
- 35.Nishida M, MacKinnon R. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell. 2002;111:957–65. doi: 10.1016/s0092-8674(02)01227-8. [DOI] [PubMed] [Google Scholar]
- 36.Ford CE, Skiba NP, Bae H, Daaka Y, Reuveny E, Shekter LR, Rosal R, Weng G, Yang C, Iyengar R, Miller RJ, Jan LY, Lefkowitz RJ, Hamm HE. Molecular Basis for Interactions of G Protein βγ Subunits with Effectors. Science. 1998;280:1271–1274. doi: 10.1126/science.280.5367.1271. [DOI] [PubMed] [Google Scholar]
- 37.Kozasa T. Purification of G protein subunits from Sf9 insect cells using hexahistidine-tagged alpha and beta gamma subunits. Methods Mol Biol. 2004;237:21–38. doi: 10.1385/1-59259-430-1:21. Review. [DOI] [PubMed] [Google Scholar]
- 38.Aryal P, Dvir H, Choe S, Slesinger PA. A discrete alcohol pocket involved in GIRK channel activation. Nat Neurosci. 2009;12:988–995. doi: 10.1038/nn.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ivanina T, Rishal I, Varon D, Mullner C, Frohnwieser-Steinecke B, Schreibmayer W, Dessauer CW, Dascal N. Mapping the Gbetagamma-binding sites in GIRK1 and GIRKsubunits of the G protein-activated K+ channel. J Biol Chem. 2003;278:29174–29183. doi: 10.1074/jbc.M304518200. [DOI] [PubMed] [Google Scholar]
- 40.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJG. Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science. 2005;310:1686–1690. doi: 10.1126/science.1118890. [DOI] [PubMed] [Google Scholar]
- 41.Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 42.Fieller EC. The Biological Standardization of Insulin. Supplement to the Journal of the Royal Statistical Society. 1940;7:1. [Google Scholar]
- 43.Knol MJ, Pestman WR, Grobbee DE. The (mis)use of overlap of confidence intervals to assess effect modification. Eur J Epidemiol. 2011;26:253–254. doi: 10.1007/s10654-011-9563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolfenden R, Andersson L, Cullis PM, Southgate CCB. Affinities of amino acid side chains for solvent water. Biochemistry. 1981;20:849–855. doi: 10.1021/bi00507a030. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Q, Kawano T, Nakata H, Nakajima Y, Nakajima S, Kozasa T. Interaction of G protein beta subunit with inward rectifier K(+) channel Kir3. Mol Pharmacol. 2003;64:1085– 1091. doi: 10.1124/mol.64.5.1085. [DOI] [PubMed] [Google Scholar]
- 46.Mirshahi T, Robillard L, Zhang H, Hébert TE, Logothetis DE. Gbeta residues that do not interact with Galpha underlie agonist-independent activity of K+ channels. J Biol Chem. 2002;277:7348–7355. doi: 10.1074/jbc.M109999200. [DOI] [PubMed] [Google Scholar]
- 47.Camacho CJ, Weng Z, Vajda S, DeLisi C. Free energy landscapes of encounter complexes in protein-protein association. Biophys J. 1999;76:1166–1178. doi: 10.1016/S0006-3495(99)77281-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Camacho CJ, Vajda S. Protein docking along smooth association pathways. PNAS. 2001;98:10636–10641. doi: 10.1073/pnas.181147798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whorton MR, MacKinnon R. X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature. 2013;498:190–197. doi: 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Permeation pathway, gates, and “closed” and “open” crystal forms of the intracellular G-loop gate of a GIRK1 chimera.
Fig. S2. Comparison of our best-scoring computational model with an NMR model of the footprint of Gβγ on GIRK1 residues.
Fig. S3. Gβ- and GIRK1-interacting residues in the best scoring and largest cluster models
Fig. S4. Critical elements of the “open” versus “closed” G-loop conformations of the channel structure
Fig. S5. Largest cluster model details
Fig. S6. Effect of channel mutations on mean currents relative to the G1*
PDB file S1. PDB coordinates of the best-scoring model
PDB file S2. PDB coordinates of the largest cluster model