

Oxidative stress enhances dendritic cells' capability of inducing CD4+ T cell proliferation and production of proinflammatory cytokines and PI3Kγ-dependent migration, but reduces generation of T-regulatory cells.
Keywords: H2O2, T cell activation, migration
Abstract
Whereas DC have increasingly been recognized for their role in activating the inflammatory cascades during IRIs, the mechanisms by which oxidative stress enhances DC activation remain to be explored. We examined the role of oxidative stress on two important features of DC: T cell activation and trafficking. Bone marrow-derived OS-DC were compared with untreated DC. DC exposed to oxidative stress augmented allogeneic T cell proliferation and showed increased migration in a chemotaxis chamber. These results were confirmed by using hypoxanthine and xanthine oxidase as another inducer of oxidative stress. We used OT-II and OT-I mice to assess the effect of oxidative stress on DC activation of OVA-specific CD4+ and CD8+ T cells, respectively. Oxidative stress increased DC capacity to promote OVA-specific CD4+ T cell activity, demonstrated by an increase in their proliferation and production of IFN-γ, IL-6, and IL-2 proinflammatory cytokines. Whereas oxidative stress increased the DC ability to stimulate IFN-γ production by OVA-specific CD8+ T cells, cellular proliferation and cytotoxicity were not affected. Compared with untreated DC, oxidative stress significantly reduced the capacity of DC to generate Tregs, which were restored by using anti-IL-6. With regard to DC trafficking, whereas oxidative stress increased DC expression of p-Akt and p-NF-κB, targeting PI3Kγ and NF-κB pathways abrogated the observed increase in DC migration. Our data propose novel insights on the activation of DC by oxidative stress and provide rationales for targeted therapies, which can potentially attenuate IRI.
Introduction
IRI has emerged as the leading cause of organ damage in medicine. Activation of cellular and humoral inflammatory cascades plays a critical role in the pathogenesis of IRI [1]. Therefore, there has been major interest in better understanding the mechanisms that regulate such inflammatory cascades [2]. Whereas all leukocyte subsets have been implicated in the pathogenesis of IRI, DC, in particular, have been shown to play an important role in orchestrating these inflammatory responses [3]. DC are professional APCs, which reside in peripheral tissues [4]. They can interact readily with danger molecules, which are generated during IRI, via their TLR system [5, 6]. DC can also play a pivotal role in regulating the function of effector T cells via antigen presentation and secretion of cytokines. Prior studies have indicated that DC accumulate in ischemic sites [7, 8] and produce proinflammatory cytokines and chemokines capable of instigating inflammatory responses [9, 10]. In transplantation, DC also play a critical role in linking innate immune responses during IRI, occurring early after transplantation, with subsequent activation of alloimmune reactions [6, 11]. Whereas several studies have highlighted the presence and potential role of DC in the pathogenesis of IRI [12–15], regulatory mechanisms, by which oxidative stress increases the activity of DC, remain unexplored. These studies would not only help to elucidate the process of DC activation by IRI but would also allow the identification of novel molecular targets that regulate this activation.
In this report, we studied the effect of oxidative stress on two important aspects of DC function: antigen presentation and trafficking. Hence, we examined the effect of oxidative stress in presenting OVA peptide to CD4+ and CD8+ T cells and in generating Tregs. We also evaluated the role of PI3Kγ in regulating DC trafficking following oxidative stress. Among various classes of PI3Ks, the PI3Kγ subunit is expressed primarily in leukocytes [16, 17], and its potential role in mediating cell trafficking has been suggested recently [18, 19].
MATERIALS AND METHODS
Mice
C57BL/6 (WT), BALB/c, C57BL/6-Tg (TcraTcrb)1100Mjb/J (OT-I), and C57BL/6-Tg(TcraTcrb)425Cbn/J (OT-II) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). OT-I and OT-II are OVA-specific TCR transgenic mice, where OT-I CD8+ T cells recognize OVA257–264 MHC class-I restricted peptide, and OT-II CD4+ T cells recognize OVA323–339 MHC class-II restricted peptide. PI3Kγ−/− mice (on C57BL/6 background) were a generous gift from Dr. Bao Lu [20] and were maintained in our animal facility. All procedures were in compliance with the standards set forth in the Guidelines for the Care and Use of Laboratory Animals (Harvard University, Boston, MA, USA).
DC culture
Bone marrow-derived DC were generated, as described previously, with minor modifications [5, 12]. Mononuclear cells from the bone marrow contents of femurs and tibiae of C57BL/6 (or PI3Kγ−/−) mice were seeded in bacteriological Petri dishes with 10 ml RPMI complete medium containing 10% FCS (Gemini Bioproducts, West Sacramento, CA, USA), 1% L-glutamine and penicillin-streptomycin (both from Cambrex, East Rutherford, NJ, USA), and 50 ng/ml IL-4 and 20 ng/ml murine rGM-CSF (both from PeproTech, Rocky Hill, NJ, USA). On Day 3, 10 ml fresh medium containing 20 ng/ml GM-CSF was added to each plate. On Day 6, 10 ml medium was collected and centrifuged. Cells were resuspended in 10 ml fresh medium, supplemented with 10 ng/ml GM-CSF, and added back to plates. DC were harvested at Day 7, and some cultures were exposed to oxidative stress (OS-DC or OS2-DC), as described below.
Induction of oxidative stress
OS-DC were incubated in 24-well plates [10, 12, 21]. Based on the maximal dose and time effects on maturation of DC, oxidative stress was induced using 500 μM H2O2, unless specified otherwise. In some experiments, hypoxanthine and xanthine oxidase were used as an additional source of oxidative stress, where OS2-DC were incubated with 2.5 mM hypoxanthine (Sigma-Aldrich, St. Louis, MO, USA) and 0.005 U/ml xanthine oxidase (Sigma-Aldrich) for 24 h, as recommended by Lassen et al. [22]. In all experiments, control DC were identically incubated in the absence of H2O2 or hypoxanthine and xanthine oxidase. Later, DC were washed to remove H2O2 (or hypoxanthine and xanthine oxidase) traces and were subjected to flow cytometry, lysed and used for protein detection by Western blot, or plated for immunological assays, as described below.
Reagents and antibodies
Anti-mouse CD11c, CD4, CD8, Annexin V, and 7-AAD were purchased from BD Biosciences (San Jose, CA, USA), whereas anti-mouse CCR7, granzyme B, perforin, IL-6, and FOXP3 were purchased from eBioscience (San Diego, CA, USA). Monoclonal rabbit (Ser473) p-Akt were obtained from Cell Signaling Technology (Danvers, MA, USA). For intracytoplasmic staining, cells were restimulated with PMA (5 ng/ml; Sigma-Aldrich) and ionomycin (500 ng/ml; Sigma-Aldrich), 5 h before staining; monensin (600 ng/ml; BD Bioscience) was added at the same time to block protein secretion. Antibody dilutions ranged from 1/100 to 1/500. Cells were run on a FACSCalibur (BD Biosciences), and data were analyzed using FlowJo software (version 6.3.2; Tree Star, Ashland, OR, USA).
A 0.1-μM OVA-I and a 0.05-μM OVA-II (both from Genscript, Piscataway, NJ, USA) were used to explore the effect of OS-DC on each of OVA-specific CD8+ and CD4+ T cell, respectively. To inhibit PI3Kδ, a 2-μM PI3Kδ inhibitor (IC87114; EMD Millipore, Billerica, MA, USA), which selectively antagonizes PI3Kδ in a concentration range of 0.3–10 μM [23], was added to DC cultures, 2 h before 24-h induction of oxidative stress. To inhibit NF-κB activation, a 0.5-nM 6-amino-4-(4-phenoxyphenylethylamino) quinazoline-neutralizing antibody (EMD Biosciences, Gibbstown, NJ, USA) was added to DC cultures, 2 h before 12-h induction of oxidative stress. The effectiveness for this NF-κBi was confirmed by p-65 expression on OS-DC, with and without NF-κBi, using Western blot.
Ultrastructural studies
Freshly isolated DC were fixed in 2.5% glutaraldehyde in PBS and stored at 4°C. Cells were then washed in PBS, postfixed in 1% osmium tetroxide in PBS for 1 h, dehydrated in acetone, and embedded in Epon. Ultrathin sections were stained with lead citrate and uranyl acetate [24] and analyzed using transmission electron microscopy (JEOL, Tokyo, Japan).
MLR and Treg assay
Polyclonal MLR.
A total of 2.5 × 105 whole splenocytes from BALB/c mice was cocultured with 5 ×104 fully MHC-mismatched control DC, OS-DC, or OS2-DC from C57BL/6 mice for 72 h, followed by pulsing with 1 μCi tritiated thymidine (PerkinElmer, Waltham, MA, USA). Tritium uptake was assessed 16 h later, using a MicroBeta FilterMate-96 Harvester and a 1450 MicroBeta TriLux (both from PerkinElmer).
Peptide-specific MLR.
OT-II CD4+ and OT-I CD8+ splenocytes were isolated by magnetic bead separation (Miltenyi Biotec, Auburn, CA, USA) and labeled with CFSE. OS-DC and control DC from C57BL/6 mice were harvested, following 3 h incubation with OVA-II or OVA-I peptide. A total of 1.0 × 105 CFSE-labeled OT-II CD4+ or OT-I CD8+ T cells was cocultured in 96-well plates with 1.0 × 104 DC (OS-DC and control DC), pretreated with OVA-II or OVA-I, respectively. After 72 h, CD4+ and CD8+ T cell proliferation or surface and intracellular staining was assessed by flow cytometry.
Treg assay.
CD4+ splenocytes from BALB/c mice were isolated, and a total of 1.0 × 105 CD4+ splenocytes was cocultured in 96-well plates with 1.0 × 104 OS-DC or control DC from C57BL/6 mice in the presence of soluble TGF-β (4 ng/ml). To neutralize IL-6 production, a 10-μg/ml anti-IL-6 (eBioscience) was added to some cultures upon mixing OS-DC and CD4+ cells. A 10-μg/ml IgG1/κ isotype control (eBioscience) was also added to some wells as negative controls for anti-IL-6. After 4 days in culture, cells were assessed by flow cytometry.
ELISPOT assay
To assess production of murine IFN-γ, IL-4, and IL-10, ELISPOT assay was performed, according to the manufacturer's instructions (BD Biosciences). Wells of the ImmunoSpot plates (EMD Millipore) were precoated with cytokine primary antibodies. A total of 1.0 × 105 OT-II CD4+ or OT-I CD8+ splenocytes, isolated by magnetic bead separation, was cocultured with 1.0 × 104 DC (OS-DC or control DC) from C57BL/6 mice pretreated with OVA-II or OVA-I, respectively. Following incubation (24 h for IFNγ and 72 h for IL-4- and Il-10-coated plates), biotinylated antibodies, Streptavidin-HRP, and aminoethyl carbazole (Sigma-Aldrich) were added, and spots were counted using an ImmunoSpot analyzer (Cellular Technology, Cleveland, OH, USA).
Luminex assay
An 11-plex MAP mouse cytokine assay was performed, according to the manufacturer's instructions (EMD Millipore). Supernatants harvested from in vitro experiments were assessed for production of IL-1β, IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, IL-17, IFN-γ, and TNF-α. Supernatants were incubated with cytokine-conjugated beads, and biotinylated beads and streptavidin were added later for detection and quantification.
CTL killing assay
Cytotoxic killing capability was assessed using the LIVE/DEAD Viability/Cytotoxicity Kit (Molecular Probes, Invitrogen Detection Technologies, Life Technologies, Grand Island, NY, USA), according to the manufacturer's recommendations. Briefly, a total of 1.0 × 105 OT-I CD8+ T cells was cocultured in 96-well plates, with 1.0 × 104 DC (OS-DC and control DC) prepulsed with OVA-I. After 4 days, different concentrations of OVA-I-primed cells from the aforementioned MLRs were added to 5 × 104 C57BL/6 DC, pretreated with OVA-I peptide in V-shaped, 96-well plate “experimental wells”. DC, in complete medium or supplemented with 1% H2O2, were used as negative and positive controls, respectively. After 4 h of incubation, the cells were harvested and stained with CD11c and with intracellular esterase (calcein acetomethoxy) and plasma membrane integrity (ethidium homodimer). Samples were then run on flow cytometry. Dying DC were defined as CD11c+ cells that stained positively for ethidium homodimer and/or negatively for calcein acetomethoxy. Dying DC, following incubation with medium alone, were subtracted from DC dying following incubation with CD8+ T cells, and the percent of killed DC was determined. A similar experience was also repeated using fully allogeneic cells (CD8+ splenocytes isolated from BALB/c mice and DC from C57BL/6 mice).
Transendothelial migration in a transwell flow chamber
Transendothelial migration was performed, as described previously [25]. Briefly, HUVECs were plated onto the underside of 0.4 μm fibronectin-coated transwell inserts (Corning, Corning, NY, USA) and were grown to complete confluence. HUVEC-transwell inserts were then placed into the flow chamber and mounted on the stage of an overhead phase-contrast microscope (Eclipse E-600; Nikon, Japan), connected to a digital device camera (ELMO Manufacturing, Nagoya, Japan). The insert was rinsed and filled with transendothelial migration media [25]. A total of 1.5 × 106 OS-DC and control DC was perfused at defined shear stress, generated by a syringe pump (Harvard Apparatus, Holliston, MA, USA). Transmigrated cells were defined as cells that adhered to and transmigrated underneath the endothelial monolayer barrier, seen as cells moving out of the focal plane, as described previously [25]. Transmigration events were recorded in real-time, analyzed off-line, and calculated as a percentage of adherent DC.
CCL21 transwell chemotaxis assay
The ability of DC to undergo CCL21-induced migration was assessed using the in vitro transwell chemotaxis assay, as described previously, with minor modifications [26]. A total of 5.0 × 105 DC, suspended in 100 μl complete medium, was placed in the upper chamber of a 5-μ transwell plate (Corning), whereas 600 μl complete medium alone or containing 1 μg/mL murine rCCL21 (PeproTech) was added to the bottom chamber. After 120 min of incubation, the cells in the bottom chamber were counted. DC, migrating in response to the buffer alone, were subtracted from DC migrating in response to CCL21. The percentages of migrating DC were determined.
Western blot analysis
DC were lysed, and lysates were prepared as described previously [5]. Membranes were blocked in 5% nonfat milk in TBST (0.05% Tween 20) for 1 h at room temperature and then incubated overnight at 4°C with rabbit antibodies [anti-p65 NF-κB, anti-Akt (total), anti-p-Akt (Ser473); Cell Signaling Technology]. The blots were then washed and incubated with HRP-conjugated anti-rabbit IgG antibodies (Amersham Biosciences, Piscataway, NJ, USA) for 2 h. Blots were developed using the ECL method. Anti-ERK (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti-β-tubulin (Abcam, Cambridge, MA, USA) antibodies were used for loading controls on stripped membranes. All antibodies were used in 1/1000 concentration.
Statistics
Data were expressed as the mean ± sd. Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Student's t-test was used for comparison of means between two groups. To compare multiple groups, ANOVA test was used, followed by Newman-Keuls multiple comparison test. Differences were reported to be significant when P ≤ 0.05.
RESULTS
Oxidative stress induces maturation of DC
Mononuclear cells isolated from bone marrow of C57BL/6 mice were cultured with GM-CSF and IL-4. At Day 7, >80% of cells were CD11c+ [27, 28]; the vast majority were myeloid DC, whereas <5% of the total DC expressed CD8 (lymphoid DC marker) or B220 (plasmacytoid DC marker). To address the dose effect of oxidative stress on DC phenotype, we treated these DC with 5, 50, or 500 μM H2O2 for 24 h and compared them with untreated DC. There was no change in DC subtypes following exposure to different concentrations of H2O2 (data not shown). As shown in Fig. 1A, the expression of CD86, CD80, and CD40 enhanced following exposure to H2O2 (ANOVA, each P<0.001). The highest expression of each of these markers was observed following exposure to 500 μM H2O2 (Newman-Keuls test, P<0.05; 500 μM OS-DC vs. each of the other groups). We also assessed the effect of oxidative stress on DC phenotype over time. DC were treated with 500 μM H2O2 for 4, 6, 12, and 24 h and compared with control DC. The highest expression of CD86, CD80, and CD40 was observed following 24 h of treatment with 500 μM H2O2 (Fig. 1B). We then carried out ultrastructural studies on control and OS-DC, following 24 h exposure of 500 μM H2O2, to assess if such phenotypic changes would also be accompanied by morphologic changes. Compared with control DC, OS-DC showed an increase in cell size, open- and active-appearing chromatin, and more prominent cellular projections (Fig. 1D); these morphologic changes characterize the maturation process [29, 30]. To ensure that our treatment did not cause excessive DC death, we have used flow cytometry to assess DC viability. The percentage of viable DC, defined as CD11c+ cells, which stained negatively for both Annexin V and 7-AAD, was similar in OS-DC and controls, up to 24 h following treatment with 500 μM H2O2 (Supplemental Fig. 1).
Figure 1. Oxidative stress enhances DC maturation.
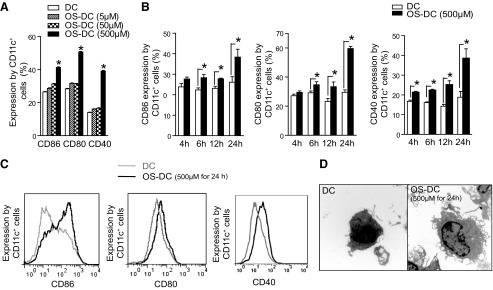
(A) DC were incubated for 24 h in medium alone or in the presence of different concentrations of H2O2, and then stained with CD11c, CD86, CD80, and CD40 and assessed by flow cytometry. There was a dose-dependent increase in the expression of all of these surface-costimulatory molecules on CD11c+ cells following treatment with H2O2, with the highest expression at 500 μM treatment of H2O2 (*P<0.05, n≥3 in each group). (B) There was also a time-dependent increase in the expression of each of CD86, CD80, and CD40 on 500 μM OS-DC compared with control DC. The highest expression was observed at 24 h post-H2O2 treatment (*P≤0.05, n≥3 in each group). (C) Representative histograms showing CD86, CD80, and CD40 expression on CD11c+ cells from control DC (gray curves) and 24-h, 500-μM OS-DC (black curves). (D) Ultrastructural studies were performed on cell suspension from control DC and 500 μM OS-DC for 24 h. By comparison, the latter were larger in size, had bigger nuclei with more open chromatin, and had more prominent protrusions (electron microscopy; original magnification, ×8000).
Oxidative stress increases DC alloactivation and trafficking
The effects of oxidative stress on DC activation of allogeneic splenocytes and on DC trafficking were studied. In a fully mismatched MLR, C57BL/6 OS-DC were found to increase the proliferation of BALB/c splenocytes more effectively than control DC, as measured by tritium uptake (Fig. 2A). We then assessed the trafficking of DC in a chemotaxis chamber in response to the CCL21 chemokine. OS-DC showed an increase in migration compared with control DC (Fig. 2B). In addition to H2O2, the effect of oxidative stress on DC alloactivation and trafficking was examined using hypoxanthine and xanthine oxidase as a second source of oxidative stress [22]. Again, OS2-DC had increased allostimulation capability and enhancement of transwell migration compared with control DC (Fig. 2C and D).
Figure 2. Oxidative stress increases DC alloactivation and in vitro transwell trafficking.

(A and B) DC were incubated for 24 h in medium alone (control DC) or in the presence of 500 μM H2O2 (OS-DC). (A) Alloactivation was assessed in a fully MHC-mismatched MLR, where 5 × 104 control DC or OS-DC from C57BL/6 mice were cocultured with 2.5 × 105 whole splenocytes from BALB/c for 72 h. An increase in alloreactive proliferation (shown by tritium uptake) was observed when splenocytes were cultured with OS-DC compared with control DC (*P=0.045, n=4 in each group). (B) In vitro trafficking was assessed by adding 5.0 × 105 DC to the upper chamber of a 5-μ transwell CCL21 chemotaxis assay. After 120 min of incubation, OS-DC showed increased migration into the lower chamber compared with control DC (*P=0.006, n=7 in each group). (C and D) DC were incubated for 24 h in medium alone (control DC) or in the presence of 2.5 mM hypoxanthine and 0.005 U/ml xanthine oxidase (OS2-DC) as a second source of oxidative stress to confirm our previous observations. (C) In a fully MHC-mismatched MLR, an increase in tritium uptake was observed when splenocytes were cultured with OS2-DC compared with control DC (*P=0.008, n≥3 in each group). (D) In a CCL21 chemotaxis assay, OS2-DC showed increased trafficking compared with control DC (*P=0.02, n≥8 in each group).
Oxidative stress increases the DC ability to activate CD4+ cells
To dissect the effect of oxidative stress on DC activation of OVA-specific CD4+ T cells, we used transgenic OT-II mice (C57BL/6 background). OS-DC and control DC from C57BL/6 mice were incubated with the OVA-II peptide for 3 h and cocultured with OVA-specific CD4+ T cells isolated from the spleens of OT-II mice. Compared with control DC, OS-DC were found to increase OVA-specific CD4+ T cell proliferation significantly (Fig. 3A). OS-DC were also found to enhance IFN-γ secretion (Fig. 3B), increase proinflammatory IL-6 (384±43 vs. 592±37 pg/ml; P<0.001) and IL-2, but decrease IL-9 secretion in the MLR (Fig. 3C). Intracytoplasmic staining showed an increase in the MFI of IL-6 in OS-DC compared with control DC, as well as in the CD4+ T cells cultured with OS-DC compared with CD4+ T cells cultured with control DC (Fig. 3D).
Figure 3. OS-DC increases peptide-specific activation of CD4+ T cells.
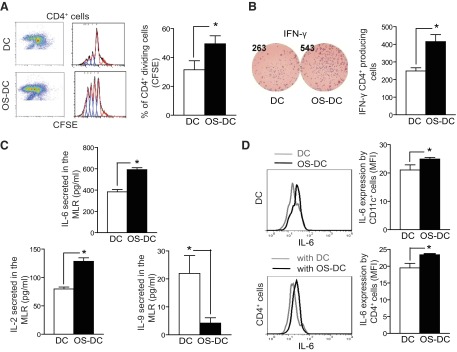
DC were incubated for 24 h in medium alone (control DC) or in the presence of 500 μM H2O2 (OS-DC). A total of 1.0 × 104 OS-DC or control DC from C57BL/6 mice was pretreated with OVA-II peptide and cocultured in a MLR with 1.0 × 105 OVA-specific CD4+ cells (isolated from OT-II spleens using microbeads). (A) After 72 h, flow cytometry evaluation revealed an increase in proliferation of CFSE-labeled CD4+ T cells, cocultured with OS-DC, compared with those cocultured with control DC (*P=0.005; n=4 each group). (B) With the use of the ELISPOT assay, IFN-γ was assessed after 24 h of coculturing; an increase in IFN-γ secretion was noted when CD4+ T cells were cocultured with OS-DC compared with control DC (*P=0.009; n=4 each group). (C) After 72 h of coculturing, the supernatant was collected from the MLR, and cytokine production was measured using Luminex. An increase in IL-6 (*P<0.001) and IL-2 (*P=0.005) but a decrease in IL-9 levels (*P=0.04) was observed when CD4+ T cells were cocultured with OS-DC compared with control DC (n=4 in each group). (D) To determine the source of increased IL-6 secretion, cells were collected from the MLR after 72 h of coculturing and stimulated with PMA and ionomycin in the presence of monensin. Cells were then labeled with surface staining for CD11c and CD4 and with intracellular staining for IL-6. An increase in IL-6 secretion, as measured by MFI, was observed in OS-DC compared with control DC (*P=0.05, n=3). Similarly, an increase in IL-6 secretion, as measured by MFI, was observed in CD4+ cells that were cocultured with OS-DC compared with those cocultured with control DC (*P=0.04, n=3).
Oxidative stress enhances IL-6 secretion by DC and decreases Treg generation
To examine the effect of oxidative stress on cytokine production from DC, bone marrow-derived DC were collected at Day 7 of culture and exposed to oxidative stress. As shown in Fig. 4A, among various cytokines examined in our 11-plex kit, IL-6 was increased significantly in the supernatant of OS-DC compared with controls. Notably, the levels of IFN-γ were undetectable in the supernatant of both groups (data not shown). We then assessed the ability of DC to generate Tregs following oxidative stress in a fully allogeneic MLR with TGF-β, with or without anti-IL-6 treatment. As shown in Fig. 4B, there was a significant difference in the percentage of Tregs among the different groups after 4 days (ANOVA, P=0.003); the percentage of Tregs generated by OS-DC (11±6%) was lower than those generated by control DC (20±2%) and control DC treated with anti-IL-6 (19±2%). Moreover, treatment with anti-IL-6 significantly restored the percentage of Tregs generated by OS-DC (OS-DC with anti-IL-6: 21±2%; Newman-Keuls test, P<0.05; OS-DC vs. each DC, DC with anti-IL-6, and OS-DC with anti-IL-6). The IgG isotype did not have effects on the percentage of Treg generation by control DC or by OS-DC (Fig. 4B). Ki67 stain was used to assess the effect of anti-IL-6 on T cell proliferation in the above Treg assay; no difference in the percentage of CD4+FOXP3−Ki67+ (non-Treg proliferation) was found between CD4 cocultured with OS-DC and CD4 cocultured with OS-DC in the presence of anti-IL-6 (data not shown).
Figure 4. OS-DC suppresses Treg generation via IL-6-dependent mechanism.

(A) DC were incubated for 24 h in medium alone (control DC) or in the presence of 500 μM H2O2 (OS-DC). Cytokine profile was assessed in the supernatant of DC culture using Luminex assay; IL-6 levels were increased in OS-DC compared with control DC (*P<0.001; n=4 each group). The levels of other cytokines, including IL-4, IL-1β, TNF-α, and IL-9, did not differ significantly. (B) To assess the effect on Treg generation, 1.0 × 104 OS-DC or control DC (from C57BL/6 mice) were cocultured for 4 days with 1.0 × 105 fully mismatched CD4+ T cells (isolated from BALB/c spleens) in the presence of TGF-β, with or without treatment with anti-IL-6. The percentage of CD4+FOXP3+ T cells generated when CD4+ cells were cocultured with OS-DC (11±6%) was lower than CD4+FOXP3+ T cells generated when CD4+ cells were cocultured with control DC (20±2%). Anti IL-6 therapy restored formation of Tregs in the OS-DC group (21±2%; *P<0.05; n≥3 in each group). (C) Representative flow plots showing FOXP3 expression (gated on CD4+), following coculturing with control DC, control DC with anti-IL-6, OS-DC, and OS-DC with anti-IL-6.
Oxidative stress did not increase CD8+ T cell proliferation and cytotoxicity
To assess the effect of oxidative stress on DC activation of OVA-specific CD8+ T cells, OS-DC and control DC from C57BL/6 mice were incubated with OVA-I peptide for 3 h and cocultured with OVA-specific CD8+ T cells isolated from the spleens of OT-I mice (C57BL/6 background). Compared with control DC, OS-DC were found to enhance IFN-γ secretion significantly by CD8+ T cells in an ELISPOT assay (Fig. 5A). However, OS-DC did not have a significant effect on OVA-specific CD8+ T cell proliferation (data not shown) or on their cytoplasmic expression of granzyme B and perforin (Fig. 5B). We then assessed the effect of OS-DC in promoting the cytotoxicity of CD8+ T cells in a killing assay. For this, we extracted OVA-I-reacting cells from the above MLR (CD8+ T cells from OT-I and DC from C57BL/6) and cocultured them again with DC preincubated with OVA-I peptide (target cells) at different E:T ratios. As shown in Fig. 5C, no difference in target cell killing was observed comparing both groups. The same experiment was also repeated in a fully allogeneic system using CD8+ T cells isolated from BALB/c spleens and DC from C57BL/6 mice. Again, no difference in the percentage of target cell killing was noted (data not shown).
Figure 5. OS-DC do not significantly affect cytotoxic activity of peptide-specific CD8+ T cells.
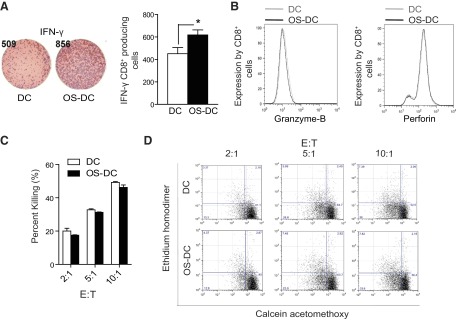
DC from C57BL/6 mice were incubated for 24 h in medium alone (control DC) or in the presence of 500 μM H2O2 (OS-DC), pretreated with OVA-I peptide, and cocultured in a MLR with OVA-specific CD8+ T cells (isolated from OT-I spleens using microbeads). (A) With the use of ELISPOT, IFN-γ was assessed after 24 h. An increase in IFN-γ secretion was observed when CD8+ T cells were cocultured with OS-DC compared with control DC (*P=0.04, n=6 each group). (B) Representative histograms showing no difference in the CD8+ T cell intracellular expression of granzyme B and perforin between those cocultured with OS-DC (black curves) or with control DC (gray curves). (C) To test the cytotoxicity of OVA-I CD8+ T cells after priming with DC control or OS-DC pulsed with OVA peptide, OVA-I-primed cells, from the aforementioned MLR (effector cells), were reincubated after 4 days with DC pulsed with OVA-I (target cells) in different E:T ratios (2:1, 5:1, and 10:1) for 4 h. Target DC were then evaluated for cell death using calcein acetomethoxy and ethidium homodimer. No difference in the percentage of killing of target DC (CD11c+) was noted, whether incubated with CD8+ cells primed with OS-DC or control DC (n=4 in each group). (D) Representative flow plots demonstrating target cell death (gated on CD11c+) from the previous experiment are shown.
Oxidative stress enhances DC transendothelial migration and surface expression of CCR7
The ability of DC to activate T cells depends on their efficient migration to the lymphoid tissue. We first assessed the effect of oxidative stress on DC transendothelial migration in the presence of shear stress [25]. Both OS-DC and control DC were perfused at a defined flow rate, mimicking physiological shear. DC transmigration across HUVEC monolayers were recorded in real-time and viewed offline. As shown in Fig. 6A, there was a higher percentage of transmigrating OS-DC compared with controls. We then assessed the effect of oxidative stress on CCR7 expression by DC. With the use of flow cytometry, we found a time-dependent increase in CCR7 expression on OS-DC compared with controls; the highest expression of CCR7 was observed following 24 h of exposure to oxidative stress (Fig. 6B).
Figure 6. Oxidative stress increases DC transendothelial migration and CCR7 expression.
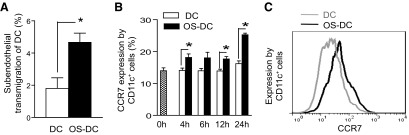
(A) Control DC and OS-DC (incubated for 24 h with 500 μM H2O2) were perfused under physiologic shear stress in a HUVEC-transwell flow chamber. A higher percentage of transendothlial migration was observed in OS-DC compared with controls (*P=0.026; n≥3 in each group). (B) DC were incubated in medium alone (DC) or in the presence of 500 μM H2O2 (OS-DC) for different time-points, and CCR7 expression on CD11c+ cells was assessed by flow cytomety. CCR7 expression was increased in OS-DC compared with control DC, with the highest expression observed at 24 h post-H2O2 treatment (*P=0.02 at 4 h, *P=0.07 at 6 h, *P=0.005 at 12 h, and *P<0.001 at 24 h, n=5 each group). (C) Representative histograms showing CCR7 expression on CD11c+ cells from control DC (gray curve) and 500 μM OS-DC for 24 h (black curve).
Targeting the PI3Kγ pathway decreases the migration of DC following oxidative stress
The PI3K/Akt pathway, downstream of CCR7 [31], has received increasing attention for its role in inflammation and cellular migration. We first investigated the effect of oxidative stress on p-Akt expression. With the use of flow cytometry, we found an up-regulation of p-Akt on OS-DC compared with control DC (Fig. 7A). To dissect the effect of PI3Kγ on DC migration, we used DC generated from PI3Kγ−/− mice (C57BL/6 background). We first assessed p-Akt levels from protein extracted from C57BL/6 DC (WT DC) and PI3Kγ−/− DC, following oxidative stress, using Western blot. A significant decrease in the ratio of p-Akt:total Akt was observed in OS- PI3Kγ−/− DC compared with OS-WT DC (Fig. 7B). We then assessed the role of PI3Kγ in a transwell migration assay in response to CCL21 stimulation in DC obtained from WT and PI3Kγ−/− mice. As shown in Fig. 7C, there was a significant difference in trafficking among the four different groups (ANOVA, P<0.001). Both groups exposed to OS showed increased trafficking compared with control DC (OS-WT DC vs. WT DC and OS-PI3Kγ−/− DC vs. PI3Kγ−/− DC; Newman-Keuls tests, each P<0.05). However, OS-PI3Kγ−/− DC showed decreased trafficking compared with OS-WT DC (Newman-Keuls tests, P<0.05). No significant difference in CCR7 expression was noted between OS-WT DC and OS-PI3Kγ−/− DC (data not shown).
Figure 7. Oxidative stress activates the PI3Kγ pathway and increases trafficking of DC.
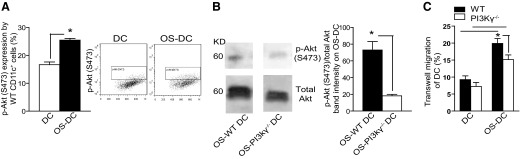
(A) DC from C57BL/6 mice were incubated for 24 h in medium alone (control DC) or in the presence of 500 μM H2O2 (OS-DC). Intracellular expression of p-Akt (S473) was assessed in control DC and OS-DC by gating on CD11c+ cells and using flow cytometry. There was a significant increase in p-Akt expression on OS-DC compared with control DC (*P=0.002, n=4 in each group). (B) Western blot analysis on lysates of OS-WT DC and OS-PI3Kγ−/− DC (quantification performed using ImageJ software) revealed a decrease in the ratio of serine p-Akt:total Akt in OS-PI3Kγ−/− DC compared with OS-WT DC (*P=0.03; n=4 in each group). (C) Trafficking of control DC and OS-DC from WT and PI3Kγ−/− mice was assessed using transwell CCL21 chemotaxis assay. OS- PI3Kγ−/− DC showed decreased trafficking compared with OS-WT DC (*P<0.05, n≥10).
We then investigated whether PI3Kδ, a key subunit also expressed mainly on leukocytes [17], plays a role in transwell migration of DC following oxidative stress. We pretreated some OS-DC, derived from WT mice and PI3Kγ−/− mice, with the PI3Kδ inhibitor and used them in our in vitro transwell assay. The addition of the PI3Kδ inhibitor did not have any effect on migration of each OS-WT DC or OS- PI3Kγ−/− DC (data not shown).
DC migration following oxidative stress is also influenced by NF-κB
As NF-κB is downstream of CCR7 and PI3K/Akt [31], we evaluated whether NF-κB could play any role in DC migration following oxidative stress. With the use of Western blot, an activation of p-NF-κB was observed in OS-DC compared with control DC (Fig. 8A). The effect of NF-κB on DC trafficking was then assessed in control DC and OS-DC (treated with 500 μM H2O2 for 12 h), with or without pretreatment with NF-κBi. At the used concentration (0.5 nM), NF-κBi did not have a significant effect on DC viability, as measured by Annexin V and 7-AAD (data not shown). As shown in Fig. 8B, there was a significant difference in trafficking among the four different groups (ANOVA, P=0.001). OS-DC showed increased trafficking compared with each control DC, control DC pretreated with NF-κBi, and OS-DC pretreated with NF-κBi (Newman-Keuls test, each P<0.05). No significant difference in CCR7 expression was noted between OS-DC and OS-DC with NF-κBi (data not shown).
Figure 8. Oxidative stress activates NF-κB and increases DC trafficking.

(A) Western blot analysis (quantification performed using ImageJ software) showed increase activation of p-NF-κB in OS-DC compared with control DC (*P=0.002, n=3 in each group). (B) Control DC and OS-DC (incubated for 12 h with 500 μM H2O2), with or without pretreatment with NF-κBi, were used in the transwell CCL21 chemotaxic assay. OS-DC showed increased trafficking compared with each of DC, DC pretreated with NF-κBi, and OS-DC pretreated with NF-κBi (*P<0.05, n=6 in each group).
DISCUSSION
Oxidative stress is a potent instigator of inflammation and subsequent tissue damage in IRI [32, 33], which is associated with sharp increase in reactive oxygen species over a short period of time in animal models [34, 35], as well as in humans [36]. Whereas the effector arms of IRI have been studied extensively, the upstream cellular pathways, which control inflammatory cascades, are poorly understood. In transplantation, IRI has been shown to represent an important link between innate immunity and alloimmune responses [11]. Among various leukocyte subsets, DC have the capability to orchestrate a network of inflammatory pathways during IRI [37]. DC are not only the main subsets of resident leukocytes in the tissue [38–40], but they also possess the TLR system. These features render them suitable to interact with endogenous danger signals occurring during IRI [41–43]. Early evidence on the role of DC in IRI came from studies that showed the accumulation of DC at sites of ischemic injury [8]. We have shown previously that ischemic injury enhances DC immunogenicity through TLR4 activation [5]. Furthermore, DC initiate inflammatory responses through the production of proinflammatory cytokines [10, 15]. Whereas these aforementioned studies have helped unveil a role for DC in the pathogenesis of IRI and inflammation, there is a need to identify the mechanisms by which oxidative stress leads to DC activation. Such mechanistic studies have the potential to identify novel targets to develop innovative therapies and reduce the burden of IRI.
We first showed that H2O2 treatment increased the surface expression of CD86, CD80, and CD40 costimulatory molecules and induced morphologic changes manifested by larger cell size and more prominent protrusions. These changes are characteristic of DC maturation [29, 30]. H2O2 treatment also enhanced DC in vitro alloreactivity and trafficking, an observation that was also confirmed by using hypoxanthine and xanthine oxidase as a second inducer of oxidative stress [22]. We examined the impact of oxidative stress on the production of inflammatory cytokines, which play a critical role in the pathogenesis of IRI [44]. Our data indicate that oxidative stress significantly increased the production of IL-6 by DC. IL-6 is a proinflammatory cytokine, which has gained significant attention for its role in inflammatory conditions [45, 46]. We then examined the potential role of oxidative stress on the capacity of DC to promote Tregs. The interaction of DC and T cells plays a central role in the homeostasis of Tregs [11]. We demonstrated that OS-DC were less efficient than untreated DC in promoting Treg generation. As Treg generation appears to be negatively affected by IL-6 [47, 48], the implication of anti-IL-6 was sought. Blocking IL-6 reversed the inhibitory effect of OS-DC on Treg generation. Together, these data may explain, in part, the higher immunogenicity of DC-bearing ischemic organs [5, 49]. The pretreatment of donor organs with anti-IL-6 may be potentially beneficial in improving the post-transplantation outcomes of grafts harvested from marginal donors.
To provide mechanistic insight on the effect of oxidative stress on peptide presentation by DC, we used OT-II and OT-I transgenic mice. TCR of these mice react specifically with OVA MHC-II- and -I-restricted residues, respectively. Our data indicate that oxidative stress predominantly enhanced the activity of CD4+ T cells. OS-DC increased OVA-specific CD4+ T cell proliferation and augmented IFN-γ, IL-6, and IL-2 secretion. The effects of OS-DC on OVA-specific CD8+ T cells were less significant. Although OS-DC increased secretion of IFN-γ, there was no increase in proliferation, granzyme B, perforin expression, or cytotoxic killing capacity of CTLs. Whereas the proliferation of CTLs is not influenced significantly by IFNγ [50], IFNγ produced by CTLs could potentially perpetuate the inflammatory cascades during IRI.
The capacity of DC to traffic to sites of injury and lymphoid tissue is a necessary element to activate T cells and generate an immune response efficiently. We have tested the impact of oxidative stress on DC trafficking in a chemotaxis chamber and under shear stress. Compared with untreated DC, OS-DC showed an increase in their migration capability. OS-DC also revealed a time-dependent increase in the surface expression of CCR7, a molecule that plays a critical role in regulating DC trafficking [51]. We then sought to assess the signaling pathway downstream of CCR7, including PI3K/Akt and NF-κB [31]. Within PI3K class I, which is the most clinically relevant of the PI3K classes [52], PI3Kα and -β are expressed on all cells, whereas PI3Kγ and -δ are only expressed on leukocytes, including DC [16, 17]. The preferential expression of PI3Kγ on leukocytes has prompted investigators to take great strides in generating PI3Kγ inhibitors for inflammatory diseases [52]. Previous studies revealed that PI3K/Akt is involved in DC trafficking following LPS stimulation [18] and that DC trafficking in PI3Kγ−/− mice, in response to LPS, is reduced when compared with WT DC [19]. However, the role of PI3Kγ and -δ pathways in DC trafficking following oxidative stress remained to be explored. Our data reveal that OS-DC from PI3Kγ−/− mice showed lower expression of p-Akt and decreased in vitro transwell migration when compared with OS-WT DC. In contrast to PI3Kγ, the use of the PI3Kδ inhibitor did not have any effect on chemotaxis-driven DC migration. These findings indicate that oxidative stress leads to activation of DC through the p-Akt/PI3Kγ pathway.
Finally, in WT mice, we have demonstrated that oxidative stress induced NF-κB activation and enhanced in vitro migration of DC. However, the increase in DC migration was abrogated following treatment with NF-κBi.
In summary, our data indicate that oxidative stress increases DC activation of CD4+ T cells more than CD8+ cytotoxic T cells. OS-DC produce more IL-6, which results in less Treg generation. Finally, oxidative stress activates PI3Kγ and increases DC migration. These findings might help guide future targeted therapies to reduce the deleterious effects of IRI, including post-transplantation IRI.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported, in part, by U.S. National Institutes of Health Grant R01-AI091930-03 (to R.A.), by a 2012 American Society of Transplantation/Pfizer Clinical Science Fellowship grant (to I.B.), by a 2013 American Heart Association Fellow-to-Faculty Transition Award (to J.A.), and by the National Heart, Lung, and Blood Institute (NHLBI) Program of Excellence in Glycosciences (PEG) Grant PO1 HL107146 (to R.S.). This work was presented at the 2012 United States and Canadian Academy of Pathology (USCAP) annual meeting in Vancouver, BC, Canada, and was the winner of the Stowell-Orbison Award and the Renal Pathology Society Pathologists in Training Best Poster Award.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- 7-AAD
- 7-aminoactinomycin D
- DC
- dendritic cell(s)
- FOXP3
- forkhead box protein 3
- H2O2
- hydrogen peroxide
- IRI
- ischemia reperfusion injury
- MFI
- mean fluorescence intensity
- NF-κBi
- NF-κB inhibitor
- OS-DC
- DC treated with hydrogen peroxide
- OS2-DC
- DC treated with hypoxanthine and xanthine oxidase
- OVA
- ovalbumin
- OVA-I
- ovalbumin257—264 MHC class I-restricted peptide
- OVA-II
- ovalbumin323—339 MHC class II-restricted peptide
- p-Akt
- phosphorylated Akt
- p-NF-κB
- phosphorylated NF-κB
- PI3Kγ−/−
- PI3Kγ-deficient
- Treg
- regulatory T cell
AUTHORSHIP
I.B. and J.A. participated in research design, performance of the research, data analysis, and writing the manuscript. M.M. participated in performance of the research and data analysis. R.A., R.M., F.R., and C.W. participated in performance of the research. J.Y.L. participated in performance of the research and writing the manuscript. P.F., R.S., T.I., and R.A. participated in research design and writing the manuscript.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES
- 1. Black S. C., Rodger I. W. (1996) Methods for studying experimental myocardial ischemic and reperfusion injury. J. Pharmacol. Toxicol. Methods 35, 179–190 [DOI] [PubMed] [Google Scholar]
- 2. Eltzschig H. K., Eckle T. (2011) Ischemia and reperfusion—from mechanism to translation. Nat. Med. 17, 1391–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nelson P. J. (2007) Renal ischemia-reperfusion injury: renal dendritic cells loudly sound the alarm. Kidney Int. 71, 604–605 [DOI] [PubMed] [Google Scholar]
- 4. Bonasio R., von Andrian U. H. (2006) Generation, migration and function of circulating dendritic cells. Curr. Opin. Immunol. 18, 503–511 [DOI] [PubMed] [Google Scholar]
- 5. Jurewicz M., Takakura A., Augello A., Naini S. M., Ichimura T., Zandi-Nejad K., Abdi R. (2010) Ischemic injury enhances dendritic cell immunogenicity via TLR4 and NF-κ B activation. J. Immunol. 184, 2939–2948 [DOI] [PubMed] [Google Scholar]
- 6. Land W. G. (2012) Emerging role of innate immunity in organ transplantation part II: potential of damage-associated molecular patterns to generate immunostimulatory dendritic cells. Transplant. Rev. (Orlando) 26, 73–87 [DOI] [PubMed] [Google Scholar]
- 7. Bosco M. C., Puppo M., Blengio F., Fraone T., Cappello P., Giovarelli M., Varesio L. (2008) Monocytes and dendritic cells in a hypoxic environment: spotlights on chemotaxis and migration. Immunobiology 213, 733–749 [DOI] [PubMed] [Google Scholar]
- 8. Kostulas N., Li H. L., Xiao B. G., Huang Y. M., Kostulas V., Link H. (2002) Dendritic cells are present in ischemic brain after permanent middle cerebral artery occlusion in the rat. Stroke 33, 1129–1134 [DOI] [PubMed] [Google Scholar]
- 9. Rutault K., Alderman C., Chain B. M., Katz D. R. (1999) Reactive oxygen species activate human peripheral blood dendritic cells. Free Radic. Biol. Med. 26, 232–238 [DOI] [PubMed] [Google Scholar]
- 10. Verhasselt V., Goldman M., Willems F. (1998) Oxidative stress up-regulates IL-8 and TNF-α synthesis by human dendritic cells. Eur. J. Immunol. 28, 3886–3890 [DOI] [PubMed] [Google Scholar]
- 11. Land W. G. (2005) The role of postischemic reperfusion injury and other nonantigen-dependent inflammatory pathways in transplantation. Transplantation 79, 505–514 [DOI] [PubMed] [Google Scholar]
- 12. Jurewicz M., Ueno T., Azzi J., Tanaka K., Murayama T., Yang S., Sayegh M. H., Niimi M., Abdi R. (2011) Donor antioxidant strategy prolongs cardiac allograft survival by attenuating tissue dendritic cell immunogenicity(dagger). Am. J. Transplant. 11, 348–355 [DOI] [PubMed] [Google Scholar]
- 13. Kim B. S., Lim S. W., Li C., Kim J. S., Sun B. K., Ahn K. O., Han S. W., Kim J., Yang C. W. (2005) Ischemia-reperfusion injury activates innate immunity in rat kidneys. Transplantation 79, 1370–1377 [DOI] [PubMed] [Google Scholar]
- 14. Loverre A., Capobianco C., Stallone G., Infante B., Schena A., Ditonno P., Palazzo S., Battaglia M., Crovace A., Castellano G., Ranieri E., Schena F. P., Gesualdo L., Grandaliano G. (2007) Ischemia-reperfusion injury-induced abnormal dendritic cell traffic in the transplanted kidney with delayed graft function. Kidney Int. 72, 994–1003 [DOI] [PubMed] [Google Scholar]
- 15. Mashreghi M. F., Klemz R., Knosalla I. S., Gerstmayer B., Janssen U., Buelow R., Jozkowicz A., Dulak J., Volk H. D., Kotsch K. (2008) Inhibition of dendritic cell maturation and function is independent of heme oxygenase 1 but requires the activation of STAT3. J. Immunol. 180, 7919–7930 [DOI] [PubMed] [Google Scholar]
- 16. Camps M., Ruckle T., Ji H., Ardissone V., Rintelen F., Shaw J., Ferrandi C., Chabert C., Gillieron C., Francon B., Martin T., Gretener D., Perrin D., Leroy D., Vitte P. A., Hirsch E., Wymann M. P., Cirillo R., Schwarz M. K., Rommel C. (2005) Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 11, 936–943 [DOI] [PubMed] [Google Scholar]
- 17. Ghigo A., Hirsch E. (2008) Isoform selective phosphoinositide 3-kinase γ and δ inhibitors and their therapeutic potential. Recent Pat. Inflamm. Allergy Drug Discov. 2, 1–10 [DOI] [PubMed] [Google Scholar]
- 18. Agrawal A., Agrawal S., Cao J. N., Su H., Osann K., Gupta S. (2007) Altered innate immune functioning of dendritic cells in elderly humans: a role of phosphoinositide 3-kinase-signaling pathway. J. Immunol. 178, 6912–6922 [DOI] [PubMed] [Google Scholar]
- 19. Del Prete A., Vermi W., Dander E., Otero K., Barberis L., Luini W., Bernasconi S., Sironi M., Santoro A., Garlanda C., Facchetti F., Wymann M. P., Vecchi A., Hirsch E., Mantovani A., Sozzani S. (2004) Defective dendritic cell migration and activation of adaptive immunity in PI3Kγ-deficient mice. EMBO J. 23, 3505–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barbier M., Attoub S., Calvez R., Laffargue M., Jarry A., Mareel M., Altruda F., Gespach C., Wu D., Lu B., Hirsch E., Wymann M. P. (2001) Tumour biology. Weakening link to colorectal cancer? Nature 413, 796. [DOI] [PubMed] [Google Scholar]
- 21. Tang D., Shi Y., Kang R., Li T., Xiao W., Wang H., Xiao X. (2007) Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol. 81, 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lassen S., Lech M., Rommele C., Mittruecker H. W., Mak T. W., Anders H. J. (2010) Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J. Immunol. 185, 1976–1983 [DOI] [PubMed] [Google Scholar]
- 23. Sadhu C., Masinovsky B., Dick K., Sowell C. G., Staunton D. E. (2003) Essential role of phosphoinositide 3-kinase δ in neutrophil directional movement. J. Immunol. 170, 2647–2654 [DOI] [PubMed] [Google Scholar]
- 24. Berkman W. A., Chowdhury L., Brown N. L., Padleckas R. (1983) Value of electron microscopy in cytologic diagnosis of fine-needle biopsy. Am. J. Roentgenol. 140, 1253–1258 [DOI] [PubMed] [Google Scholar]
- 25. Lee J. Y., Buzney C. D., Poznansky M. C., Sackstein R. (2009) Dynamic alterations in chemokine gradients induce transendothelial shuttling of human T cells under physiologic shear conditions. J. Leukoc. Biol. 86, 1285–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fiorina P., Jurewicz M., Vergani A., Augello A., Paez J., Ricchiuti V., Tchipachvili V., Sayegh M. H., Abdi R. (2008) Phenotypic and functional differences between wild-type and CCR2−/− dendritic cells: implications for islet transplantation. Transplantation 85, 1030–1038 [DOI] [PubMed] [Google Scholar]
- 27. Wobben R., Husecken Y., Lodewick C., Gibbert K., Fandrey J., Winning S. (2013) Role of hypoxia inducible factor-1α for interferon synthesis in mouse dendritic cells. Biol. Chem. 394, 495–505 [DOI] [PubMed] [Google Scholar]
- 28. Inaba K., Swiggard W. J., Steinman R. M., Romani N., Schuler G., Brinster C. (2009) Isolation of dendritic cells. Curr. Protoc. Immunol. Chapter 3, Unit 3.7 [DOI] [PubMed] [Google Scholar]
- 29. Xing F., Wang J., Hu M., Yu Y., Chen G., Liu J. (2011) Comparison of immature and mature bone marrow-derived dendritic cells by atomic force microscopy. Nanoscale Res. Lett. 6, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeng X., Wang T., Zhu C., Xing X., Ye Y., Lai X., Song B., Zeng Y. (2012) Topographical and biological evidence revealed FTY720-mediated anergy-polarization of mouse bone marrow-derived dendritic cells in vitro. PLoS One 7, e34830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Randolph G. J., Ochando J., Partida-Sanchez S. (2008) Migration of dendritic cell subsets and their precursors. Annu. Rev. Immunol. 26, 293–316 [DOI] [PubMed] [Google Scholar]
- 32. Baud L., Ardaillou R. (1993) Involvement of reactive oxygen species in kidney damage. Br. Med. Bull. 49, 621–629 [DOI] [PubMed] [Google Scholar]
- 33. McCord J. M. (1985) Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med. 312, 159–163 [DOI] [PubMed] [Google Scholar]
- 34. Bolli R., Patel B. S., Jeroudi M. O., Lai E. K., McCay P. B. (1988) Demonstration of free radical generation in “stunned” myocardium of intact dogs with the use of the spin trap α-phenyl N-tert-butyl nitrone. J. Clin. Invest. 82, 476–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bolli R., Jeroudi M. O., Patel B. S., DuBose C. M., Lai E. K., Roberts R., McCay P. B. (1989) Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc. Natl. Acad. Sci. USA 86, 4695–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grech E. D., Dodd N. J., Jackson M. J., Morrison W. L., Faragher E. B., Ramsdale D. R. (1996) Evidence for free radical generation after primary percutaneous transluminal coronary angioplasty recanalization in acute myocardial infarction. Am. J. Cardiol. 77, 122–127 [DOI] [PubMed] [Google Scholar]
- 37. Li L., Okusa M. D. (2010) Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin. Nephrol. 30, 268–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Banchereau J., Steinman R. M. (1998) Dendritic cells and the control of immunity. Nature 392, 245–252 [DOI] [PubMed] [Google Scholar]
- 39. Soos T. J., Sims T. N., Barisoni L., Lin K., Littman D. R., Dustin M. L., Nelson P. J. (2006) CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 70, 591–596 [DOI] [PubMed] [Google Scholar]
- 40. Steptoe R. J., Patel R. K., Subbotin V. M., Thomson A. W. (2000) Comparative analysis of dendritic cell density and total number in commonly transplanted organs: morphometric estimation in normal mice. Transpl. Immunol. 8, 49–56 [DOI] [PubMed] [Google Scholar]
- 41. Nace G., Evankovich J., Eid R., Tsung A. (2012) Dendritic cells and damage-associated molecular patterns: endogenous danger signals linking innate and adaptive immunity. J. Innate Immun. 4, 6–15 [DOI] [PubMed] [Google Scholar]
- 42. Tsung A., Sahai R., Tanaka H., Nakao A., Fink M. P., Lotze M. T., Yang H., Li J., Tracey K. J., Geller D. A., Billiar T. R. (2005) The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201, 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhai Y., Shen X. D., O'Connell R., Gao F., Lassman C., Busuttil R. W., Cheng G., Kupiec-Weglinski J. W. (2004) Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J. Immunol. 173, 7115–7119 [DOI] [PubMed] [Google Scholar]
- 44. Sharma H. S., Das D. K. (1997) Role of cytokines in myocardial ischemia and reperfusion. Mediators Inflamm. 6, 175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fonseca J. E., Santos M. J., Canhao H., Choy E. (2009) Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun. Rev. 8, 538–542 [DOI] [PubMed] [Google Scholar]
- 46. Navarro-Millan I., Singh J. A., Curtis J. R. (2012) Systematic review of tocilizumab for rheumatoid arthritis: a new biologic agent targeting the interleukin-6 receptor. Clin. Ther. 34, 788–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wan S., Xia C., Morel L. (2007) IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J. Immunol. 178, 271–279 [DOI] [PubMed] [Google Scholar]
- 48. Pasare C., Medzhitov R. (2003) Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036 [DOI] [PubMed] [Google Scholar]
- 49. Liang Y., Christopher K., Finn P. W., Colson Y. L., Perkins D. L. (2007) Graft produced interleukin-6 functions as a danger signal and promotes rejection after transplantation. Transplantation 84, 771–777 [DOI] [PubMed] [Google Scholar]
- 50. Gajewski T. F., Schell S. R., Nau G., Fitch F. W. (1989) Regulation of T-cell activation: differences among T-cell subsets. Immunol. Rev. 111, 79–110 [DOI] [PubMed] [Google Scholar]
- 51. Willimann K., Legler D. F., Loetscher M., Roos R. S., Delgado M. B., Clark-Lewis I., Baggiolini M., Moser B. (1998) The chemokine SLC is expressed in T cell areas of lymph nodes and mucosal lymphoid tissues and attracts activated T cells via CCR7. Eur. J. Immunol. 28, 2025–2034 [DOI] [PubMed] [Google Scholar]
- 52. Ruckle T., Schwarz M. K., Rommel C. (2006) PI3Kγ inhibition: towards an “aspirin of the 21st century”? Nat. Rev. Drug Discov. 5, 903–918 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.