

Abstract
Azathioprine (AZA) is widely used in clinical practice for preventing graft rejection in organ transplantations and various autoimmune and dermatological diseases with documented unpredictable hepatotoxicity. The potential molecular cytotoxic mechanisms of AZA towards isolated rat hepatocytes were investigated in this study using “Accelerated Cytotoxicity Mechanism Screening” techniques. The concentration of AZA required to cause 50% cytotoxicity in 2 hrs at 37°C was found to be 400 μM. A significant increase in AZA-induced cytotoxicity and reactive oxygen species (ROS) formation was observed when glutathione- (GSH-) depleted hepatocytes were used. The addition of N-acetylcysteine decreased cytotoxicity and ROS formation. Xanthine oxidase inhibition by allopurinol decreased AZA-induced cytotoxicity, ROS, and hydrogen peroxide (H2O2) formation and increased % mitochondrial membrane potential (MMP). Addition of N-acetylcysteine and allopurinol together caused nearly complete cytoprotection against AZA-induced hepatocyte death. TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl), a known ROS scavenger and a superoxide dismutase mimic, and antioxidants, like DPPD (N,N′-diphenyl-p-phenylenediamine), Trolox (a water soluble vitamin E analogue), and mesna (2-mercaptoethanesulfonate), also decreased hepatocyte death and ROS formation. Results from this study suggest that AZA-induced cytotoxicity in isolated rat hepatocytes may be partly due to ROS formation and GSH depletion that resulted in oxidative stress and mitochondrial injury.
1. Introduction
Azathioprine (AZA), prodrug of 6-mercaptopurine, is widely used as an immunosuppressant for several diseases such as inflammatory bowel disease (IBD) and autoimmune diseases and following transplantation to avoid organ rejection [1–4]. In most cases, hepatotoxicity is an unpredictable side effect of AZA, whose molecular and pathogenic mechanisms remain unknown [5]. It has even been reported that 3.5% of 173 adult IBD patients developed hepatitis as a consequence of AZA treatment [6]. A variety of histopathologic findings have been observed in AZA-induced hepatotoxicity. Nodular regenerative hyperplasia, venoocclusive disease, peliosis hepatis, sinusoidal dilatation, and perisinusoidal fibrosis have been reported [7–13]. Cholestasis, with or without associated hepatocyte necrosis, has also been reported for these thiopurine drugs in clinical studies [7, 14]. The molecular mechanisms of AZA-induced cytotoxicity using the “Accelerated Cytotoxicity Mechanism Screening” (ACMS) techniques were investigated in this study.
The ACMS methods determine the molecular cytotoxic mechanisms of drugs/xenobiotics when incubated at 37°C for 2 to 3 hours using freshly isolated hepatocytes from Sprague-Dawley male rats. ACMS is a useful tool for identifying the bioactivation or detoxifying pathways of a drug/xenobiotic by comparing the effects of specific enzyme modulators on cell viability induced by the drug/xenobiotic being investigated. A major assumption with ACMS is that high dose/short time (in vitro) exposure simulates low dose/long time (in vivo) exposure [15]. With 24 halobenzenes, it was found that the relative lethal concentrations required to cause 50% cytotoxicity in 2 hrs at 37°C (LC50, according to ACMS) that was determined using hepatocytes isolated from phenobarbital-induced Sprague-Dawley rats in vitro correlated with hepatotoxicity in vivo at 24 to 54 hrs [16]. Moreover, using these techniques, the molecular hepatocytotoxic mechanisms found in vitro for seven classes of xenobiotics/drugs were found to be similar to the rat hepatotoxic mechanisms reported in vivo [17]. Our laboratory successfully used ACMS techniques to investigate molecular mechanisms of drugs/xenobiotics-induced cytotoxicity in isolated rat hepatocytes. Recent examples include chlorpromazine [18]; isoniazid [19]; amodiaquine [20]; and polychlorinated biphenyls [21].
AZA has been reported to conjugate with reduced glutathione (GSH) to form 6-mercaptopurine (6-MP), catalyzed by glutathione S-transferases [22–25]. Previous studies performed with rat hepatocyte primary cultures showed that toxic concentrations of AZA (25–250 μM) led to profound intracellular GSH depletion, mitochondrial injury, metabolic activity reduction, decreased adenosine 5′-triphosphate (ATP) levels, and cell death due to necrosis, not apoptosis. Toxic effects were acute and dose-dependent. Hepatocyte death was prevented by GSH or N-acetylcysteine, Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, a vitamin E analogue), high dose of allopurinol (acts as an antioxidant), cyclosporine A, and glycine [24, 25]. Similar effects were observed by Menor and colleagues [5] where AZA (150 μM) decreased the viability of rat hepatocytes and induced intracellular GSH depletion, metabolic activity reduction, and lactate dehydrogenase release. However, the cell death was not accompanied by DNA laddering, procaspase-3 cleavage, or cytochrome c release. AZA caused mitochondrial dysfunction and activation of stress-activated protein kinase pathways leading to necrotic cell death in intact isolated rat mitochondria [5].
Clinically relevant concentrations of AZA (0.5–5 μM) were also found to be toxic to rat hepatocyte cultures and involved oxidative stress, mitochondrial injury, and ATP depletion that led to cell death by necrosis. Allopurinol (xanthine oxidase inhibitor) and Trolox together provided near complete hepatocyte protection from AZA [10]. Xanthine oxidase is proposed to be involved in several steps of AZA metabolism such as in the direct metabolism of AZA to form an inactive metabolite, 1-methyl-4-nitrothioimidazole, in the conversion of AZA to 6-MP, and in the formation of 6-thiouric acid from 6-MP [10, 24, 25]. The possibility that xanthine oxidase may play a role in AZA-induced tissue injury has been raised by the observation that patients taking allopurinol, a xanthine oxidase inhibitor, experienced less nephrotoxicity during rejection episodes after renal transplantation [26].
Thiopurine S-methyltransferase (TMPT) converts 6-MP to 6-methyl mercaptopurine (6-MMP) and elevated 6-MMP levels were reported to be associated with hepatotoxicity (reviewed in [27, 28]). However, several studies reported AZA-induced hepatotoxicity had no relationship with 6-MMP levels [29–31]. AZA-induced myelosuppression and skin reactions were related to thiopurine S-methyltransferase (TPMT) polymorphisms [2, 32]. However, TPMT polymorphisms did not appear to be involved in AZA-induced hepatotoxicity [33].
In this study, we investigated different toxicity routes of AZA towards isolated rat hepatocytes using ACMS techniques. We hypothesize that AZA causes cytotoxicity towards isolated rat hepatocytes by depleting hepatocyte GSH and producing reactive oxygen species (ROS). We also hypothesize that xanthine oxidase is also involved in AZA-induced oxidative stress in rat hepatocytes.
2. Materials and Methods
2.1. Chemicals
Type II collagenase was purchased from Worthington Biochemical Corp. (Lakewood, New Jersey, USA). 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES) was purchased from Boehringer-Mannheim Ltd. (Montreal, Canada). Azathioprine and all other chemicals were purchased from Sigma-Aldrich Corp. (Oakville, Ontario, Canada).
2.2. Animal Treatment and Hepatocyte Preparation
Male Sprague-Dawley rats weighing 275–300 g (Charles River Laboratories International Inc., USA) were used for experimental purposes and carried out in compliance with the Guide to the Care and Use of Experimental Animals [34]. Rats were housed in ventilated plastic cages. There were 12 air changes per hr, 12 hr light photoperiod (lights on at 08:00 hr), and an environmental temperature of 21–23°C with a 50–60% relative humidity. The animals were fed a normal standard chow diet and water ad libitum. Hepatocytes were isolated from rats by collagenase perfusion of the liver [35]. Isolated hepatocytes (10 mL, 106 cells/mL) were suspended in Krebs-Henseleit buffer (pH 7.4) containing 12.5 mM HEPES in continually rotating 50 mL round bottom flasks, under an atmospheric condition of 95% O2 and 5% CO2 in a 37°C water bath for 30 min prior to the addition of chemicals.
GSH-depleted hepatocytes were obtained by preincubating the hepatocytes with 200 μM 1-bromoheptane for 30 min [36]. 1-Bromoheptane rapidly conjugates hepatocyte GSH without affecting hepatocyte viability. GSH precursors, N-acetylcysteine (1 mM) or L-cysteine (1 mM), were added 30 min prior to the addition of AZA or other agents. Xanthine oxidase-inhibited hepatocytes were obtained by preincubating hepatocytes with 20 μM allopurinol for 30 min [37]. The concentrations of enzyme modulators/antioxidants/ROS scavenger used in the experiments did not affect hepatocyte viability.
2.3. Cell Viability
Hepatocyte viability was assessed microscopically by plasma membrane disruption as determined by the trypan blue (0.1% w/v) exclusion test [35]. Hepatocyte viability was determined every 30 min during a 3 hr incubation period. Only cell preparations with viability of 80 to 90% were used.
2.4. ROS Formation Assay
Hepatocyte hydroxyl, peroxyl, and other ROS generations were determined using 2′, 7′-dichlorofluorescein diacetate (DCFD) which can permeate hepatocytes and be deacetylated by intracellular esterases to form nonfluorescent dichlorofluorescein. Dichlorofluorescein is oxidized by intracellular ROS to form the highly fluorescent dichlorofluorescein. ROS formation was assayed by withdrawing 1 mL hepatocyte samples at 30 min, which were then centrifuged for 1 min at 5000 ×g. The cells were resuspended in Krebs-Heinseleit buffer and 1.6 μM DCFD was added. The cells were incubated at 37°C for 10 min and the fluorescent intensity (FI) of dichlorofluorescein was measured using a SPECTRAmax Gemini XS spectrofluorometer (Molecular Devices, LLC, CA, USA) set at 490 nm excitation and 520 nm emission wavelengths [38].
2.5. Mitochondrial Membrane Potential (MMP) Assay
The uptake of the cationic fluorescent dye, rhodamine 123, has been used for the estimation of MMP according to Andersson and colleagues [39]. Aliquots (500 μL) of the cell suspension at 30 minutes were separated from the incubation medium by centrifugation at 5000 ×g for 1 min. The cell pellet was resuspended in 2 mL of fresh incubation medium containing 1.5 μM rhodamine 123 and incubated at 37°C in a thermostatic bath for 10 min with gentle shaking. Hepatocytes were then separated by centrifugation and the amount of rhodamine 123 remaining in the incubation medium was measured at 490 nm excitation and 520 nm emission wavelengths using a SPECTRAmax Gemini XS spectrofluorometer (Molecular Devices, LLC, CA, USA). The capacity of mitochondria to take up the rhodamine 123 was calculated as the difference in fluorescence intensity between control and treated cells [39] and was expressed as % MMP [40].
2.6. Hydrogen Peroxide (H2O2) Measurement
H2O2 was measured in hepatocytes by taking samples at 30 min by adding FOX 1 reagent. The FOX 1 reagent consisted of 25 mM sulfuric acid, 250 μM ferrous ammonium sulfate, 100 μM xylenol orange, and 100 mM sorbitol. At the above time point, 50 μL of hepatocytes suspension was added to 950 μL of the FOX 1 reagent and incubated for 30 min at room temperature. Samples were then spectrophotometrically analyzed at 560 nm using a SPECTRAmax Plus 384 spectrophotometer (Molecular Devices, LLC, CA, USA). The extinction coefficient 2.35 × 105 M−1 cm−1 was used to measure the concentration of H2O2 [41].
2.7. Cellular GSH and Oxidized Glutathione (GSSG) Determination
GSH and GSSG (the disulfide dimer of GSH) in hepatocytes were determined by commercial kits from Cayman Chemical, MI, USA, according to manufacturer's instruction which utilizes an optimized enzymatic recycling method [42].
2.8. Statistical Analysis
The SPSS software package (version 14.0, SPSS Inc., Chicago, USA) was used to analyze the data. Values were expressed as mean ± standard error of the mean (SEM) from 3 independent experiments. Statistical analysis was performed using a one-way analysis of variance (ANOVA) and Tukey's post hoc test to assess significance between control and treatment groups in these experiments. P < 0.05 was considered significant.
3. Results and Discussion
A concentration and time dependent increase in cytotoxicity and ROS formation and a decrease in % MMP were observed with AZA (100–500 μM) compared to control hepatocytes (Figure 1) incubated for 3 hrs. Incubation of isolated rat hepatocytes for 2 hrs at 37°C with 400 μM AZA induced an approximate 50% loss in hepatocyte viability as measured by the trypan blue exclusion assay (LC50, according to ACMS). We use this LC50 value to investigate potential cytotoxic mechanisms of drug or xenobiotic under investigation. Although this in vitro study is limited for use at high concentrations of the drug, ACMS techniques assume that the drug metabolic/toxic pathways at cytotoxic drug concentrations in vitro at 2 hrs are similar to those that occur in vivo in 24 to 36 hrs [15].
Figure 1.
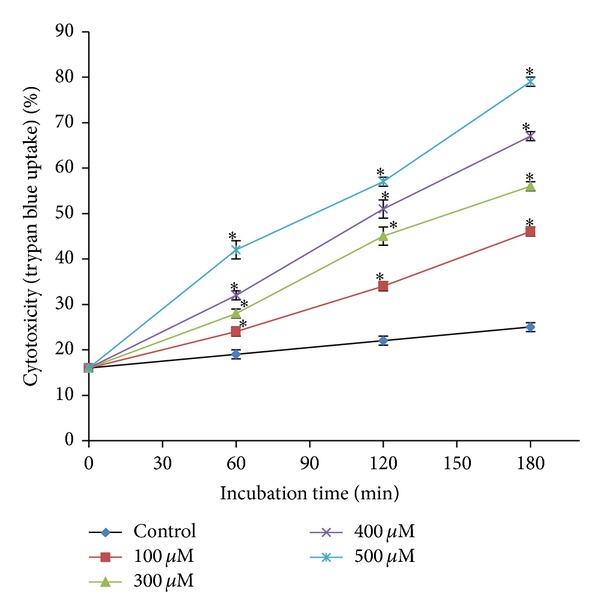
Concentration-response curve of AZA (100–500 μM) towards isolated rat hepatocytes to determine ACMS LC50. ∗Significant compared to control hepatocytes.
GSH and xanthine oxidase dependence of AZA in isolated rat hepatocytes are presented in Figure 2. AZA treatment (400 μM) significantly depleted hepatocyte GSH and increased GSSG levels (data not shown). A significant increase in AZA-induced cytotoxicity and ROS formation were observed when hepatocyte GSH was depleted by using 1-bromoheptane whereas addition of N-acetylcysteine (1 mM, a cysteine precursor which generates GSH) prevented AZA-induced cytotoxicity (Table 1), ROS, and H2O2 generation and increased % MMP and hepatocyte GSH which indicates that GSH was required for AZA detoxification. N-Acetylcysteine has been used as a tool for investigating the role of ROS in numerous biological and pathological processes. The usefulness of N-acetylcysteine in different diseases including cardiovascular diseases, cancer, and chemical/metal toxicity has been reviewed in Zafarullah and colleagues [43]. Addition of L-cysteine (1 mM) also had similar effects (data not shown) confirming the potential role of GSH in AZA-induced cytotoxicity. A similar depletion of GSH levels and mitochondrial toxicity during AZA metabolism were observed in previous studies in primary cultures of rat hepatocytes with both toxic and clinically relevant AZA concentrations [5, 10, 25]. However, human liver parenchymal cells were reported to be much less sensitive than rat hepatocytes to thiopurine treatments [1]. Protective effects of N-acetylcysteine against AZA-induced hepatotoxicity have been reported in several in vitro [5, 25, 44] and in vivo [45] studies which clearly indicates a potential role of GSH in AZA-induced cytotoxicity.
Figure 2.
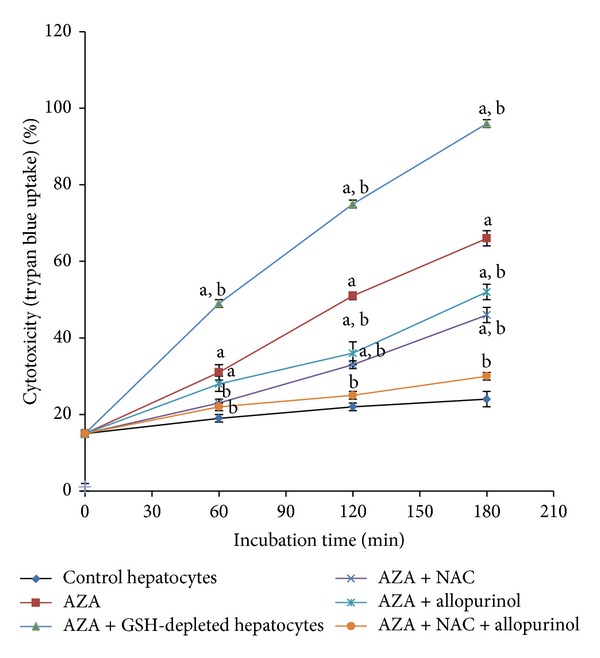
Glutathione and xanthine oxidase dependence of AZA towards rat hepatocytes. NAC, N-acetylcysteine; asignificant compared to control hepatocytes; bsignificant compared to AZA (400 μM).
Table 1.
AZA-induced oxidative stress with GSH depletion and protection with a GSH precursor, a xanthine oxidase inhibitor, various antioxidants, and a radical scavenger.
| Addition | ROS (FI unit) | MMP (%) | H2O2 (nmoles/106 cells) |
|---|---|---|---|
| Incubation time | 30 min | 30 min | 30 min |
|
| |||
| Control | 102 ± 1 | 100 | 6.34 ± 0.07 |
| +400 μM AZA | 139 ± 3a | 86 ± 1a | 8.11 ± 0.08a |
| +GSH-depleted hepatocytes | 174 ± 5a,b | 75 ± 1a,b | 9.21 ± 0.13a,b |
| +1 mM NAC | 124 ± 3a,b | 89 ± 1a | 6.97 ± 0.04a,b |
| +20 μM allopurinol | 132 ± 3a | 92 ± 1a,b | 7.75 ± 0.19a |
| +1 mM NAC + 20 μM allopurinol | 107 ± 1b | 97 ± 2b | 6.54 ± 0.14b |
| +1 mM mesna | 121 ± 3a,b | 93 ± 2a,b | 7.10 ± 0.18a,b |
| +1 mM Trolox | 113 ± 3b | 94 ± 2a,b | 7.12 ± 0.02a,b |
| +200 μM TEMPOL | 123 ± 4a,b | 93 ± 1a,b | 7.46 ± 0.18a,b |
| +2 μM DPPD | 124 ± 2a,b | 92 ± 2a,b | 7.26 ± 0.06a,b |
Data are presented as mean ± SEM (n = 3). All modulating agents were noncytotoxic compared to control hepatocytes at concentrations used. Refer to Section 2 for a description of the experiments performed and experimental conditions. FI, fluorescence intensity; NAC, N-acetylcysteine; mesna, 2-mercaptoethanesulfonate; Trolox, 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid; TEMPOL, 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl; DPPD, N,N′-diphenyl-p-phenylenediamine; asignificant compared to control (only hepatocytes); bsignificant compared to 400 μM AZA.
Xanthine oxidase is involved in several stages of AZA metabolic pathways (Figure 3) and is well-known to produce ROS [10, 46]. Xanthine oxidase is exemplified by numerous studies in which inhibition of xanthine oxidase attenuated symptoms of several vascular diseases including congestive heart failure, sickle cell anemia, and diabetes [47–49]. When we inhibited xanthine oxidase by preincubating hepatocytes with 20 μM allopurinol, AZA-induced cytotoxicity was significantly prevented (Figure 2). A significant decrease in ROS and H2O2 formation and an increase in % MMP were observed compared to control hepatocytes with AZA-treated and xanthine oxidase-inhibited hepatocytes (Table 1). Xanthine oxidase inhibition by allopurinol was also found to be protective in primary rat hepatocytes [10]. A recent case study reported that the addition of allopurinol with appropriate AZA dose reduction may correct AZA-induced hepatotoxicity and can induce remission [50]. In AZA or 6-MP nonresponders, addition of allopurinol also demonstrated safety and efficacy for long-term maintenance in IBD patients [51, 52]. In our study, the combined addition of N-acetylcysteine (1 mM) and allopurinol (20 μM) showed nearly complete protection against AZA-induced cytotoxicity in rat hepatocytes. The combination of these two agents for improved AZA therapeutic efficacy and liver enzymes needs future clinical experimentation.
Figure 3.
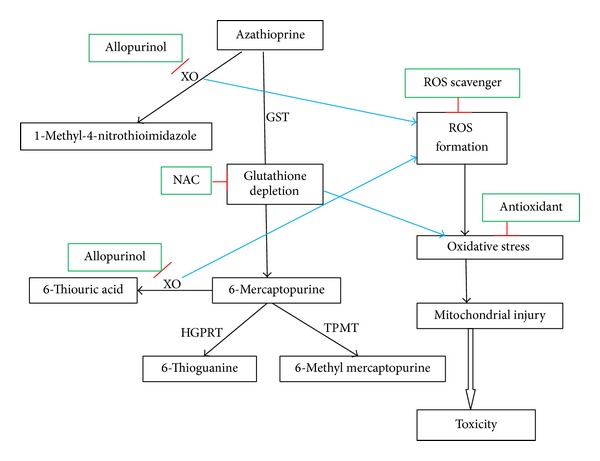
Proposed routes of AZA-induced cytotoxicity in isolated rat hepatocytes. XO, xanthine oxidase; GST, glutathione S-transferase; NAC, N-acetylcysteine; HGPRT, hypoxanthine guanine phosphoribosyl transferase; TPMT, thiopurine S-methyl transferase.
A significant decrease in AZA-induced cytotoxicity (Table 2) and ROS formation (Table 1) in isolated rat hepatocytes was achieved by the ROS scavenger and the superoxide dismutase mimic TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl, 200 μM) suggesting the involvement of ROS. Potent antioxidants, like Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, 1 mM), DPPD (N,N′-diphenyl-p-phenylenediamine, 2 μM), and mesna (2-mercaptoethanesulfonate, 1 mM), also significantly decreased AZA-induced cytotoxicity (Table 2), ROS, and H2O2 formation and increased % MMP (Table 1) suggesting the involvement of oxidative stress in AZA-induced cytotoxicity in hepatocytes. Possible routes of cytotoxicity of AZA in isolated rat hepatocytes are presented in Figure 3.
Table 2.
Effects of a ROS scavenger and various antioxidants on AZA-induced cytotoxicity in isolated rat hepatocytes.
| Addition | Cytotoxicity (trypan blue uptake) (%) | ||
|---|---|---|---|
| Incubation time | 60 min | 120 min | 180 min |
|
| |||
| Control hepatocytes | 19 ± 1 | 22 ± 1 | 24 ± 2 |
| +400 μM AZA | 31 ± 2a | 51 ± 1a | 66 ± 2a |
| +1 mM mesna | 23 ± 1b | 33 ± 1a,b | 45 ± 1a,b |
| +1 mM Trolox | 24 ± 1b | 35 ± 2a,b | 43 ± 1a,b |
| +200 μM TEMPOL | 24 ± 1b | 38 ± 2a,b | 49 ± 2a,b |
| +2 μM DPPD | 26 ± 1a | 37 ± 1a,b | 47 ± 1a,b |
Data are presented as mean ± SEM (n = 3); all modulating agents were noncytotoxic compared to control hepatocytes at concentrations used. Refer to Section 2 for a description of the experiments performed and experimental conditions. Mesna, 2-mercaptoethanesulfonate; Trolox, 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid; TEMPOL, 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl; DPPD, N,N′-diphenyl-p-phenylenediamine. aSignificant compared to control (only hepatocytes); bsignificant compared to 400 μM AZA.
4. Conclusions
Data obtained from the ACMS technique suggests that AZA toxicity towards isolated rat hepatocytes involves two distinct pathways (i) a xanthine oxidase-catalyzed production of an inactive metabolite (1-methyl-4-nitrothioimidazole), (ii) glutathione S-transferase- (GST-) catalyzed pathway leading to GSH depletion followed by xanthine oxidase-catalyzed formation of inactive metabolites. Addition of a GSH precursor, N-acetylcysteine and a xanthine oxidase inhibitor, and allopurinol together significantly reversed cytotoxicity which raises the possibility of using these two agents therapeutically. Several antioxidants also prevented hepatocyte death suggesting that antioxidant therapy may be of use therapeutically to prevent or decrease AZA-induced hepatotoxicity. In vivo animal and clinical studies are warranted to test their therapeutic effectiveness against AZA-induced hepatotoxicity.
Acknowledgments
This research has been funded by a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant. Abdullah Al Maruf is funded by a Connaught International Scholarship for doctoral students awarded by University of Toronto.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Petit E, Langouet S, Akhdar H, Nicolas-Nicolaz C, Guillouzo A, Morel F. Differential toxic effects of azathioprine, 6-mercaptopurine and 6-thioguanine on human hepatocytes. Toxicology in Vitro. 2008;22(3):632–642. doi: 10.1016/j.tiv.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Lennard L, Welch JC, Lilleyman JS. Thiopurine drugs in the treatment of childhood leukaemia: the influence of inherited thiopurine methyltransferase activity on drug metabolism and cytotoxicity. British Journal of Clinical Pharmacology. 1997;44(5):455–461. doi: 10.1046/j.1365-2125.1997.t01-1-00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubinsky MC. Azathioprine, 6-mercaptopurine in inflammatory bowel disease: pharmacology, efficacy, and safety. Clinical Gastroenterology and Hepatology. 2004;2(9):731–743. doi: 10.1016/s1542-3565(04)00344-1. [DOI] [PubMed] [Google Scholar]
- 4.El-Azhary RA. Azathioprine: current status and future considerations. International Journal of Dermatology. 2003;42(5):335–341. doi: 10.1046/j.1365-4362.2003.01823.x. [DOI] [PubMed] [Google Scholar]
- 5.Menor C, Fernández-Moreno MD, Fueyo JA, et al. Azathioprine acts upon rat hepatocyte mitochondria and stress-activated protein kinases leading to necrosis: protective role of N-acetyl-L-cysteine. Journal of Pharmacology and Experimental Therapeutics. 2004;311(2):668–676. doi: 10.1124/jpet.104.069286. [DOI] [PubMed] [Google Scholar]
- 6.Rietdijk ST, Bertelsman J, Hommes DW, Vogels E, Reitsma PH, van Deventer SJH. Genetic polymorphisms of the thiopurine S-methyltransferase (TPMT) locus in patients treated with azathioprine for inflammatory bowel disease. Gastroenterology. 2001;120, supplement, article 622A [Google Scholar]
- 7.Gisbert JP, González-Lama Y, Maté J. Thiopurine-induced liver injury in patients with inflammatory bowel disease: a systematic review. American Journal of Gastroenterology. 2007;102(7):1518–1527. doi: 10.1111/j.1572-0241.2007.01187.x. [DOI] [PubMed] [Google Scholar]
- 8.Vernier-Massouille G, Cosnes J, Lemann M, et al. Nodular regenerative hyperplasia in patients with inflammatory bowel disease treated with azathioprine. Gut. 2007;56(10):1404–1409. doi: 10.1136/gut.2006.114363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daniel F, Cadranel J, Seksik P, et al. Azathioprine induced nodular regenerative hyperplasia in IBD patients. Gastroenterologie Clinique et Biologique. 2005;29(5):600–603. doi: 10.1016/s0399-8320(05)82136-0. [DOI] [PubMed] [Google Scholar]
- 10.Tapner MJ, Jones BE, Wu WM, Farrell GC. Toxicity of low dose azathioprine and 6-mercaptopurine in rat hepatocytes. Roles of xanthine oxidase and mitochondrial injury. Journal of Hepatology. 2004;40(3):454–463. doi: 10.1016/j.jhep.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 11.Russmann S, Zimmermann A, Krähenbühl S, Kern B, Reichen J. Veno-occlusive disease, nodular regenerative hyperplasia and hepatocellular carcinoma after azathioprine treatment in a patient with ulcerative colitis. European Journal of Gastroenterology and Hepatology. 2001;13(3):287–290. doi: 10.1097/00042737-200103000-00013. [DOI] [PubMed] [Google Scholar]
- 12.Read AE, Wiesner RH, LaBrecque DR. Hepatic veno-occlusive disease associated with renal transplantation and azathioprine therapy. Annals of Internal Medicine. 1986;104(5):651–655. doi: 10.7326/0003-4819-104-5-651. [DOI] [PubMed] [Google Scholar]
- 13.Degott C, Rueff B, Kreis H, Duboust A, Potet F, Benhamou JP. Peliosis hepatis in recipients of renal transplants. Gut. 1978;19(8):748–753. doi: 10.1136/gut.19.8.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romagnuolo J, Sadowski DC, Lalor E, Jewell L, Thomson ABR. Cholestatic hepatocellular injury with azathioprine: a case report and review of the mechanisms of hepatotoxicity. The Canadian Journal of Gastroenterology. 1998;12(7):479–483. doi: 10.1155/1998/294752. [DOI] [PubMed] [Google Scholar]
- 15.O'Brien PJ, Siraki AG. Accelerated cytotoxicity mechanism screening using drug metabolising enzyme modulators. Current Drug Metabolism. 2005;6(2):101–109. doi: 10.2174/1389200053586082. [DOI] [PubMed] [Google Scholar]
- 16.Chan K, Jensen NS, Silber PM, O'Brien PJ. Structure-activity relationships for halobenzene induced cytotoxicity in rat and human hepatoctyes. Chemico-Biological Interactions. 2007;165(3):165–174. doi: 10.1016/j.cbi.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 17.O'Brien PJ, Chan K, Silber PM. Human and animal hepatocytes in vitro with extrapolation in vivo. Chemico-Biological Interactions. 2004;150(1):97–114. doi: 10.1016/j.cbi.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 18.MacAllister SL, Young C, Guzdek A, Zhidkov N, O'Brien PJ. Molecular cytotoxic mechanisms of chlorpromazine in isolated rat hepatocytes. Canadian Journal of Physiology and Pharmacology. 2013;91(1):56–63. doi: 10.1139/cjpp-2012-0223. [DOI] [PubMed] [Google Scholar]
- 19.Tafazoli S, Mashregi M, O'Brien PJ. Role of hydrazine in isoniazid-induced hepatotoxicity in a hepatocyte inflammation model. Toxicology and Applied Pharmacology. 2008;229(1):94–101. doi: 10.1016/j.taap.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Tafazoli S, O'Brien PJ. Amodiaquine-induced oxidative stress in a hepatocyte inflammation model. Toxicology. 2009;256(1-2):101–109. doi: 10.1016/j.tox.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Chan K, Lehmler HJ, Sivagnanam M, Feng CY, Robertson L, O'Brien PJ. Cytotoxic effects of polychlorinated biphenyl hydroquinone metabolites in rat hepatocytes. Journal of Applied Toxicology. 2010;30(2):163–171. doi: 10.1002/jat.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplowitz N. Interaction of azathioprine and glutathione in the liver of the rat. Journal of Pharmacology and Experimental Therapeutics. 1977;200(3):479–486. [PubMed] [Google Scholar]
- 23.Deleve LD, Wang X, Kuhlenkamp JF, Kaplowitz N. Toxicity of azathioprine and monocrotaline in murine sinusoidal endothelial cells and hepatocytes: The role of glutathione and relevance to hepatic venoocclusive disease. Hepatology. 1996;23(3):589–599. doi: 10.1002/hep.510230326. [DOI] [PubMed] [Google Scholar]
- 24.Lee AU, Farrell GC. Comparative toxicity of azathioprine to rat hepatocytes and sinusoidal endothelial cells in culture. In: Wisse E, Knook DL, de Zanger R, Fraser R, editors. Cells of the Hepatic Sinusoid. Vol. 7. Leiden, The Netherlands: Kupffer Cell Foundation; 1999. pp. 108–109. [Google Scholar]
- 25.Lee AU, Farrell GC. Mechanism of azathioprine-induced injury to hepatocytes: roles of glutathione depletion and mitochondrial injury. Journal of Hepatology. 2001;35(6):756–764. doi: 10.1016/s0168-8278(01)00196-9. [DOI] [PubMed] [Google Scholar]
- 26.Chocair P. R ., Duley JA, Cameron JS, et al. Purine and Pyrimidine Metabolism in Man VIII. Vol. 370. Berlin, Germany: Springer; 1994. Does low-dose allopurinol, with azathioprine, cyclosporin and prednisolone improve renal transplant immunosuppression? pp. 205–208. (Advances in Experimental Medicine and Biology). [DOI] [PubMed] [Google Scholar]
- 27.Bradford K, Shih DQ. Optimizing 6-mercaptopurine and azathioprine therapy in the management of inflammatory bowel disease. World Journal of Gastroenterology. 2011;17(37):4166–4173. doi: 10.3748/wjg.v17.i37.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katsanos KH, Tsianos EV. Azathioprine/6-mercaptopurinetoxicity: the role of the TPMT gene. Annals of Gastroenterology. 2007;20(4):251–264. [Google Scholar]
- 29.Andrejic J, Rojas-Balcazar J, Dennis M, Berkelhammer C. Azathioprine-induced hypersensitivity hepatitis: tolerance to 6-mercaptopurine. Inflammatory Bowel Diseases. 2010;16(11):1828–1829. doi: 10.1002/ibd.21240. [DOI] [PubMed] [Google Scholar]
- 30.McGovern DPB, Travis SPL, Duley J, Shobowale-Bakre EM, Dalton HR. Azathioprine intolerance in patients with IBD may be imidazole-related and is independent of TPMT activity. Gastroenterology. 2002;122(3):838–839. doi: 10.1053/gast.2002.32124. [DOI] [PubMed] [Google Scholar]
- 31.Gupta P, Gokhale R, Kirschner BS. 6-Mercaptopurine metabolite levels in children with inflammatory bowel disease. Journal of Pediatric Gastroenterology and Nutrition. 2001;33(4):450–454. doi: 10.1097/00005176-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Snow JL, Gibson LE. The role of genetic variation in thiopurine methyltransferase activity and the efficacy and/or side effects of azathioprine therapy in dermatologic patients. Archives of Dermatology. 1995;131(2):193–197. [PubMed] [Google Scholar]
- 33.King PD, Perry MC. Hepatotoxicity of chemotherapeutic and oncologic agents. Drug induced liver disease. Gastroenterology Clinics of North America. 1995;24(4):969–990. [PubMed] [Google Scholar]
- 34.Olfert E, Cross B, McWilliam A. Guide to the Care and Use of Experimental Animals. 1st edition. Ottawa, Canada: Canadian Council on Animal; 1993. [Google Scholar]
- 35.Moldéus P, Högberg J, Orrenius S. Isolation and use of liver cells. Methods in Enzymology. 1978;52:60–71. doi: 10.1016/s0076-6879(78)52006-5. [DOI] [PubMed] [Google Scholar]
- 36.Khan S, O'Brien PJ. 1-Bromoalkanes as new potent nontoxic glutathione depletors in isolated rat hepatocytes. Biochemical and Biophysical Research Communications. 1991;179(1):436–441. doi: 10.1016/0006-291x(91)91389-t. [DOI] [PubMed] [Google Scholar]
- 37.Shaw S, Jayatilleke E. The role of aldehyde oxidase in ethanol-induced hepatic lipid peroxidation in the rat. Biochemical Journal. 1990;268(3):579–583. doi: 10.1042/bj2680579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Possel H, Noack H, Augustin W, Keilhoff G, Wolf G. 2,7-Dihydrodichlorofluorescein diacetate as a fluorescent marker for peroxynitrite formation. FEBS Letters. 1997;416(2):175–178. doi: 10.1016/s0014-5793(97)01197-6. [DOI] [PubMed] [Google Scholar]
- 39.Andersson BS, Aw TY, Jones DP. Mitochondrial transmembrane potential and pH gradient during anoxia. The American Journal of Physiology. 1987;252(4) part 1:C349–C355. doi: 10.1152/ajpcell.1987.252.4.C349. [DOI] [PubMed] [Google Scholar]
- 40.MacAllister SL, Maruf AA, Wan L, Chung E, O'Brien P. Modeling xenobiotic susceptibility to hepatotoxicity using an in vitro oxidative stress inflammation model. Canadian Journal of Physiology and Pharmacology. 2013;91(3):236–240. doi: 10.1139/cjpp-2012-0255. [DOI] [PubMed] [Google Scholar]
- 41.Ou P, Wolff SP. A discontinuous method for catalase determination at 'near physiological' concentrations of H2O2 and its application to the study of H2O2 fluxes within cells. Journal of Biochemical and Biophysical Methods. 1996;31(1-2):59–67. doi: 10.1016/0165-022x(95)00039-t. [DOI] [PubMed] [Google Scholar]
- 42.Eyer P, Podhradský D. Evaluation of the micromethod for determination of glutathione using enzymatic cycling and Ellman's reagent. Analytical Biochemistry. 1986;153(1):57–66. doi: 10.1016/0003-2697(86)90061-8. [DOI] [PubMed] [Google Scholar]
- 43.Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cellular and Molecular Life Sciences. 2003;60(1):6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Y, Shen C, Yin J, Yu J, Meng Q. Azathioprine hepatotoxicity and the protective effect of liquorice and glycyrrhizic acid. Phytotherapy Research. 2006;20(8):640–645. doi: 10.1002/ptr.1920. [DOI] [PubMed] [Google Scholar]
- 45.Raza M, Ahmad M, Gado A, Al-Shabanah OA. A comparison of hepatoprotective activities of aminoguanidine and N-acetylcysteine in rat against the toxic damage induced by azathioprine. Comparative Biochemistry and Physiology C: Toxicology and Pharmacology. 2003;134(4):451–456. doi: 10.1016/s1532-0456(03)00022-x. [DOI] [PubMed] [Google Scholar]
- 46.Kelley EE, Khoo NKH, Hundley NJ, Malik UZ, Freeman BA, Tarpey MM. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radical Biology and Medicine. 2010;48(4):493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butler R, Morris AD, Belch JJ, Hill A, Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35(3):746–751. doi: 10.1161/01.hyp.35.3.746. [DOI] [PubMed] [Google Scholar]
- 48.Desco M, Asensi M, Márquez R, et al. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002;51(4):1118–1124. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- 49.Farquharson CAJ, Butler R, Hill A, Belch JJF, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002;106(2):221–226. doi: 10.1161/01.cir.0000022140.61460.1d. [DOI] [PubMed] [Google Scholar]
- 50.Al-Shamma S, Eross B, McLaughlin S. Use of a xanthine oxidase inhibitor in autoimmune hepatitis. Hepatology. 2013;57(3):1281–1282. doi: 10.1002/hep.26198. [DOI] [PubMed] [Google Scholar]
- 51.Leung Y, Sparrow MP, Schwartz M, Hanauer SB. Long term efficacy and safety of allopurinol and azathioprine or 6-mercaptopurine in patients with inflammatory bowel disease. Journal of Crohn's and Colitis. 2009;3(3):162–167. doi: 10.1016/j.crohns.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Sparrow MP, Hande SA, Friedman S, Cao D, Hanauer SB. Effect of allopurinol on clinical outcomes in inflammatory bowel disease nonresponders to azathioprine or 6-mercaptopurine. Clinical Gastroenterology and Hepatology. 2007;5(2):209–214. doi: 10.1016/j.cgh.2006.11.020. [DOI] [PubMed] [Google Scholar]