

Abstract
The small G proteins Rac1 and RhoA regulate actin cytoskeleton, cell shape, adhesion, migration, and proliferation. Recent studies in our laboratory have shown that NADPH oxidase Nox4-derived ROS are involved in transforming growth factor (TGF)-β1-induced rat kidney myofibroblast differentiation assessed by the acquisition of an α-smooth muscle actin (α-SMA) phenotype and expression of an alternatively spliced fibronectin variant (Fn-EIIIA). Rac1 and RhoA are essential in signaling by some Nox homologs, but their role as effectors of Nox4 in kidney myofibroblast differentiation is not known. In the present study, we explored a link among Rac1 and RhoA and Nox4-dependent ROS generation in TGF-β1-induced kidney myofibroblast activation. TGF-β1 stimulated an increase in Nox4 protein expression, NADPH oxidase activity, and abundant α-SMA and Fn-EIIIA expression. RhoA but not Rac1 was involved in TGF-β1 induction of Nox4 signaling of kidney myofibroblast activation. TGF-β1 stimulated active RhoA-GTP and increased Rho kinase (ROCK). Inhibition of RhoA with small interfering RNA and ROCK using Y-27632 significantly reduced TGF-β1-induced stimulation of Nox4 protein, NADPH oxidase activity, and α-SMA and Fn-EIIIA expression. Treatment with diphenyleneiodonium, an inhibitor of NADPH oxidase, did not decrease RhoA activation but inhibited TGF-β1-induced α-SMA and Fn-EIIIA expression, indicating that RhoA is upstream of ROS generation. RhoA/ROCK also regulated polymerase (DNA-directed) δ-interacting protein 2 (Poldip2), a newly discovered Nox4 enhancer protein. Collectively, these data indicate that RhoA/ROCK is upstream of Poldip2-dependent Nox4 regulation and ROS production and induces redox signaling of kidney myofibroblast activation and may broader implications in the pathophysiology of renal fibrosis.
Keywords: fibrosis, NADPH oxidase, oxidative stress, GTPase, myofibroblast differentiation, Rho kinase, transforming growth factor-β1, polymerase (DNA-directed) δ-interacting protein 2
transforming growth factor (TGF)-β1 is the predominant growth factor responsible for mesenchymal cell synthesis of the matrix during fibrosis (3, 14, 46). A hallmark of mesenchymal cell activation is the acquisition of a myofibroblast phenotype, whereby fibroblasts acquire the expression of smooth muscle proteins, most notably α-smooth muscle actin (α-SMA), and synthesize the extracellular matrix. TGF-β1 regulates differentiation of fibroblasts to a myofibroblast phenotype (3, 11, 54) and subsequent matrix synthesis, particularly of the alternatively spliced isoform of fibronectin (Fn-EIIIA) (3, 6, 54).
The signaling pathways for TGF-β1-induced myofibroblast differentiation are complex and not completely understood. The fibroblast-to-myofibroblast transition is differentially regulated by the cononical Smad pathway (28). In mesangial cells and fibroblasts, TGF-β/Smad signaling (Smad2/3) is tightly controlled by MAPK (Ras/MEK/ERK) signaling cascades (3, 24, 66). Also, ERK and Akt/PKB act as alternative pathways in TGF-β1 signaling of matrix proteins (15, 24, 25, 30), making these three signaling proteins important transduction molecules in myofibroblast differentiation.
ROS are also known to be second messengers in cell activation (19). Recent studies have indicated that NADPH oxidase-generated ROS, particularly from the Nox4 homolog, appears to be responsible for TGF-β signaling of myofibroblast activation and differentiation (3, 10). We have recently observed TGF-β1-induced myofibroblast differentiation and expression of Fn-EIIIA signals through a redox pathway involving Nox4 and ERK in kidney fibroblasts (5).
A variety of studies also have indicated that a small G protein signaling pathway involving Rho family GTPases (RhoA and Rac1) act as molecular switches for a variety of cellular functions that could be involved in myofibroblast differentiation (29). Of these, RhoA and its downstream effector Rho kinase (ROCK) are known to regulate myofibroblast differentiation (26, 50). The RhoA/ROCK pathway constitutes an important mediator for a variety of cell functions, including actin cytoskeleton reorganization, regulation of cell shape, adhesion, migration, and proliferation (26, 51). As with TGF-β1 and Nox4, a close association exists between RhoA/ROCK and myofibroblast activation by regulating the actin cytoskeleton, stress fiber formation, and α-SMA expression in progenitor cells, pericytes, and fibroblasts (20, 26, 47, 51). RhoA/ROCK may also be involved in the regulation of NADPH oxidase signaling (39, 53, 64); however, if or where RhoA/ROCK functions in TGF-β1 signaling of renal myofibroblast differentiation is not known. Also, the small G protein Rac1 is necessary for Nox1 and Nox2 activation and is a component of the active enzymatic complex; however, control of Nox4 function through Rac1 is not clear and remains controversial (3, 8, 18). Nox4 does not require the classical cytoplasmic subunits or Rac1, and activation of Nox4 is undisturbed in cells where Rac1 is knocked down, supporting the notion that Nox4 is Rac independent (41) and suggesting a role for other G protein transduction pathways, perhaps through RhoA. In addition, polymerase (DNA-directed) δ-interacting protein 2 (Poldip2), a Nox4 enhancing protein, induces cytoskeletal changes, including strengthening of focal adhesions and stress fiber formation in vascular smooth muscle cells, suggesting interplay among these signaling proteins (39, 57). A role for RhoA and Poldip2 interactions with Nox4 has not been explored in TGF-β1-induced myofibroblast activation. The present study investigated a role for Rac1 and RhoA/ROCK in Poldip2/Nox4 signaling in TGF-β1-induced kidney myofibroblast differentiation and cell migration.
MATERIALS AND METHODS
Kidney fibroblast cell culture.
Normal rat kidney fibroblast cells (NRK-49F) were obtained from the American Type Culture Collection (ATCC; Rockville, MD). Cells were grown in RPMI medium containing 10% FCS, antibiotics, and antimycotics in a humidified incubator at 37°C under 5% CO2. Cells were grown to 80% confluence and then rendered quiescent by replacing the complete medium with serum-free medium for a period of 24 h, as previously described (5). Quiescent cells were treated with TGF-β1 (1 ng/ml) and then analyzed for Rac1, RhoA/ROCK, and NADPH oxidase signaling pathways. In experiments using chemical inhibitors of the various signaling pathways, compounds were added 0.5–2 h before the addition of TGF-β1 and incubated until the time of harvest. The following inhibitors were used: NSC-23766 (50 μM) for Rac1 (Calbiochem, EMD Biosciences, San Diego, CA), small interfering (si)RNA targeting RhoA (siRhoA; Dharmacon Products, Thermo Scientific, Lafayette, CO), ROCK inhibitor Y-27632 (10 μM, Calbiochem), NADPH oxidase inhibitor diphenyleneiodonium (100 nM, Sigma-Aldrich, St. Louis, MO), and siRNA targeting Poldip2 (siPoldp2; Life Technologies-Invitrogen, Grand Island, NY). Previous studies have indicated that Nox4 signaling is an early event, occurring within 5 min of stimulation by TGF-β1, possibly due to translational mechanisms (5); thus, readout times for signaling proteins Rac1, RhoA, Nox4, and Poldip2 ranged from 5 min to 6 h. Fibroblast-to-myofibroblast differentiation was assessed by the expression of α-SMA and Fn-EIIIA detected by immunoblot analysis and immunohistochemistry at 24 or 48 h after stimulation with TGF-β1.
Adenoviruses.
NH2-terminal Myc-tagged Poldip2 (AdPoldip2) (39) or active wild-type Nox4 (40) and adenovirus with empty vector or noninfected fibroblasts served as controls. NRK-49F cells were infected for 1 h with recombinant adenovirus in serum-free culture medium and incubated for another 2 days in the same medium without virus before being harvested for protein extraction.
Western blot analysis.
Immunoblot analysis was performed as previously described (5, 13). Cells were lysed in RIPA buffer and centrifuged at 4°C at 14,000 rpm for 5 min, and the supernatant was retained and quantified for protein using the Bio-Rad DC protein assay method (Bio-Rad Laboratories, Herculus, CA). After samples had been boiled in sample buffer for 10 min, equal amounts of protein lysate were loaded onto SDS-PAGE gels and electrophoretically separated. Separated proteins were transferred to polyvinylidene difluoride membranes followed by blockade with 5% nonfat dry milk. Protein bands were detected by enhanced chemiluminescence using standard ECL detection methods as recommended by the manufacturer (Amersham Pharmacia Biotech). Signals were visualized by film radiography or using a Bio-Rad Chemidoc XRS photo documentation system (Bio-Rad Laboratories) and represented by column graphs as average intensity percentages of TGF-β1 intensity. The antibodies used were targeted against α-SMA (clone 1A4, Sigma Chemical), Fn-EIIIA (clone IST-9, Abcam, Cambridge, MA), Nox4 [H-300, Santa Cruz Biotechnology, Santa Cruz, CA, or produced in our laboratory (17)], Poldip2 (Santa Cruz Biotechnology), Rac1 (Cytoskeleton, Denver, CO), RhoA (Millipore, Temecula, CA), ROCK, and phosphorylated myosin phosphatase targeting protein-1 (MYPT-1; Santa Cruz Biotechnology). Western blot data were normalized to GAPDH (anti-GAPDH, Novus Biologicals, Littelton, CO) or total nonphosphorylated protein.
Rac1 and RhoA activation assays.
Activation of Rac1 and RhoA was determined using GTP pulldown assay kits according to the manufacturers' instructions (Rac1: Cytoskeleton and RhoA: Millipore). After cells were lysed, active GTP-Rac1 was pulled down from 150 μg of cleared lysate using 10 μg of PAK-PBD beads. Similarly, active GTP-RhoA was bound using 15 μg of glutathione-agarose bound glutathione-S-transferase-tagged rhotekin-RBD. For both assays, samples were incubated at 4°C on a rotator for 1 h, washed three times in PBS, resuspended in Laemmli buffer, and then resolved with 12.5% SDS-PAGE. Lysates and precipitates were analyzed by immunoblot analysis using mouse monoclonal antibody specifically against Rac1 or RhoA.
ROS detection by 2′,7′-dichlorodihydrofluorescein assay.
Kidney fibroblasts were cultured in a multiwell LAB-TEK no. 1 coverslide chambers (Nalge Nunc, Naperville, IL) and then made quiescent at 30–40% confluence as described above. To explore a role for RhoA/ROCK in endogenous ROS generation, cells were treated with Y-27632 (10 μM) 1 h before TGF-β1 stimulation, and 2′,7′-dichlorodihydrofluorescein (DCF) fluorescence was examined as an indicator of ROS generation, as previously described (5, 17). The peroxide-sensitive fluorescent probe DCF diacetate (10 μM, Molecular Probes, Eugene, OR) was added to cells under various conditions, and photographs were taken 30 min after the addition of TGF-β1 using an Olympus Fluoview confocal laser scanning inverted microscope equipped with a 40A planfluor objective. DCF fluorescence was detected with an excitation wavelength of 488 nm, and DCF emission was detected using a 510- to 550-nm band-pass filter.
NADPH oxidase assay.
NADPH oxidase was measured by the lucigenin-enhanced chemiluminescence method as previously described (5, 17). Cells were treated with NSC-23766, Y-27632, or vehicle and then stimulated with TGF-β1 for 30 min. Cells were washed in PBS and homogenized in lysis buffer containing 20 mM KH2PO4 (pH 7.0), 1 mM EGTA, 1 mM PMSF, 10 μg/ml aprotinin, and 0.5 μg/ml leupeptin. Homogenate (100 μl) was added to 900 μl of 50 mM potassium phosphate buffer (pH 7.0) containing 1 mM EGTA, 150 mM sucrose, 5 μM lucigenin (Sigma Chemical) as an electron acceptor, and 100 μM NADPH (Roche Diagnostics, Indianapolis, IN) as an electron donor. Photon emission expressed as relative light units was measured every 30 s for 5 min in a Modulus luminometer (Turner BioSystems, Promega Biosystems, Sunnyvale, CA). There was no measurable activity in the absence of NADPH. Protein content was measured using Bio-Rad protein assay reagent. NADPH oxidase activity was expressed as the rate of relative chemiluminescence units per milligram of protein per minute. H2O2 is the major product of Nox4, and thus measuring superoxide underestimates its activity.
Immunofluorescence microscopy.
Immunofluorescence microscopy was used to examine the relative roles of Rac1 and RhoA/ROCK in myofibroblast α-SMA and Fn-EIIIA expression. Quiescent kidney fibroblast cells grown in multiwell plastic chamber slides were treated with NSC-23766 or Y-27632 followed by TGF-β1 and then incubated for 24 or 48 h. At the termination of the study time, cells were washed twice with ice-cold PBS and fixed in methanol at −20°C for 5 min. After a brief rinse, cells were blocked with 0.1% BSA in PBS and then stained with Cy3-conjugated mouse anti-α-SMA. Fn-EIIIA was detected using indirect immunofluorescence as previously described (5). Cy3-conjugated donkey anti-mouse served as secondary antibody (Chemicon and Millipore). Stained cells were washed with PBS, mounted with coverslips, viewed by epifluorescence, and photographed using an Olympus AX70 Research microscope equipped with a DP-70 digital camera (Melville, NY).
siRNA transfection.
To specifically examine a role for RhoA in kidney fibroblast ROS generation, cells were transfected with siGenome smart pool rat RhoA or a nontargeting RNA pool as a control using DharmaFECT 2 transfection agent (Thermo/Dharmacon Research, Lafayette, CO). Likewise, a role for Poldip2 in kidney fibroblast activation was examined using Stealth siRNA targeting rat Poldip2. Nontargeting siRNA served as a control. Cells were grown to 60% confluence in six-well culture plates, and the medium was then replaced with antibiotic/antimycotic-free RPMI containing siRNA in DharmaFECT 2 reagent at a concentration of 100 nM. After an incubation period of 72 h, transfected cells were made quiescent and then treated with TGF-β1. Subsequently, NADPH oxidase activity assay, Nox4, Poldip2, α-SMA, and Fn-EIIIA were examined as described above.
Statistical analysis.
All experiments were repeated at least three times. Experiments were analyzed by one-way ANOVA followed by Newman-Keuls multiple-comparison post hoc test performed by GraphPad Prism 5 software (GraphPad Prism, San Diego, CA). Significance was assigned at P ≤ 0.05.
RESULTS
TGF-β1-induced activation of kidney myofibroblast differentiation is not Rac1 dependent.
Cellular signaling by several NADPH oxidase homologs requires the cytosolic subunit Rac1; however, an essential role for this small G protein in TGF-β-induced Nox4 signaling of kidney myofibroblast differentiation is less certain (see the discussion above). To determine if Rac1 regulates myofibroblast activation, Rac1 GTP loading after stimulation with TGF-β1 was examined at time intervals that we have previously reported to stimulate an early increase in Nox4 protein expression in kidney myofibroblasts (5). Short-term treatment of cells with TGF-β1 had no effect on the level of active Rac1-GTP, as determined by a pulldown assay (Fig. 1, A and B). The Rac1 inhibitor NSC-23766 substantially reduced active Rac1-GTP levels (Fig. 1B) but had no effect on TGF-β1-induced increases in Nox4 protein (Fig. 1C) or NADPH oxidase activity as assayed by lucigenin-enhanced chemiluminescence (Fig. 1D). *P < 0.05 vs. control. #P < 0.05 vs. TGF-β1 according to ANOVA.
Fig. 1.

Transforming growth factor (TGF)-β1-induced myofibroblast signaling via Nox4 is independent of Rac-GTP. A: TGF-β1 had no effect on active Rac-GTP from 5 to 60 min, a time course known to stimulate myofibroblast differentiation (5) (see other figures). In additional experiments, cells were incubated with or without NSC-23766 (50 μM), an inhibitor of Rac, 2 h before the addition of TGF-β1 or diluent. B–D: Rac-GTP loading (B), Nox4 protein (C), and NADPH oxidase activity (D) were examined after 30 min of incubation and compared with diluent controls. NSC-23766 reduced active Rac-GTP but had no effect on Nox4 expression (C) or NADPH oxidase activity (D). Cont, control. *P < 0.05 vs. control. #P < 0.05 vs. TGF-β1 according to ANOVA.
As observed in a previous study (5), TGF-β1 stimulated kidney fibroblasts to transition to a myofibroblast phenotype as assessed by increased expression of α-SMA and Fn-EIIIA. In resting fibroblasts, there was negligible expression of α-SMA and Fn-EIIIA as assessed by immunoblot analysis (Fig. 2, A and B) and immunohistochemistry (Fig. 2, C and G). TGF-β1 substantially increased both α-SMA and Fn-EIIIA (Fig. 2, A, B, D, and H). TGF-β1 also induced myofibroblast transition characterized by the acquisition of α-SMA into well-defined stress fibers as well as Fn-EIIIA substrata expression viewed by immunofluorescence relative to controls. NSC-23766 did not reduce TGF-β1-induced increased expression of α-SMA and Fn-EIIIA by immunoblot analysis or immunohistochemistry (Fig. 2, A, B, E, and I). These results indicate that Rac1 does not participate in TGF-β1-induced Nox4-mediated kidney myofibroblast differentiation.
Fig. 2.

TGF-β1-induced kidney myofibroblast differentiation is not regulated through Rac-1. Kidney fibroblasts were stimulated with TGF-β1 or diluent with or without pretreatment with NSC-23766, as described in Fig. 1. Myofibroblast differentiation was examined 2 days later by detection of α-smooth muscle actin (α-SMA) and alternatively spliced fibronectin variant (Fn-EIIIA) protein using Western blot analysis (A and B) and immunofluorescence microscopy (C–J). TGF-β1 stimulated increased α-SMA protein incorporation in stress fibers by immunohistochemistry (D). Incubation of cells with NSC-23766 did not reduce TGF-β1-induced increased expression of α-SMA or Fn-EIIIA, as shown in Western blots (A and B) or by immunohistochemistry (E and I). Similarly, α-SMA-positive stress fibers were not altered by NSC-23766 (E). *P < 0.05 vs. control according to ANOVA.
TGF-β1 rapidly stimulates RhoA and ROCK activation.
Because RhoA GTPase is a known target of TGF-β1 (20, 48), we then focused on a role for this G protein in signaling kidney myofibroblast differentiation. We observed that TGF-β1 stimulated Rho GTP loading as early as 5 min after the addition of TGF-β1 to kidney fibroblasts (Fig. 3A). TGF-β1 continued increasing the level of RhoA-GTP up to 1 h, peaking at 30 min. Total RhoA remained unaffected by TGF-β1. Similarly, protein expression of ROCK, an immediate effector of activated Rho-GTP, increased at 5 min after the addition of TGF-β1, again peaking at 30 min (Fig. 3B). Additionally, TGF-β1 stimulated phosphorylation of MYPT-1, the immediate substrate for ROCK, used as an indicator of its activity, closely correlating with the activation of RhoA and ROCK (Fig. 3C).
Fig. 3.
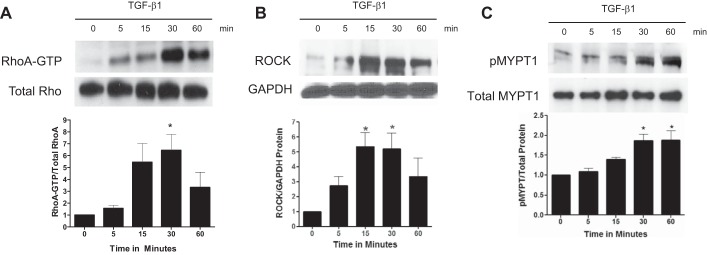
TGF-β1 rapidly activates the RhoA/Rho kinase (ROCK) signaling pathway. A: TGF-β1 stimulated myofibroblast signaling of active RhoA-GTP from 5 to 60 min. B and C: TGF-β1 also stimulated the downstream effector ROCK (B) and phosphorylation of the ROCK substrate myosin phosphatase targeting protein-1 (MYPT-1; C), as detected by immunoblot analysis of lysates collected 5–30 min after the addition of TGF-β1 or diluent. pMYPT-1, phosphorylated MYPT-1. *P < 0.05 vs. control according to ANOVA.
RhoA/ROCK mediates TGF-β1-induced myofibroblast activation assessed by α-SMA and Fn-EIIIA expression.
The above experiments indicated that the activation of the RhoA/ROCK pathway is an early event in kidney fibroblast activation by TGF-β1. To further examine a role for this pathway in kidney myofibroblast differentiation, cells were exposed to siRhoA before the addition of TGF-β1. The results showed that downregulation of RhoA protein with siRhoA inhibited RhoA expression relative to nontargeting siRNAs (Fig. 4D). siRhoA significantly reduced the expression of both α-SMA and Fn-EIIIA relative to controls as detected by immunoblot analysis (Fig. 4, A–C). Similarly, the effect of ROCK inhibitor Y-27632 on myofibroblast differentiation was examined (Fig. 5, A–F). Y-27632 inhibited TGF-β1-stimulated increases in both α-SMA and Fn-EIIIA expression by Western blot analysis (Fig. 5, A–C), which was substantiated by immunofluorescence histochemistry (Fig. 5, D–F). These results indicate that RhoA/ROCK plays a role in myofibroblast activation by TGF-β1.
Fig. 4.

TGF-β1-induced myofibroblast differentiation requires RhoA signaling. Cells were exposed to small interfering (si)RNA to RhoA (siRhoA) or nontargeting (nt)RNA for 72 h. TGF-β1 or diluent was then added, and lysates were collected 24 h later to examine markers of myofibroblast differentiation by immunoblot analysis. TGF-β1-enhanced α-SMA and Fn-EIIIA protein expression was reduced by siRhoA relative to ntRNA (A–C). D: verification of the effectiveness of siRhoA to inhibit RhoA protein relative to ntRNA. *P < 0.05 vs. ntRNA control. #P < 0.05 vs. ntRNA + TGF-β1 according to ANOVA.
Fig. 5.

TGF-β1-induced myofibroblast differentiation is blocked by ROCK inhibition. A–C: addition of Y-27632 (10 μM), an inhibitor of ROCK, 1 h before TGF-β1 also blocked myofibroblast differentiation, as illustrated by the reduced expression of α-SMA and Fn-EIIIA relative to controls. D, E, H, and I: TGF-β1 stimulated an increase in α-SMA protein expression incorporation in stress fibers (E) as well as Fn-EIIIA protein (I) relative to controls (D and H), as illustrated by immunofluorescence histochemistry. F and J: The ROCK inhibitor Y-27632 blocked α-SMA (F) and Fn-EIIIA (J) protein expression. F and G: diluent had no effect on the expression of α-SMA or Fn-EIIIA. *P < 0.05 vs. control. #P < 0.05 vs. TGF-β1 according to ANOVA.
Role of RhoA/ROCK in ROS-mediated myofibroblast activation through NADPH oxidase.
Our previous study (5) indicated that TGF-β1-induced kidney myofibroblast differentiation is dependent on Nox4-derived ROS. To determine a role for the RhoA/ROCK pathway in TGF-β1-induced Nox4/ROS stimulation of myofibroblast differentiation, the effects of siRhoA and Y-27632 on Nox4 protein expression and NADPH oxidase activity were investigated. As previously shown, TGF-β1 stimulated a significant increase in Nox4 protein and NADPH oxidase activity (Fig. 6, A–F). TGF-β1-induced increases in Nox4 protein expression were significantly reduced by siRhoA (Fig. 6, A and B) as well as by Y-27632 (Fig. 6, D and E). Similarly, TGF-β1-enhanced NADPH oxidase activity was abrogated by siRhoA (Fig. 6C) as well as Y-27632 (Fig. 6F). TGF-β1 stimulated a significant increase in intracellular ROS, as revealed by increased DCF fluorescence detected by confocal microscopy (Fig. 6G). Y-27632 nearly completely inhibited DCF fluorescence (Fig. 6G). Diphenyleneiodonium, an inhibitor of NADPH oxidase and other flavin-containing oxidases, had no effect on RhoA-GTP loading by TGF-β1 (Fig. 6, H and I) but inhibited NADPH oxidase activity (Fig. 6J), indicating that NADPH oxidase-derived ROS are downstream of RhoA. Overall, the above data show that RhoA/ROCK regulates TGF-β1-induced myofibroblast activation through Nox4-dependent ROS generation.
Fig. 6.

The RhoA/ROCK pathway signals Nox4-derived ROS. A and C: kidney fibroblasts were treated with siRhoA or ntRNA for 72 h followed by stimulation TGF-β1 or diluent, and Nox4 expression (A) and NADPH oxidase activity (C) were then examined 30 min later. A–C: siRhoA inhibited the TGF-β1-induced increased expression of Nox4 protein (A and B) and NADPH oxidase activity (C). D–F: similarly, cells treated with the ROCK inhibitor Y-27632 before the addition of TGF-β1 blocked the increased expression of Nox4 protein (D and E) and NADPH oxidase activity (F) relative to diluent controls. A–F: inhibitors alone had no effect on basal levels of Nox4 protein or NADPH oxidase activity. G: ROS generation, detected by 2′,7′-dichlorodihydrofluorescein (DCF) fluorescence, was also inhibited by Y-27632 within 30 min after incubation with TGF-β1. H–J: inhibition of Nox4 using diphenyleneiodonium (DPI) had no effect on TGF-β1-induced activation of RhoA-GTP (H and I) but reduced NADPH oxidase activity (J), indicating that RhoA is upstream of Nox4. *P < 0.05 vs. control. #P < 0.05 vs. TGF-β1 according to ANOVA.
Role of Poldip2/Nox4 in TGF-β1-induced myofibroblast activation.
A role for Poldip2, a known regulator of Nox4, in kidney myofibroblast differentiation was examined by transduction of fibroblasts with AdPoldip2 and compared with control adenovirus without construct. Likewise, the effect of Nox4 on Poldip2 expression and myofibroblast differentiation was examined using Nox4 adenovirus. The results showed that overexpression of Poldip2 protein examined 2 days after infection enhanced basal expression of Nox4 (Fig. 7, A–C) and enhanced NADPH oxidase activity (Fig. 7D) relative to controls. Similarly, overexpression of Poldip2 protein via AdPoldip2 enhanced the basal expression of myofibroblast differentiation markers α-SMA and Fn-EIIIA (Fig. 7, A, E, and F). Conversely, we found that overexpression of Nox4 protein via AdNox4 enhanced basal expression of Poldip2 (Fig. 7, G–I) and enhanced NADPH oxidase activity (Fig. 7J) relative to controls. Similarly, overexpression of Nox4 protein enhanced the basal expression of myofibroblast differentiation markers α-SMA and Fn-EIIIA (Fig. 7, G, K, and L). We also found that treatment of cells with TGF-β1 increased Poldip2 and Nox4 protein expression (Fig. 8, A–C) and NADPH oxidase activity (Fig. 8D) relative to controls as early as 30 min after stimulation. Knockdown of Poldip2 using siPoldip2 inhibited TGF-β1-mediated increases in Nox4 protein and NADPH oxidase activity as well as downstream markers of myofibroblast activation (α-SMA and Fn-EIIIA) relative to nontargeting controls (Fig. 8, A, E, and F).
Fig. 7.

Polymerase (DNA-directed) δ-interacting protein 2 (Poldip2) regulates myofibroblast differentiation via Nox4. A–F: kidney fibroblasts were infected with AdPoldip2 or adenovirus with green fluorescent protein (AdGFP) over a 48-h period, and the relative effects of overexpression of Poldip2 on Nox4 protein (A–C), NADPH oxidase activity (D), and α-SMA and Fn-EIIIA (A, E, and F) expression were examined. Increased expression of Poldip2, as determined by Myc and Poldip protein by immunoblot analysis, resulted in enhanced expression of Poldip2 and Nox4 protein and NADPH oxidase activity as well as the myofibroblast differentiation markers α-SMA and Fn-EIIIA relative to control. G–L: infection of kidney fibroblasts with AdNox4 significantly increased Nox4 and Poldip2 protein (G–I), NADPH oxidase activity (J), and the myofibroblast differentiation markers α-SMA and Fn-EIIIA (G, K, and L) relative to control (AdGFP). *P < 0.05 vs. AdGFP vs. control.
Fig. 8.

TGF-β1-induced myofibroblast differentiation is regulated via signaling via Poldip2 and Nox4. A–D: kidney fibroblasts were treated with siRNA to Poldip2 (siPoldip2) or ntRNA for 72 h followed by TGF-β1 or diluent, and Poldip2 and Nox4 expression (A–C) and NADPH oxidase activity (D) were then examined 30 min later. siPoldip2 inhibited TGF-β1-induced increases in Poldip2 and Nox4 protein expression as well as NADPH oxidase activity relative to control (ntRNA). siPoldip2 also inhibited TGF-β1-induced myofibroblast differentiation, as illustrated by reductions in α-SMA (A and E) and Fn-EIIIA (A and F) relative to ntRNA. *P < 0.05 vs. ntRNA control. #P < 0.05 vs. ntRNA + TGF-β1 according to ANOVA.
RhoA regulates Poldip2/Nox4 in TGF-β1-induced myofibroblast activation.
To explore a role for the RhoA/ROCK pathway in the regulation of Poldip2/Nox4 by TGF-β1, cells were transfected with siRhoA or treated with Y-27632 before the addition of TGF-β1. The results showed that inhibition of either RhoA or ROCK resulted in a reduction in TGF-β1-induced increases in Poldip2 protein (Fig. 9, A and B). Overexpression of Poldip2 in kidney fibroblasts infected with AdPoldip2 had no effect on RhoA-GTP loading or expression of ROCK or phosphorylated MYPT-1 protein relative to cells infected with adenovirus with green fluorescent protein, indicating that Poldip2 is downstream of RhoA/ROCK signaling (Fig. 9, C–G). Also, inhibition of ROCK using Y-27632 had no effect on AdPoldip2-induced overexpression of α-SMA or Fn-EIIIA proteins (Fig. 9, H–J). These results indicate the specificity of Poldip2 in myofibroblast differentiation by bypassing the Rho/ROCK pathway. Collectively, the results above indicate that the RhoA/ROCK pathway plays a significant role in regulating myofibroblast activation through modulation of Poldip2 and Nox4.
Fig. 9.

RhoA/ROCK regulate TGF-β1-induced Poldip2 protein expression. A: kidney fibroblasts were treated with siRhoA or ntRNA for 72 h followed by TGF-β1 or diluent, and lysates were then collected 30 min later for immunoblot analysis of Poldip2 protein. siRhoA inhibited TGF-β1-induced increases in Poldip2 protein relative to ntRNA control. B: in additional experiments, kidney fibroblasts were treated with the ROCK inhibitor Y-27632 before the addition of TGF-β1 or diluent, and Poldip2 protein expression was determined by immunoblot analysis. Inhibition of ROCK reduced TGF-β1-induced increases in Poldip2 protein expression relative to controls. C–G: overexpression of Poldip2 in kidney fibroblasts infected with AdPoldip2 had no effect on RhoA-GTP loading or expression of ROCK and pMYPT1 relative to cells exposed to AdGFP, indicating that Poldip2 is downstream of RhoA/ROCK signaling. H–J: inhibition of ROCK using Y-27632 had no effect on AdPoldip2-induced overexpression of α-SMA (H and I) or Fn-EIIIA (H and J), showing the specificity of Poldip2 in myofibroblast differentiation. *P < 0.05 vs. control. #P < 0.05 vs. TGF-β1 according to ANOVA.
DISCUSSION
TGF-β1 signaling of myofibroblast differentiation is known to occur through two pathways: Smad and ERK (1, 42). Details of these transduction pathways have proved to be complex, may vary in different cell types, and remain incompletely understood (3, 28). Our previous study (5) showed that TGF-β1-induced kidney myofibroblast differentiation, characterized by α-SMA and Fn-EIIIA expression, is regulated by Nox4-derived ROS via a signaling cascade involving Smad3, Nox4-generated ROS, and ERK1/2. The present study shows that Rac1 is not involved in downstream Nox4 regulation of kidney myofibroblast differentiation. Rather, the small GTPase RhoA/ROCK pathway regulates Nox4-dependent ROS generation and subsequent downstream cellular events in TGF-β1-induced kidney myofibroblast activation. TGF-β1 rapidly activates RhoA and ROCK, which, in turn, phosphorylates MYPT-1, an indicator of ROCK activity. siRhoA and the ROCK inhibitor Y-27632 both abolished TGF-β1-induced increases in Nox4 protein and NADPH oxidase activity as well as expression markers of myofibroblast differentiation (α-SMA and Fn-EIIIA). These results and those of our previous study collectively indicate that TGF-β1 induces kidney myofibroblast differentiation as assessed by α-SMA expression and Fn-EIIIA synthesis via a redox signaling cascade involving the TGF-β1 receptor (TGF-β1R)1/RhoA/ROCK/Nox4/Poldip2 pathway (Fig. 10). As in our previous studies (3, 5), activation of these signaling events occurred within minutes, consistent with translational mechanisms in Nox4 signaling. A novel role for Poldip2 in signaling of kidney myofibroblast differentiation was indicated by the finding that Poldip2 enhanced Nox4 protein and NADPH oxidase activity and subsequent α-SMA and Fn-EIIIA expression. Conversely, siPoldip2 inhibited TGF-β1-induced Nox4 upregulation, ROS generation, and myofibroblast differentiation. This demonstrates that Poldip2 is required for TGF-β1-induced Nox4-dependent ROS production and subsequent myofibroblast activation and acts as an upstream regulator of Nox4 expression and ROS release. Here, we provide the first evidence that Poldip2 is a downstream effector of RhoA in that siRhoA and Y27643 inhibited Poldip2 protein expression and myofibroblast differentiation.
Fig. 10.

Proposed signaling cascade involving RhoA/ROCK/Poldip2/Nox4 in TGF-β1-induced kidney myofibroblast activation.
It is well established that TGF-β1 binds TGF-βR type II (TGF-βRII) and subsequently phosphorylates a type I receptor (TGF-βRI) subunit, forming a heterodimeric complex (1, 42) that signals through a canonical pathway involving the Smad family of transcriptional activators in myofibroblasts. TGF-βR1 phosphorylates Smad2/3, which subsequently complexes with Smad4 and translocates to the nucleus, where the dimer binds to the promoter region of the α-SMA gene (3, 28). ROS play a significant role in the progression of fibrotic disease (23, 32, 52). ROS have been identified as intermediates in TGF-β signaling of mesangial cells or fibroblasts during fibrosis or progressive disease, which has received considerable recent interest (3). Indeed, ROS in the form of superoxide and hydrogen peroxide are directly linked to transmodulation of fibroblasts to α-SMA myofibroblasts (67) and are important mediators of fibrogenic cellular responses including hypertrophy, migration, proliferation, apoptosis, and regulation of the extracellular matrix (5, 17, 23, 56, 60). NADPH oxidases are important sources of ROS involved in both normal physiological functions and oxidative stress (3). Of the NADPH oxidases of the Nox family, the Nox4 homolog is now recognized as a principal source of ROS generation and subsequent redox-dependent pathological processes in progressive fibrotic disease (3). TGF-β1 is a major regulator of Nox4 protein expression and Nox4-dependent ROS production in a number of cell types, including smooth muscle cells, endothelial cells, hepatocytes, and fibroblasts (3). The results of the present study in kidney fibroblasts also agree with our previous studies and other published studies indicating a close relationship between ROS generated by NADPH oxidase and α-SMA expression and a myofibroblast phenotype in adventitial fibroblast and vascular smooth muscle cells (3, 5, 9, 10, 55). We (5) have previously shown that kidney myofibroblasts express NADPH oxidase homologs Nox2 and Nox4, with the latter appearing to be the most important in the generation of ROS in response to TGF-β1 and downstream signaling of ERK1/2 and α-SMA and Fn-EIIIA expression.
The mechanisms by which Nox enzymes are regulated in cardiovascular or renal cells and how they generate ROS are not fully understood. A number of regulatory subunits of Nox enzymes have been identified. Importantly, p22phox is required as a docking and maturation subunit for the homologs Nox1, Nox2, and Nox4 (3, 7). Activation mechanisms for Nox1 are similar to those of Nox2 but involve complex formation with regulatory cytosolic subunits Nox organizer 1, Nox activator 1, and the small G protein Rac1 (3, 7, 35, 36). The small GTPases act as molecular switches, and it is well established that the Nox family of NADPH oxidases is principally regulated through a Rac-dependent pathway (8, 18, 29). Nox1 and Nox2 are tightly regulated by Rac1 through its binding to phox and Nox organizer 1 or Nox activator 1 subunits in Nox2 and Nox1 assembly. A role for Rac1 in Nox4 activation is uncertain. Although it has been suggested that Rac1 is implicated in the control of Nox4 function in mesangial and endothelial cells (8, 18), several studies in heterologous systems have shown that Nox4 does not require the recruitment of regulatory cytoplasmic subunits and Rac1 to be active. Nox4-mediated ROS generation is undisturbed in Rac1 knockdown (41) or chemical inhibition (4), supporting the notion that Nox4 is Rac1 independent and suggests alternate transduction pathways. Unlike mesangial cells, in which Rac promotes TGF-β1-stimulated collagen type 1 (30) and Rac1 appears to be involved in Nox4 signaling (18), kidney myofibroblast do not use this GTPase in TGF-β1 signaling of Nox4 activation of α-SMA and Fn-EIIIA. TGF-β1 did not stimulate Rac1 activity, and inhibition of Rac1 had no effect on TGF-β1-induced Nox4-dependent ROS production or α-SMA and Fn-EIIIA expression in kidney myofibroblasts.
Rather, our data indicate that TGF-β1/Nox4 signaling of myofibroblast differentiation implicates RhoA GTPase and ROCK. Of particular interest are experiments identifying the RhoA/ROCK system as an important signaling system regulating the actin cytoskeleton and stress fiber formation (39). The RhoA/ROCK system plays an important role in regulating the myofibroblast phenotype in mesenchymal progenitor cells, pericytes, and fibroblasts (12, 21, 34, 58). However, studies showing an association of ROS signaling and the RhoA/ROCK pathway in vitro are limited (29, 39, 64, 65). Studies in endothelial cells have indicated that Rac1 is necessary for downstream activation of RhoA and subsequent Nox4 activation and cell proliferation, whereas Rac1-dependent Nox2 activity was responsible for cytoskeletal rearrangement via JNK (63, 64). In vascular smooth muscle cells, RhoA was found to be downstream of Nox4 and associated with focal contacts and cell migration (39). Our experiments in kidney fibroblasts showed that RhoA is upstream of Nox4, as in endothelial cells, but similar to smooth muscle cells, in that TGF-β1-induced kidney fibroblast differentiation to a myofibroblast phenotype is associated with the formation of α-SMA incorporated in stress fibers. The present study placing RhoA/ROCK upstream of Nox oxidase is consistent with reports showing that the ROCK inhibitor fasudil significantly reduced angiotensin II-induced Nox4 upregulation and ROS production in the vascular endothelium (27) as well as diabetes-mediated increases in Nox4 mRNA expression in the renal cortex (16).
An important recent discovery in smooth muscle cells shows that Poldip2 associates with Nox4 and stabilizes focal adhesions and stress fiber formation through RhoA (39) and is involved in the maintenance of a constitutively expressed smooth muscle phenotype. To date, our experiments with renal fibroblasts are the only other studies in which a Poldip2/Nox4 interaction has been identified and show regulation of the Nox4/Poldip2 interaction in response to TGF-β1. Interestingly, our experiments showed that TGF-β1 induces a reverse order of signaling events in kidney myofibroblasts compared with smooth muscle cells, where Poldip2 and Nox4 are downstream of RhoA. A RhoA/Poldip2/Nox4 sequence of transduction may indicate a novel outside-in pathway for ROS signaling at the level of the substratum. The differences between vascular smooth muscle cells and kidney myofibroblasts may reflect different states of stress fiber and matrix expression in the two cell types. Smooth muscle cells constitutively express α-SMA, whereas kidney fibroblast differentiation into myofibroblasts appears to be regulated by TGF-β1 via RhoA/ROCK/Poldip2/Nox4 interactions. Poldip2 has been shown to associate with Nox4 and control ROS production by Nox4 (39, 57). Here, we show that Poldip2 controls Nox4 expression and NADPH oxidase activity. Moreover, our data suggest that TGF-β1-mediated upregulation of Nox4 protein and NADPH oxidase activity depends on the induction of Poldip2 expression by TGF-β1. A TGF-β1-dependent increase in Poldip2 most likely translates to an increase in Nox4 expression since Poldip2 controls the expression of the oxidase. It is also conceivable that the increased Poldip2 levels promoted by TGF-β1 are also a means to enhance Nox4-dependent ROS production. Importantly, all these events take place downstream of RhoA/ROCK activation by TGF-β1. Also, we show, for the first time, that Nox4 can regulate Poldip2 expression, suggesting a functional association or positive feedback loop in the activation of these two proteins.
The above experiments indicate that RhoA/ROCK-dependent regulation of Poldip2/Nox4 is involved in TGF-β1-induced kidney myofibroblast activation in vitro. A number of studies have also indicated that RhoA/ROCK may be a key initiator in oxidative stress during disease where ROCK inhibitors blocked the progression of cardiac hypertrophy, liver fibrosis, diabetic nephropathy, renal fibrosis, and glomerulosclerosis (16, 31, 43–45). Of interest are the observations that oxidative stress or Nox1 and Nox4 are inhibited by ROCK inhibitors in several of these models (16, 45). Furthermore, myofibroblasts during renal fibrosis originate, at least in part, from a perivascular adventitium (2, 13, 38, 62). This observation is consistent with the known role of NADPH oxidase-generated ROS in adventitial fibroblast activation in aortic perivascular disease (22, 49). Thus, a role for this RhoA/ROCK/Poldip2/Nox4 pathway in interstitial myofibroblast encroachment in vivo during renal fibrosis is plausible but awaits further investigation.
GRANTS
This work was supported by grants from the Veterans Administration Merit Review Program, National Institutes of Health (NIH) Grant RO1-DK-080106, and the NIH George O'Brien Kidney Center and Morphology Core.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.M., M.P., Y.G., and J.L.B. conception and design of research; N.M. and M.P. performed experiments; N.M., M.P., K.K.G., Y.G., and J.L.B. analyzed data; N.M., M.P., K.K.G., Y.G., and J.L.B. interpreted results of experiments; N.M., M.P., and J.L.B. prepared figures; N.M., M.P., and J.L.B. drafted manuscript; N.M., M.P., K.K.G., Y.G., and J.L.B. approved final version of manuscript; K.K.G., Y.G., and J.L.B. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors thank Fredyne Springer and Christine Spencer for technical contributions and Dr. Karen Block for the rabbit anti-Nox4 antibody.
REFERENCES
- 1.Attisano L, Wrana JL. Signal transduction by the TGF-β superfamily. Science 296: 1646–1647, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Barnes JL, Glass WF. Renal interstitial fibrosis: a critical evaluation of the origin of myofibroblasts. In: Experimental Models for Renal Diseases: Pathogenesis and Diagnosis, edited by Herrera GA. Basel: Karger, 2011, p. 73–93 [DOI] [PubMed] [Google Scholar]
- 3.Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int 79: 944–956, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basuroy S, Bhattacharya S, Leffler CW, Parfenova H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-α in cerebral vascular endothelial cells. Am J Physiol Cell Physiol 296: C422–C432, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE, Barnes JL. NAD(P)H oxidase mediates TGF-β1-induced activation of kidney myofibroblasts. J Am Soc Nephrol 21: 93–102, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borsi L, Castellani P, Risso AM, Leprini A, Zardi L. Transforming growth factor-β regulates the splicing pattern of fibronectin messenger RNA precursor. FEBS Lett 261: 175–178, 1990 [DOI] [PubMed] [Google Scholar]
- 7.Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radic Biol Med 49: 687–706, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Chai D, Wang B, Shen L, Pu J, Zhang XK, He B. RXR agonists inhibit high-glucose-induced oxidative stress by repressing PKC activity in human endothelial cells. Free Radic Biol Med 44: 1334–1347, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 27: 42–48, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 97: 900–907, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 122: 103–111, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diah S, Zhang GX, Nagai Y, Zhang W, Gang L, Kimura S, Hamid MR, Tamiya T, Nishiyama A, Hitomi H. Aldosterone induces myofibroblastic transdifferentiation and collagen gene expression through the Rho-kinase dependent signaling pathway in rat mesangial cells. Exp Cell Res 314: 3654–3662, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Faulkner JL, Szcykalski LM, Springer F, Barnes JL. Origin of interstitial fibroblasts in an accelerated model of angiotensin II (Ang II)-induced renal fibrosis. Am J Pathol 167: 1193–1205, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaedeke J, Peters H, Noble NA, Border WA. Angiotensin II, TGF-β, and renal fibrosis. Contrib Nephrol 135: 153–160, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Ghosh C, Abboud HE. Tyrosine phosphorylation-dependent PI3 kinase/Akt signal transduction regulates TGF-β-induced fibronectin expression in mesangial cells. Cell Signal 16: 31–41, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Gojo A, Utsunomiya K, Taniguchi K, Yokota T, Ishizawa S, Kanazawa Y, Kurata H, Tajima N. The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur J Pharmacol 568: 242–247, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 280: 39616–39626, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Gorin Y, Ricono JM, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Nox4 mediates angiotensin II-induced activation of Akt/protein kinase B in mesangial cells. Am J Physiol Renal Physiol 285: F219–F229, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept 91: 21–27, 2000 [DOI] [PubMed] [Google Scholar]
- 20.Harvey KA, Paranavitana CN, Zaloga GP, Siddiqui RA. Diverse signaling pathways regulate fibroblast differentiation and transformation through Rho kinase activation. J Cell Physiol 211: 353–363, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Haudek SB, Gupta D, Dewald O, Schwartz RJ, Wei L, Trial J, Entman ML. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc Res 83: 511–518, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haurani MJ, Cifuentes ME, Shepard AD, Pagano PJ. Nox4 oxidase overexpression specifically decreases endogenous Nox4 mRNA and inhibits angiotensin II-induced adventitial myofibroblast migration. Hypertension 52: 143–149, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haurani MJ, Pagano PJ. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: bellwether for vascular disease? Cardiovasc Res 75: 679–689, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Hayashida T, Decaestecker M, Schnaper HW. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-β-dependent responses in human mesangial cells. FASEB J 17: 1576–1578, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Hayashida T, Wu MH, Pierce A, Poncelet AC, Varga J, Schnaper HW. MAP-kinase activity necessary for TGFβ1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J Cell Sci 120: 4230–4240, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Heasman SJ, Ridley AJ. Mamalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, Ichiki T, Takahashi S, Takeshita A. Long-term inhibition of Rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circ Res 93: 767–775, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 127: 526–537, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Hordijk PL. Regulation of NADPH oxidases. The role of Rac proteins. Circ Res 98: 453–462, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Hubchak SC, Sparks EE, Hayashida T, Schnaper HW. Rac1 promotes TGF-β-stimulated mesangial cell type I collagen expression through a PI3K/Akt-dependent mechanism. Am J Physiol Renal Physiol 297: F1316–F1323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanda T, Wakino S, Hayashi K, Homma K, Ozawa Y, Saruta T. Effect of fasudil on Rho-kinase and nephropathy in subtotally nephrectomized spontaneously hypertensive rats. Kidney Int 64: 2009–2019, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol 6: 395–423, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawamura S, Miyamoto S, Brown JH. Initiation and transduction of stretch-induced RhoA and Rac1 activation through caveolae: cytoskeletal regulation of ERK translocation. J Biol Chem 278: 31111–31117, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Kolyada AY, Riley KN, Herman IM. Rho GTPase signaling modulates cell shape and contractile phenotype in an isoactin-specific manner. Am J Physiol Cell Physiol 285: C1116–C1121, 2003 [DOI] [PubMed] [Google Scholar]
- 35.Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med 43: 319–331, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2: 329–333, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173: 1617–1627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 105: 249–259, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog NOX4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol 24: 1844–1854, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martyn KD, Frederick LM, von LK, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18: 69–82, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Massague J, Gomis RR. The logic of TGF-β signaling. FEBS Lett 580: 2811–2820, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Moriyama T, Kawada N, Nagatoya K, Takeji M, Horio M, Ando A, Imai E, Hori M. Fluvastatin suppresses oxidative stress and fibrosis in the interstitium of mouse kidneys with unilateral ureteral obstruction. Kidney Int 59: 2095–2103, 2001 [DOI] [PubMed] [Google Scholar]
- 44.Nagatoya K, Moriyama T, Kawada N, Takeji M, Oseto S, Murozono T, Ando A, Imai E, Hori M. Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction. Kidney Int 61: 1684–1695, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Nishikimi T, Akimoto K, Wang X, Mori Y, Tadokoro K, Ishikawa Y, Shimokawa H, Ono H, Matsuoka H. Fasudil, a Rho-kinase inhibitor, attenuates glomerulosclerosis in Dahl salt-sensitive rats. J Hypertens 22: 1787–1796, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Okuda S, Languino LR, Ruoslahti E, Border WA. Elevated expression of transforming growth factor-beta and proteoglycan production in experimental glomerulonephritis. Possible role in expansion of the mesangial extracellular matrix. J Clin Invest 86: 453–462, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parizi M, Howard EW, Tomasek JJ. Regulation of LPA-promoted myofibroblast contraction: role of Rho, myosin light chain kinase, and myosin light chain phosphatase. Exp Cell Res 254: 210–220, 2000 [DOI] [PubMed] [Google Scholar]
- 48.Peng F, Zhang B, Wu D, Ingram AJ, Gao B, Krepinski JC. TGF-β-induced RhoA activation and fibronectin production in mesangial cells requires caveolae. Am J Physiol Renal Physiol 295: F153–F164, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rey FE, Pagano PJ. The reactive adventitia. Fibroblast oxidase in vascular function. Arterioscler Thromb Vasc Biol 22: 1962–1971, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol 11: 471–477, 2001 [DOI] [PubMed] [Google Scholar]
- 51.Riento K, Ridley AJ. ROCKs: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4: 446–456, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol 18: 2439–2446, 2007 [DOI] [PubMed] [Google Scholar]
- 53.San Martín A, Griendling KK. Redox control of vascular smooth muscle migration. Antioxid Redox Signal 12: 625–640, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J Cell Biol 142: 873–842, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen WL, Gao PJ, Che ZQ, Ji KD, Yin M, Yan C, Berk BC, Zhu DL. NAD(P)H oxidase-derived reactive oxygen species regulate angiotensin-II induced adventitial fibroblast phenotypic differentiation. Biochem Biophys Res Commun 339: 337–343, 2006 [DOI] [PubMed] [Google Scholar]
- 56.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270: 296–299, 1995 [DOI] [PubMed] [Google Scholar]
- 57.Sutliff RL, Amanso AM, Parastatidis I, Dikalova AE, Hansen L, Datla SR, Long JS, El-Ali AM, Gleason JG, Jr, Taylor WR, Hart CM, Griendling KK, Lassègue B. Polymerase delta interacting protein 2 sustains vascular structure and function. Arterioscler Thromb Vasc Biol 33: 2154–2161, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vardouli L, Vasilaki E, Papadimitriou E, Kardassis D, Stournaras C. A novel mechanism of TGFβ-induced actin reorganization mediated by Smad proteins and Rho GTPases. FEBS J 275: 4074–4087, 2008 [DOI] [PubMed] [Google Scholar]
- 59.Venkatesan B, Mahimainathan L, Das F, Ghosh-Choudhury N, Ghosh Choudhury G. Downregulation of catalase by reactive oxygen species via PI 3 kinase/Akt signaling in mesangial cells. J Cell Physiol 211: 457–467, 2007 [DOI] [PubMed] [Google Scholar]
- 60.Weber DS, Seshiah PN, Taniyama Y, Griendling KK. Src-dependent migration of vascular smooth muscle cells by PDGF is reactive oxygen species dependent. Circulation 106: S260, 1995 [Google Scholar]
- 61.Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol 3: 950–957, 2001 [DOI] [PubMed] [Google Scholar]
- 62.Wiggins R, Goyal M, Merritt S, Killen PD. Vascular adventitial cell expression of collagen I messenger ribonucleic acid in anti-glomerular basement membrane antibody-induced crescentic nephritis in the rabbit. A cellular source for interstitial collagen synthesis in inflammatory renal disease. Lab Invest 68: 557–565, 1993 [PubMed] [Google Scholar]
- 63.Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol 30: 3553–3568, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu RF, Ma Z, Myers DP, Terada LS. HIV-1 Tat activates dual Nox pathways leading to independent activation of ERK and JNK MAP kinases. J Biol Chem 282: 37412–37419, 2007 [DOI] [PubMed] [Google Scholar]
- 65.Wu RF, Xu YC, Ma Z, Nwariaku FE, Sarosi GA, Jr, Terada LS. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol 171: 893–904, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yue J, Mulder KM. Requirement of Ras/MAPK pathway activation by transforming growth factor β for transforming growth factor β1 production in a Smad-dependent pathway. J Biol Chem 275: 30765–30773, 2000 [DOI] [PubMed] [Google Scholar]
- 67.Zalewski A, Shi Y. Vascular myofibroblasts. Lessons from coronary repair and remodeling. Arterioscler Thromb Vasc Biol 17: 417–422, 1997 [DOI] [PubMed] [Google Scholar]