

Abstract
Impaired adipogenesis renders an adipose tissue unable to expand, leading to lipotoxicity and conditions such as diabetes and cardiovascular disease. While factors important for adipogenesis have been studied extensively, those that set the limits of adipose tissue expansion remain undetermined. Feeding a Western-type diet to apolipoprotein E2 knock-in mice, a model of metabolic syndrome, produced 3 groups of equally obese mice: mice with normal glucose tolerance, hyperinsulinemic yet glucose-tolerant mice, and prediabetic mice with impaired glucose tolerance and reduced circulating insulin. Using proteomics, we compared subcutaneous adipose tissues from mice in these groups and found that the expression of PTRF (polymerase I and transcript release factor) associated selectively with their glucose tolerance status. Lentiviral and pharmacologically overexpressed PTRF, whose function is critical for caveola formation, compromised adipocyte differentiation of cultured 3T3-L1cells. In human adipose tissue, PTRF mRNA levels positively correlated with markers of lipolysis and cellular senescence. Furthermore, a negative relationship between telomere length and PTRF mRNA levels was observed in human subcutaneous fat. PTRF is associated with limited adipose tissue expansion underpinning the key role of caveolae in adipocyte regulation. Furthermore, PTRF may be a suitable adipocyte marker for predicting pathological obesity and inform clinical management.—Perez-Diaz, S., Johnson, L. A., DeKroon, R. M., Moreno-Navarrete, J. M., Alzate, O., Fernandez-Real, J. M., Maeda, N., Arbones-Mainar, J. M. Polymerase I and transcript release factor (PTRF) regulates adipocyte differentiation and determines adipose tissue expandability.
Keywords: senescence, glucose intolerance, telomeres
Obese individuals are epidemiologically associated with a wide array of metabolic risk factors (1, 2). However, evidence suggests that 20–30% of the obese population can remain as metabolically healthy individuals on the basis of a beneficial adipose tissue growth (3, 4). The subcutaneous adipose tissue is a key endocrine organ able to expand during times of caloric excess with a paramount importance in energy homeostasis (5). In addition, it has been proposed that impaired adipogenesis can cause a failure to sequester lipotoxic fatty acids (6). This deregulated adipose tissue ultimately leads to conditions such as metabolic syndrome, diabetes, dyslipidemia, and cardiovascular disease (reviewed in ref. 7).
The aim of our study was to investigate the biogenesis of subcutaneous adipose tissue, to determine the factors that set the limits of its expansion. This study employed a murine model of diet-induced obesity, the dyslipidemic apolipoprotein E2 (APOE2) knock-in mice (8, 9), whose endogenous murine Apoe gene has been substituted by the human APOE*2 gene (10). These mice also present additional features of the metabolic syndrome such as fatty liver susceptibility (11), hypercoagulant activity (12), and aortic plaque deposition (10). With 2-dimensional fluorescence difference gel electrophoresis (2D-DIGE) and mass spectrometry analyses we identified that protein expression of polymerase I and transcript release factor (PTRF) in the subcutaneous adipose tissue is selectively associated with the glucose tolerance status of the obese mice. We demonstrated that PTRF overexpression compromises adipocyte differentiation in vitro and that PTRF mRNA levels are directly associated with cellular senescence in human adipose tissue.
MATERIALS AND METHODS
Mice
Male mice homozygous for replacement of the endogenous Apoe gene with the human APOE*2 on a C57BL/6 background (10) were fed a Western-type diet (WD) containing 21% (w/w) fat and 0.2% (w/w) cholesterol (TD88137; Teklad. Madison, WI, USA) over 12 wk, starting at 8 wk of age. At the end of the intervention period, mice were denied access to food for 4 h and euthanized. Subcutaneous adipose tissue, aortic root, and liver were collected and frozen in liquid N2. Plasma concentrations of nonesterified free fatty acids (NEFAs), glucose, and cholesterol were determined using kits from Wako (Richmond, VA, USA). Triglyceride and insulin concentrations were determined using kits from Stanbio (San Antonio, TX, USA) and Crystal Chem (Chicago, IL, USA), respectively. Plasma adiponectin was measured using an ELISA as described previously (13). Oral glucose tolerance tests (OGTTs) were performed as described previously (6). The bases of the mouse hearts were collected and processed for aortic cross-sectional analysis as described previously (14). Livers were homogenized, and triglyceride concentration determined as described previously (15). The animals were handled under protocols approved by the Institutional Animal Care and Use Committees of the University of North Carolina at Chapel Hill.
Proteomics
Proteomics analysis was performed with the internal control methodology using 2D-DIGE as described previously (16). Quantitative differential protein expression analysis was performed with the DeCyder 2D software (GE Healthcare, Piscataway, NJ, USA). After determination of differential protein expression using as selection criteria the Student's t test with P < 0.05, selected protein spots were subjected to protein identification by matrix-assisted laser desorption/ionization tandem time-of-flight (MALDI-TOF/TOF) mass spectrometry at the University of North Carolina Proteomics Core Facility as described previously (17).
Cells
3T3-L1 murine preadipocytes (no. CL-173; American Type Culture Collection, Rockville, MD, USA), a gift from Dr. Moreno-Aliaga (Universidad de Navarra, Pamplona, Spain), and human embryonic kidney 293 (HEK 293T; CRL-3216, American Type Culture Collection), a gift from Dr. Fernandez-Vizarra (University of Cambridge, Cambridge, UK), were used. Cells were grown and preadipocytes transformed into mature adipocytes as described previously (15). Adipocytes were considered mature at 8–10 d postdifferentiation.
Lentivirus production
The open-reading frame encoding the murine Ptrf1 tagged by monomeric enhanced green fluorescent protein (EGFP; ref. 18) was excised from the pcDNA5/FRT/TO vector (27709; Addgene, Cambridge, MA, USA) with PmeI digest and blunt-end ligated into the second-generation transfer vector pWPXLd (12258; Addgene) in which the GFP sequence had been substituted by a puromycin resistance cassette (pWPXLd-ires-PuroR) (19). HIV-1-derived lentiviral particles were produced by transfecting cloroquine-pretreated HEK 293T cells with the transfer vector along with the attenuated packaging vector psPAX2 (12260; Addgene) and the vesicular stomatitis virus glycoprotein (VSV-G)-encoding plasmid pMD2.G (12259; Addgene), in combination with the transfection enhancer IBAfect (IBA, Göttingen, Germany). Lentivirus-containing supernatants of transfected cells were collected 48 h later and passed through 0.45 μm filters (ThermoFisher Scientific, San Jose, CA, USA). Viral load for each preparation was determined by Dr. Martinez-Sapiña (Department Of Microbiology, Hospital Universitario Miguel Servet, Zaragoza, Spain) using 1:1000 diluted supernatants with the COBAS TaqMan HIV-1 Test, v. 2.0 system (Roche Molecular Systems, Branchburg, NJ. USA). Similar numbers of lentiviral particles of the PTRF1-mGFP/pWPXLd-ires-PuroR or the empty pWPXLd-ires-PuroR vector were then used to transduce the preconfluent 3T3-L1 cells supplemented with polybrene (Sigma-Aldrich, St. Louis, MO, USA), and subsequently cells were selected for puromycin resistance. Two different 3T3-L1 cell lines were established, constitutively expressing either the empty pWPXLd-ires-PuroR or the PTRF1-mGFP/pWPXLd-ires-PuroR.
Western blot, lipid accumulation, and cell cycle analysis
Differentiated 3T3-L1 cells were lysed and immunoblotted for total acyl-CoA carboxylase (ACC; C83B10; Cell Signaling, Beverly, MA, USA), phospho-ACC (Ser79; 3661; Cell Signaling), PTRF (sc-133934; Santa Cruz Biotecnology, Santa Cruz, CA, USA), adipocyte fatty acid-binding protein (AFABP; sc-18661; Santa Cruz Biotecnology), caveolin-1 (sc-894; Santa Cruz Biotecnology), and actin (sc-1615; Santa Cruz Biotecnology), followed by the appropriate secondary antibodies. Lipid content was quantified by adding Nile Red (Acros, Geel, Belgium) to 3T3-L1 mature adipocytes, and fluorescence was measured (λex 485 nm, λem 572 nm) with a Synergy HT spectrophotometer (Biotek, Winooski, VT, USA) or using a FACSAria cytometer (λem 604 nm; Becton Dickinson, San Jose, CA. USA). Oil Red O stain on 3T3-L1 cells was performed as described previously (20). Cell cycle was investigated 2 d after reaching 100% confluence. Briefly, 3T3-L1 cells were induced to differentiate with the adipogenic cocktail (15) and collected at selected time points to evaluate by flow cytometry the distribution of cells at specific cell cycle stages, as described previously (21), using a FACSArray (Becton Dickinson). Each fluorescence-activated cell-sorting analysis sample contained 104 cells.
Radioisotopes
Glucose uptake was assayed with deoxy-d-glucose as described previously (22). For the isoproterenol-stimulated lipolysis, 3T3-L1 adipocytes seeded in 12-well plates at d 8 postdifferentiation were incubated overnight with 0.5 ml/well of culture medium supplemented with 1 nCi/μl [9,10-3H(N)]-palmitic acid (NET043005MC; Perkin-Elmer, Madrid, Spain). Lipolysis was initiated with the addition of 10 μM isoproterenol (Aleudrina; Reig Jofre, Barcelona. Spain) in serum-free DMEM containing 2% BSA (Sigma-Aldrich). The conditioned medium was collected at 0, 30, 60, 90, and 120 min, and then analyzed for fatty acid release in a Rackbeta liquid scintillation counter (Model 1209; LKB Wallac, Turku. Finland) after addition of Ultimate Gold scintillant (Perkin-Elmer). De novo lipogenesis was assayed in 3T3-L1 adipocytes seeded in 12-well plates at d 8 postdifferentiation and then incubated with 1 ml/well of culture medium supplemented with 1 nCi/μl [3H]-acetic acid (NET003005MC; Perkin-Elmer). After overnight incubation, lipids were extracted, and radioactivity was assayed as described above.
Human adipose tissues
Adipose tissue samples (93 visceral and 85 subcutaneous fat depots) from a group of Caucasian participants, with different degree of obesity [body mass index (BMI) between 20 and 58 kg/m2], were analyzed. Body weight had been stable for ≥3 mo before the study, and all subjects gave written informed consent. The cohort was recruited at the Endocrinology Service of the Hospital Universitari Dr. Josep Trueta (Girona, Spain) and the hospital's institutional review board approved the protocols. Exclusion criteria have been previously detailed (23). Adipose tissues were obtained during elective surgical procedures (cholecystectomy, surgery of abdominal hernia, and gastric bypass surgery) and immediately frozen in liquid nitrogen. RNA preparation, reverse transcription, and quantitative PCR, as well as measurement of telomere length, were performed as described previously (23, 24).
Statistical analysis
All statistical analyses were performed using SPSS 17.0 software (SPSS, Chicago, IL, USA). Results are expressed as means ± sem. One-way ANOVA followed by Student's t test for post hoc analysis was used unless otherwise stated. To test the strength of the correlations between variables, the Pearson product-moment correlation coefficients were calculated.
RESULTS
Metabolic effects of WD in APOE2 knock-in mice
Chronic administration of a WD is an established model of diet-induced obesity and type 2 diabetes in mice (9). A cohort of 34 male APOE2 knock-in mice was fed a WD for 12 wk, resulting in a large disparity of body weights, ranging from 25 to 55 g (39.3±7.6, mean±sd). To assess the extent of obesity, another cohort of male APOE2 knock-in mice was fed normal chow for the same amount of time, and their average body weight was 29.8 ± 1.8 (mean±sd, n=7). The metabolic status of these obese mice was assessed by measuring fasting plasma insulin and performing OGTTs. As Fig. 1A illustrates, body weight correlated with plasma insulin (r=0.59, n=34, P<0.001) rather than with glucose tolerance (r=0.17, n=34, P=0.34), suggesting an important role for insulin action on body weight. We next divided the body weight variable into quintiles and defined mice within the 2 top quintiles as obese (body weight >42 g). Interestingly, 3 different subgroups emerged within the obese group: normal glucose-tolerant (NGT) mice, with low area under the curve (AUC) from the OGTT and low plasma insulin; hyperinsulinemic (INS) mice, with low AUC and high plasma insulin; and impaired glucose tolerance (IGT) mice, with the highest AUC values but relatively low plasma insulin, likely due to damaged β cells producing less insulin.
Figure 1.

WD effects on glucose homeostasis and subcutaneous adipose proteome in APOE2 knock-in mice.A) OGTTs. Each bubble corresponds to 1 mouse; bubble size denotes glucose intolerance, measured as area under the curve (AUC) of the OGTT. B) Heat map representation of the protein levels of the 11 PTRF spots found to be differentially expressed in the proteomics analysis. Data shown are normalized to the normal glucose tolerance (NGT) group, set at 100%. Color intensity indicates growing concentration levels. INS, hyperinsulinemic; IGT, impaired glucose tolerance. C) Representative 2-dimensional gel electrophoresis gels of the 3 obese groups indicating the differential protein expression of PTRF, as identified by MALDI-TOF mass spectrometry, as described in Material and Methods.
No statistical differences were found in the average body weight among the 3 obese subgroups, despite the differences in their glucose tolerance and insulin levels (Table 1). As expected, AUC was correlated with fasting plasma glucose levels, pointing to the differences in carbohydrate metabolism among these groups. No differences in plasma lipids or adiponectin were observed on WD feeding. All groups displayed high but similar plasma cholesterol, triglycerides, and NEFAs. Neither were the net amounts of subcutaneous, visceral (gonadal), and liver fat significantly different among groups. We note that these subgroups are small, and some of these parameters might be underpowered to detect significant variations. However, the ratio of visceral to subcutaneous fat significantly increased in the IGT group compared to the NGT group, indicating a failure of subcutaneous fat to expand and subsequent lipid accumulation in the viscera in the IGT group. Indeed, this ratio was significantly correlated with the AUC, a marker of glucose intolerance, for the entire cohort of WD-fed mice, including lean as well as obese mice (r=0.490, n=34, P<0.01). According to the adipose tissue expandability hypothesis, once the capacity of subcutaneous fat to grow and store lipids is compromised, lipotoxicity and other obesity-associated metabolic complications arise (25). Consistently, IGT mice displayed greater atherosclerotic lesions, measured in cross-sectional slices at the level of the aortic sinus, providing evidence for atherogenic differences within groups with similar plasma lipids and obesity.
Table 1.
Morphometric values and plasma biochemistry of APOE2 knock-in mice fed WD for 12 wk
| Parameter | NGT | INS | IGT |
|---|---|---|---|
| n | 4 | 4 | 5 |
| Body weight (g) | 47.1 ± 2.9 | 48.9 ± 1.9 | 46.5 ± 1.2 |
| Insulin | 1.36 ± 0.07# | 2.93 ± 0.19 | 1.75 ± 0.16# |
| Glucose (mg/dl) | 130 ± 11 | 155 ± 6¶ | 205 ± 7* |
| AUC | 23,192 ± 768 | 27,401 ± 358 | 35,812 ± 2369* |
| Triglycerides (mg/dl) | 301 ± 34 | 268 ± 9 | 312 ± 28 |
| Cholesterol (mg/dl) | 526 ± 60 | 495 ± 14 | 531 ± 34 |
| NEFA (meq/L) | 0.60 ± 0.10 | 0.75 ± 0.10 | 0.62 ± 0.08 |
| Adiponectin (μg/ml) | 27.10 ± 3.43 | 27.82 ± 2.88 | 27.38 ± 1.73 |
| Visceral fat (g) | 1.96 ± 0.18 | 2.28 ± 0.21 | 2.11 ± 0.15 |
| Subcutaneous fat (g) | 2.38 ± 0.30 | 2.37 ± 0.33 | 1.98 ± 0.28 |
| Liver fat (mg triglyceride/mg protein) | 1.03 ± 0.32 | 1.06 ± 0.20 | 1.49 ± 0.34 |
| Visceral/subcutaneous ratio | 0.835 ± 0.040 | 0.985 ± 0.061 | 1.125 ± 0.115¶ |
| Aortic lesion (μm2×100) | 919 ± 177 | 1370 ± 353 | 3513 ± 2369¶ |
Values are means ± sem.
P < 0.05 vs. INS;
P < 0.05 vs. NGT;
P < 0.05 vs. NGT and INS.
Proteomic characterization of the subcutaneous adipose tissue
The subcutaneous fraction of adipose tissue of the 3 equally obese subgroups (NGT, INS, IGT) was further examined by 2D-DIGE to determine whether proteome changes were associated to tissue expandability. Quantitative differential protein expression analysis was performed on 3 individual samples from each group. An internal control was run in each gel, allowing the quantitative determination of protein expression changes. Individual subcutaneous fractions had 124 polypeptides whose expression levels were significantly up- or down-regulated in the INS and IGT mice, compared to the NGT group. The amino acid sequence of the isolated peptides determined by mass spectrometry showed that, of the 124 differentially regulated spots, 90 were identified, and 11 were fragments of the protein PTRF (Supplemental Table S2 and Fig. 1B). Among these fragments, 10 were post-translationally modified forms of the PTRF protein, and 1 (s2519) was a degraded PTRF form with smaller molecular mass and different isoelectric point (Supplemental Table S2 and Fig. 1C). The average fold change of PTRF (mean±sem relative to NGT mice) was significantly lower (0.67±3) in the INS group and significantly higher (1.43±11) in the IGT mice group. Interestingly, of the 90 known spots, 27 were identified as membrane-associated proteins, highlighting the important role of this cellular domain in the adipocyte metabolism.
PTRF is increased in insulin-resistant 3T3-L1 adipocytes
To confirm that PTRF increases in an insulin resistance context, mature 3T3-L1 adipocytes were desensitized to the effects of insulin via dexamethasone treatment (26). Dexamethasone has a direct effect on glucose transporters and, as observed in our proteomics analysis, reduced the insulin-mediated glucose-uptake of the 3T3-L1 adipocytes as PTRF protein levels increased (Fig. 2A, B). Caveolin-1, a structural protein of caveolae, mirrored PTRF increase, suggesting that insulin resistance was accompanied by a global remodeling of caveolae. In agreement with the notion that compromised adipose tissue expansion leads to maladaptive obesity, this insulin desensitization diminished the number of functional adipocytes, as demonstrated by the dexamethasone-induced reduction of the adipocyte differentiation marker AFABP (Fig. 2A, C) and the dedifferentiation of mature 3T3-L1 adipocytes over 4 d treatment (Fig. 2D).
Figure 2.

Effect of insulin resistance on PTRF levels in 3T3-L1 adipocytes. 3T3-L1 mature adipocytes were treated with 10 μm dexamethasone for 8 d. A) Representative immunoblots for PTRF, caveolin-1 (Cav1), AFABP, and actin protein levels. B, C) Fold change of PTRF (dashed trace) and insulin-mediated glucose uptake (IM-GU; solid trace) (B) and fold change of Cav1 (dashed trace) and AFABP (solid trace) (C), after actin normalization over the 8 d of dexamethasone treatment. Data (means±sem) are representative of 2 independent experiments, each performed in triplicate. *P < 0.05 vs. d 0; Mann-Whitney U test. D) Dexamethasone (10 μM) treatment induces dedifferentiation of mature 3T3-L1 adipocytes over 4 d treatment. The same area was photographed on 4 consecutive days.
Lentiviral overexpression of PTRF leads to impaired adipocyte differentiation and lipogenesis
To study the role of PTRF in adipocyte metabolism, we infected 3T3-L1 cells with lentiviral particles containing the full-length Ptrf cDNA in the pWPXLd-ires-PuroR (PTRF cells) vector and compared them to 3T3-L1 control cells infected with lentivirus carrying the empty pWPXLd-ires-PuroR vector. Cells constitutively expressing these transgenes were selected and induced to differentiate into adipocytes. Mature adipocytes (8 d postdifferentiation) similarly increased PTRF levels compared with preadipocytes in both control and PTRF lines (1.2- and 1.4-fold, respectively). However, mature PTRF-adipocytes had a 1.6- and 10-fold increase of PTRF protein and mRNA levels, respectively, compared to control adipocytes (Fig. 3A, B). We stained differentiated adipocytes with Oil Red O (Fig. 3C) and quantitated changes in intracellular lipid storage using a fluorescence spectroscopy (data not shown) and flow cytometry (Fig. 3D). Both methods showed that intracellular lipids decreased by ∼5-fold in PTRF-transduced cells. Interestingly, PTRF adipocytes were larger and had bigger lipid droplets, indicating that PTRF overexpression does affect intracellular lipid accumulation, as well as lipid storage management. To determine whether these differences appeared during the adipocyte differentiation or later, in matured adipocytes, we also investigated the early stages of the adipocyte differentiation. During the first hours of the adipocyte differentiation cells undergo a mitotic clonal expansion and adipogenic genes are expressed in a tightly regulated manner (27). PTRF and control preadipocytes treated with the adipogenic cocktail equally progressed synchronously through the cell cycle. Beginning at 12 h postdifferentition, a large fraction of both cell populations started mitosis, peaking at ≈20 h, and then reverting to G1 status by 24 h, indicating completion of DNA replication (Supplemental Fig. S3). In addition, we performed mRNA analysis at cell confluence (48 h before differentiation) and during the early stages of adipocyte differentiation. PTRF-transfected cells with were less able to stimulate primary adipogenic genes such as Cebpb and Cebpd on addition of the adipogenic cocktail during the first hours of differentiation. These findings prove that PTRF overexpression hinders all the stages of 3T3-L1 differentiation to some extent.
Figure 3.

PTRF overexpression compromises 3T3-L1 differentiation. 3T3-L1 preadipocytes were transduced with either empty lentivirus (control) or lentivirus encoding PTRF (PTRF) and subsequently differentiated into adipocytes. A) Immunoblot for PTRF expression. B) PTRF mRNA levels in adipocytes. C) Oil Red-O stained adipocytes to visualize lipid accumulation at ×1 (top panels) and ×40 view (bottom panels). D) Flow cytometric analysis of adipocytes stained with Nile Red. Percentages of subpopulations of nondifferentiated preadipocytes (red) and lipid-laden adipocytes (green) are superimposed. Data are representative of 3 replicates. E) Time-lapse analysis of CEBPβ and CEBPδ mRNA expression during early adipogenesis. Preadipocytes were stimulated to differentiate at time 0 and harvested at the indicated time points. Individual assessments are means ± sem of 5 replicates. *P < 0.05.
To elucidate the consequences of this compromised differentiation, we examined the possibility that PTRF overexpression could be altering lipogenic pathways, rendering cells unable to expand. 3T3-L1 cells were incubated overnight with [3H]-acetate, a substrate for de novo lipogenesis, and the amount of radioactivity incorporated into the cellular lipid fraction was measured. Consistent with the notion of an impaired differentiation, PTRF-transduced cells displayed ∼4 times less newly synthesized lipids than control adipocytes (Fig. 4A). The mechanism of impaired lipogenesis was investigated by analyzing ACC, a key enzyme for the biosynthesis of fatty acids (28). ACC protein levels were significantly reduced (20%) in PTRF adipocytes compared to control adipocytes. PTRF overexpression did not change ACC phosphorylation (p-ACC). However, when the ratio between p-ACC and total ACC was considered, we observed a higher ratio on PTRF transduction, compared with control cells (1.30 vs. 1.00, P=0.01; Fig. 4B). On the other hand, PTRF cells had a 30% reduction of the fatty acid carrier and marker of lipid accumulation, AFABP/aP2, compared with the control 3T3-L1 cells. In addition, at d 8 postdifferentiation, PTRF cells showed a dramatic decrease in expression of adipocyte late-expression genes, such as Pparγ and Afabp, markers of terminal differentiation (Fig. 4C). These results indicate that PTRF overexpression leads to decreased adipocyte differentiation and impaired lipid accumulation.
Figure 4.
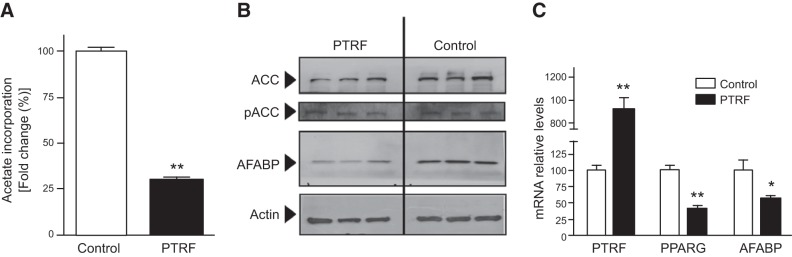
PTRF overexpression impairs lipogenesis in vitro. A) Lipogenesis in mature 3T3-L1 adipocytes, measured as the uptake of [3H]-acetate. B) Western blot analysis of components of lipogenic pathway in adipocytes. C) mRNA levels of PTRF, PPARG, and AFABP assessed by qPCR. Values are means ± sem (n=5). *P < 0.05, **P < 0.01.
Overexpressing PTRF in adipocytes stimulates lipolysis
Next, we examined the relationship between PTRF and insulin-stimulated glucose transport activity. Insulin stimulation increased [3H]-deoxyglucose uptake compared to unstimulated cells, but this phenomenon was not significantly affected by PTRF overexpression (Fig. 5A). To test the lipolytic capacity of PTRF, adipocytes were loaded with [3H]-palmitate overnight, which was incorporated into triglycerides. Subsequently, the release of radiolabeled palmitate in the absence or presence of the β-adrenergic isoproterenol was determined. No differences in lipolysis between PTRF and control cells were observed in the absence of isoproterenol (not shown). However, 234% more palmitate was released from PTRF adipocytes compared to control adipocytes after a 2 h incubation period with isoproterenol (AUC=488 vs. 1146 cpm/ng triglyceride/min for control and PTRF, respectively; Fig. 5B). These results support the important role of PTRF in regulating lipolysis, as previously suggested (29, 30).
Figure 5.
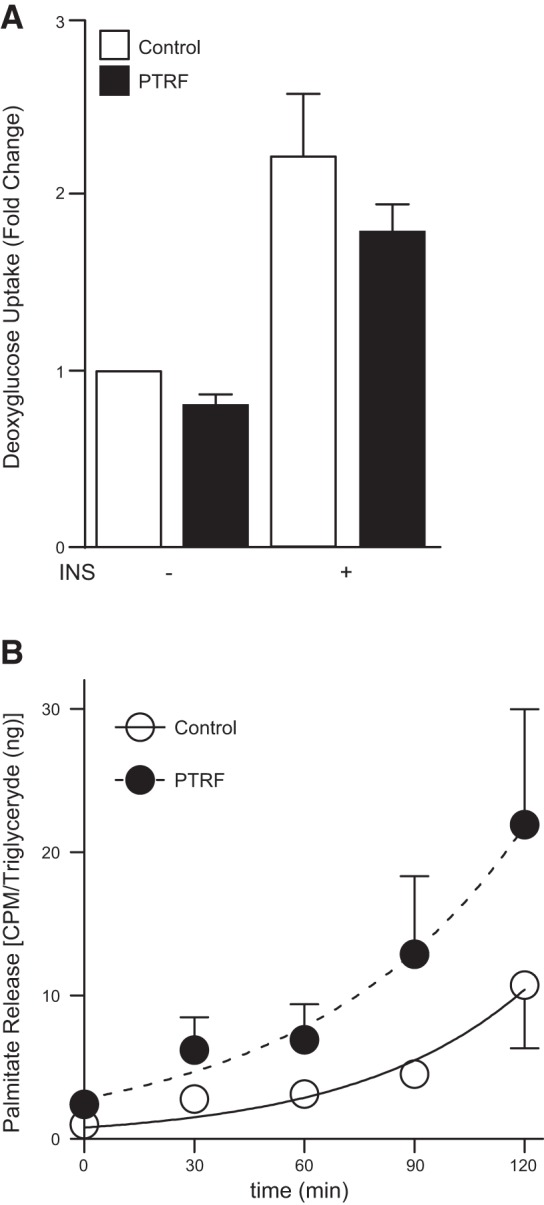
Analysis of insulin-mediated glucose uptake and isoproterenol-stimulated lipolysis from 3T3-L1 adipocytes overexpressing PTRF. A) Serum-starved 3T3-L1 adipocytes in the absence (−INS) and presence (+INS) of 500 nM insulin were incubated with [3H]-deoxyglucose, and the incorporated radioactivity was determined. B) Efflux of palmitate from the indicated 3T3-L1 serum-starved adipocytes treated with 10 μM isoproterenol for 2 h. Differences were tested with the extra sum-of-squares F test method. Data of both analyses are pooled from 5 separate experiments. P = 0.028.
PTRF expression positively associates with adipocyte remodeling and senescence markers in human adipose tissue
Next, we studied the relationship between PTRF expression and mRNA levels of selected markers of adipocyte functionality in human adipose tissue from a cohort of Caucasian participants (BMI between 20 and 58 kg/m2). As described previously (31), a positive correlation between mRNA levels of PTRF and caveolin, both caveolar components, was found in both visceral and subcutaneous adipose depots (Table 2). Adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) release free fatty acids from adipose triacylglycerol stores (32). Both ATGL and its coactivator CGI58 (33) correlated with PTRF expression in the visceral depot. Likewise, the PAT family members, perilipin A (PLIN1), adipose differentiation-related protein (ADRP), and TIP47, which ultimately control lipolysis catalyzed by HSL (34), were also strongly correlated with PTRF mRNA levels in visceral fat. In addition, expression levels of protein kinase A and its substrate PLIN1 were also associated with PTRF in subcutaneous adipose tissue, highlighting the connection between PTRF and adipocyte lipolysis.
Table 2.
Linear correlation analyses between PTRF mRNA expression and that of selected genes
| Gene | VAT, n = 93 |
SAT, n = 85 |
||
|---|---|---|---|---|
| r | P | r | P | |
| Caveolin | 0.39 | 0.005 | 0.36 | 0.01 |
| PKA | −0.09 | 0.5 | 0.43 | 0.002 |
| FSP27 | 0.42 | 0.003 | 0.13 | 0.3 |
| PLIN1 | 0.4 | 0.003 | 0.34 | 0.01 |
| ADRP (or PLIN2) | 0.33 | 0.01 | 0.27 | 0.06 |
| TIP47 (or PLIN3) | 0.44 | 0.001 | 0.28 | 0.05 |
| ATGL | 0.47 | <0.0001 | 0.17 | 0.2 |
| CGI-58 | 0.32 | 0.01 | −0.10 | 0.5 |
| P16ink4a | 0.50 | <0.0001 | 0.51 | <0.0001 |
| PAI-1 | 0.30 | 0.01 | 0.54 | <0.0001 |
r, Pearson coefficient.
Fat-specific protein 27, which plays a dual role in regulating both adipocyte metabolism and cell death (35), also paralleled PTRF expression in the visceral depots. Adipose tissue senescence may contribute to the diabetic phenotype (7), and it has been shown that PTRF is a regulator of stress-induced senescence (36). Consistent with these observations, we found a direct association between PTRF mRNA levels and the markers of senescence P16Ink4a and PAI1 (Table 2). Indeed, when analyzing telomere length, a strong negative relationship was observed between subcutaneous adipocyte telomeres and PTRF mRNA levels (Fig. 6). Furthermore, this correlation remained significant after age, sex, and BMI adjustments. Although correlation cannot prove causation, PTRF association with shorter telomeres in subcutaneous fat might suggest the presence of functionally limited adipocyte precursors, which can no longer proliferate and hence are incapable of expanding.
Figure 6.
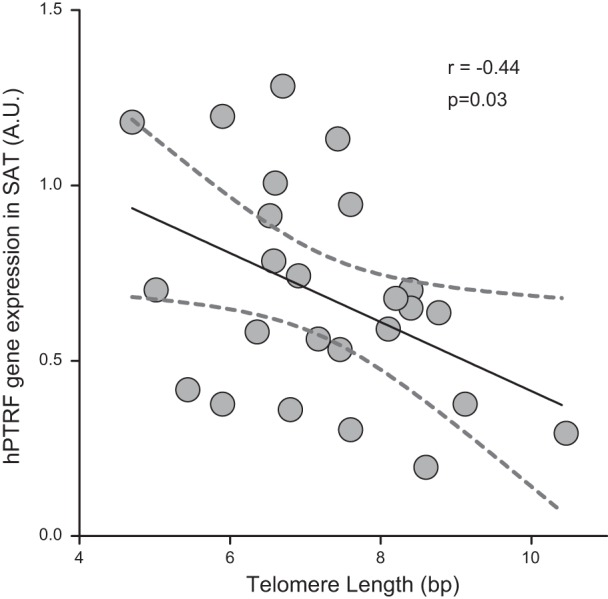
Negative correlation between telomere length and mRNA levels of PTRF in human subcutaneous adipose tissue (SAT). Graph shows linear regression lines (solid trace) and the 95% confidence bands (dashed traces). Each dot corresponds to 1 individual (n=25). r, Pearson coefficient; A.U., arbitrary units.
DISCUSSION
The accumulation of triglycerides in the adipose tissue, as long as its expansive capacity is preserved, may represent an adaptive and safe way to store energy surplus (25). Our results show that the PTRF protein is differentially regulated within the subcutaneous adipose tissue of a mouse model of metabolic syndrome, in a manner associated with glucose tolerance. PTRF appeared downregulated in hyperinsulinemic mice and upregulated in a context comparable to a prediabetic state, characterized by low plasma insulin and impaired glucose tolerance. In addition, stable PTRF overexpression in 3T3-L1 adipocytes compromised adipocyte differentiation, reduced de novo lipogenesis, and increased lipolysis. We also confirmed that PTRF was associated with markers of increased lipolysis and senescence in human adipose tissue. These data demonstrate that PTRF plays an important role in adipose tissue expansion and functionality and its up-regulation renders a defective adipocyte incapable of accommodating surplus lipids.
Similarly obese APOE2 knock-in mice displayed a broad disparity of impairments in their glucose metabolism, although there were no differences in plasma lipids or adiponectin. Only the ratio of visceral to subcutaneous fat mirrored glucose intolerance. We posit that the primary cause of the metabolic problems in these obese mice was the inability of their subcutaneous adipose tissues to expand and adequately store the surplus of lipids provided by the Western-type diet. Our subsequent proteomic analysis yielded a panoply of PTRF isoforms differentially regulated in the subcutaneous adipose tissue in a glucose-tolerant manner. PTRF was initially identified as a transcriptional enhancer of polymerase I (37) and later found at the cytoplasmic side of caveolae, where its function is critical for caveola formation (31, 38). In agreement with our results, a subset of genes associated with the structure of caveolae (PTRF among them) also appeared to be associated with adipose tissue expansion in pups as well adult mice exposed to an obesogenic environment (39). Caveolae are abundant in adipocytes and play an important role in regulating cellular metabolism (reviewed in ref. 40). Our INS subgroup of obese mice showed a 33% down-regulation of the PTRF isoforms in the adipose tissue, in the same range of the previously described 50% reduction of PTRF protein levels after acute insulin injection (41). Interestingly, a PTRF degradation product appeared increased in the INS group, suggesting that some regulation is at the level of PTRF degradation. In this sense, different truncated isoforms of PTRF have been described using vectorial proteomics (38). We note, however, that simultaneously with this increase in the degraded isoform, an additional 10 PTRF isoforms appeared downregulated, making it very difficult to dissect out the specific contributions of each individual isoform. Further studies are required to determine the contribution of individual isoforms to the globally observed effects.
Conversely, the most metabolically challenged prediabetic obese mice, the IGT group, had 10 upregulated PTRF isoforms (range 107–297%). To determine the cellular mechanisms underlying this association, we overexpressed PTRF in an in vitro model of adipocyte. PTRF localizes to a caveolae subclass that metabolizes triglycerides (29) and is shipped out to the cytosol on insulin stimulation, where it colocalizes with the HSL (30). As described previously (41), PTRF did not increase basal lipolysis but instead increased inducible lipolysis. Surprisingly, no changes were observed in glucose transporter 4 (GLUT4) protein levels on PTRF up-regulation (not shown), nor did we detect variations in insulin-stimulated deoxyglucose uptake between control adipocytes and their PTRF overexpressing counterparts. On the other hand, adipocyte hypertrophy and a slow generation rate of new adipocytes are associated to dysfunctional adipose tissue and low insulin sensitivity (42, 43). In this regard, PTRF overexpression resulted in larger adipocytes containing bigger lipid droplets, and both pharmacological and lentiviral PTRF overexpression in 3T3-L1 cells were accompanied by a diminished rate of adipocyte differentiation, as proved by lower mRNA levels of early adipogenic genes (CEBPβ, CEBPδ) and decreased lipogenic markers, such as PPARγ expression and AFABP mRNA and protein levels, as well as an increased pACC/ACC ratio along with diminished de novo lipogenesis. Considering all together, PTRF overexpression plays an important role in the adipocyte differentiation process, subsequently affecting lipid storage management.
Glucose intolerance and hyperinsulinemia were also described in a PTRF-knockout mouse (44). We must consider why the overexpression of PTRF mirrors results observed when PTRF is absent. Loss of PTRF, such as observed in the PTRF-KO mouse, or loss of caveolae for that matter, leads to a direct impairment in caveolar glucose/insulin receptor machinery and causes major damage to those pathways (41, 44–48). Our data suggest that whole-body glucose intolerance caused by PTRF up-regulation is a stepwise process, which stems from a defective adipocyte differentiation, ultimately rendering the adipose tissue dysfunctional. The mechanisms by which PTRF leads to adipose tissue senescence and inhibits differentiation have not yet been established. Overexpression of caveolin-1 increased senescence and suppressed adipogenic differentiation in human mesenquimal stem cells (50), and previous studies have shown that PTRF binds Mdm2, a negative regulator of p53 (51), into caveolae. This sequestration keeps Mdm2 away from p53 (36, 52), triggering the p53-dependent senescence and antiadipogenic pathways (53, 54). On the other hand, Mdm2 can also induce adipocyte differentiation independently of its ability to regulate p53 by promoting cAMP-mediated transcriptional activation of CREB and induction of CEBPδ expression (55). Typically, CEBPβ and CEBPδ are induced during the early stages of adipocyte differentiation, promoting further CEBPα and PPARγ expression (27). Therefore, we hypothesize that the reduced expresion of CEBPδ and CEBPβ during differentiation of PTRF-overexpressing 3T3-L1 preadipocytes is mediated by a decrease in available Mdm2 as a result of PTRF sequestration.
The effects of PTRF on human adipose tissue in the context of obesity have not yet been investigated. We found that PTRF mRNA levels were highly correlated with the levels of senescence markers in human adipose tissue. Adipose tissue expansion critically depends on the proliferation of adipocyte precursors, and senescent cells have reduced adipogenesis and fail to sequester lipotoxic fatty acids (7). PTRF is upregulated in human fibroblasts during replicative senescence (53, 56) as well as in stress-induced premature senescence (SIPS; ref. 36). Both types of senescence share a common pathway through the p53/p21 and caveolar pathways. Short telomeres reflect damaged DNA and also trigger a p53-dependent response (57), yet PTRF effects on cellular senescence and SIPS have been described as independent of telomere status (36, 53). Therefore, our results show for the first time a previously unappreciated association of telomere shortening with PTRF expression.
In summary, our data demonstrate that PTRF is linked to metabolic disorders at the whole-body level in obese mice as well as in obese individuals. The presence of adipocytes with impaired adipogenesis and a failure to maintain telomere length can explain, at least partially, the association of PTRF with the inability of the adipose tissue to expand appropriately and makes this protein a suitable marker of dysfunctional adipocytes.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Trono (École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) and Dr. Helenius (Eidgenössische Technische Hochschule Zürich, Zurich, Switzerland) for the generous gifts of the lentiviral expression system and the Ptrf1-mEGFP plasmid, respectively. The authors also acknowledge the invaluable technical assistance made by Ms. Pilar García-Sobreviela (Instituto Aragonés de Ciencias de la Salud) to the present work. Likewise, the authors thank Drs. E. Fernadez-Vizarra (University of Cambridge, Cambridge, UK), J. Osada (Universidad de Zaragoza, Zaragoza, Spain), and R. Köhler (Instituto Aragonés de Ciencias de la Salud) for their critical reading.
This work was supported by the Miguel Servet program from the Instituto de Salud Carlos III (Madrid, Spain), by the Marie-Curie Action: 303717-APOMET from the European Commission to J.M.A.-M., and by U.S. National Institutes of Health grant HL042360 to N.M. S.P.-D. was partially supported by a predoctoral fellowship from the Diputación General de Aragón (Madrid, Spain).
The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 2D-DIGE
- 2-dimensonal fluorescence difference gel electrophoresis
- ACC
- acyl-CoA carboxylase
- AFABP
- adipocyte fatty acid-binding protein
- APOE2
- apolipoprotein E2
- ATGL
- adipose triglyceride lipase
- AUC
- area under the curve
- BMI
- body mass index
- EGFP
- enhanced green fluorescent protein
- HEK
- human embryonic kidney
- HSL
- hormone-sensitive lipase
- IGT
- impaired glucose tolerance
- INS
- hyperinsulinemic
- MALDI
- matrix-assisted laser desorption/ionization
- NEFA
- nonesterified free fatty acid
- NGT
- normal glucose-tolerant
- OGTT
- oral glucose tolerance test
- PTRF
- polymerase I and transcript release factor
- TOF
- time of flight
- WD
- Western-type diet
REFERENCES
- 1. Haffner S. M. (2006) Relationship of metabolic risk factors and development of cardiovascular disease and diabetes. Obesity 4, 121S–127S [DOI] [PubMed] [Google Scholar]
- 2. Kilpelainen T. O., Zillikens M. C., Stancakova A., Finucane F. M., Ried J. S., Langenberg C., Zhang W., Beckmann J. S., Luan J., Vandenput L., Styrkarsdottir U., Zhou Y., Smith A. V., Zhao J.-H., Amin N., Vedantam S., Shin S. Y., Haritunians T., Fu M., Feitosa M. F., Kumari M., Halldorsson B. V., Tikkanen E., Mangino M., Hayward C., Song C., Arnold A. M., Aulchenko Y. S., Oostra B. A., Campbell H., Cupples L. A., Davis K. E., Döring A., Eiriksdottir G., Estrada K., Fernández-Real J. M., Garcia M., Gieger C., Glazer N. L., Guiducci C., Hofman A., Humphries S. E., Isomaa B., Jacobs L. C., Jula A., Karasik D., Karlsson M. K., Khaw K.-T., Kim L. J., Kivimäki M., Klopp N., Kühnel B., Kuusisto J., Liu Y., Ljunggren O, Lorentzon M., Luben R. N., McKnight B., Mellström D., Mitchell B. D., Mooser V., Moreno J. M., Männistö S., O'Connell J. R., Pascoe L., Peltonen L., Peral B., Perola M., Psaty B. M., Salomaa V., Savage D. B., Semple R. K., Skaric-Juric T., Sigurdsson G., Song K. S., Spector T. D., Syvänen A.-C., Talmud P. J., Thorleifsson G., Thorsteinsdottir U., Uitterlinden A. G., van Duijn C. M., Vidal-Puig A., Wild S. H., Wright A. F., Clegg D. J., Schadt E., Wilson J. F., Rudan I., Ripatti S., Borecki I. B., Shuldiner A. R., Ingelsson E., Jansson J.-O., Kaplan R. C., Gudnason V., Harris T. B., Groop L., Kiel D. P., Rivadeneira F., Walker M., Barroso I., Vollenweider P., Waeber G., Chambers J. C., Kooner J. S., Soranzo N., Hirschhorn J. N., Stefansson K., Wichmann H.-E., Ohlsson C., O'Rahilly S., Wareham N. J., Speliotes E. K., Fox C. S., Laakso M., Loos R. J. F. (2011) Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat. Genet. 43, 753–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stefan N., Kantartzis K., Machann J., Schick F., Thamer C., Rittig K., Balletshofer B., Machicao F., Fritsche A., Häring H. U. (2008) Identification and characterization of metabolically benign obesity in humans. Arch. Intern. Med. 168, 1609–1616 [DOI] [PubMed] [Google Scholar]
- 4. Wildman R. P., Muntner P., Reynolds K., McGinn A. P., Rajpathak S., Wylie-Rosett J., Sowers M. R. (2008) The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch. Intern. Med. 168, 1617–1624 [DOI] [PubMed] [Google Scholar]
- 5. Scherer P. E. (2006) Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes 55, 1537–1545 [DOI] [PubMed] [Google Scholar]
- 6. Arbones-Mainar J. M., Johnson L. A., Altenburg M. K., Maeda N. (2008) Differential modulation of diet-induced obesity and adipocyte functionality by human apolipoprotein E3 and E4 in mice. Int. J. Obes. 32, 1595–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tchkonia T., Morbeck D. E., Von Zglinicki T., Van Deursen J., Lustgarten J., Scrable H., Khosla S., Jensen M. D., Kirkland J. L. (2010) Fat tissue, aging, and cellular senescence. Aging Cell 9, 667–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hennuyer N., Tailleux A., Torpier G., Mezdour H., Fruchart J.-C., Staels B., Fiévet C. (2005) PPARalpha, but not PPARgamma, activators decrease macrophage-laden atherosclerotic lesions in a nondiabetic mouse model of mixed dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 25, 1897–902 [DOI] [PubMed] [Google Scholar]
- 9. Kuhel D. G., Konaniah E. S., Basford J. E., McVey C., Goodin C. T., Chatterjee T. K., Weintraub N. L, Hui D. Y. (2013) Apolipoprotein E2 accentuates postprandial inflammation and diet-induced obesity to promote hyperinsulinemia in mice. Diabetes 62, 382–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sullivan P. M., Mezdour H., Quarfordt S. H., Maeda N. (1998) Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J. Clin. Invest. 102, 130–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shiri-Sverdlov R., Wouters K., van Gorp P. J., Gijbels M. J., Noel B., Buffat L., Staels B., Maeda N., van Bilsen M., Hofker M. H. (2006) Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J. Hepatol. 44, 732–741 [DOI] [PubMed] [Google Scholar]
- 12. Vanschoonbeek K., Wouters K., van der Meijden P. E. J., van Gorp P. J., Feijge M. A., H., Herfs M., Schurgers L. J., Hofker M. H., de Maat M. P., Heemskerk J. W. (2008) Anticoagulant effect of dietary fish oil in hyperlipidemia: a study of hepatic gene expression in APOE2 knock-in mice. Arterioscler. Thromb. Vasc. Biol. 28, 2023–2029 [DOI] [PubMed] [Google Scholar]
- 13. Arbonés-Mainar J. M., Navarro M. A., Acín S., Guzmán M. A., Arnal C., Surra J. C., Carnicer R., Roche H. M., Osada J. (2006) Trans-10, cis-12- and cis-9, trans-11-conjugated linoleic acid isomers selectively modify HDL-apolipoprotein composition in apolipoprotein E knockout mice. J. Nutr. 136, 353–359 [DOI] [PubMed] [Google Scholar]
- 14. Arbonés-Mainar J. M., Navarro M. A., Guzmán M. A., Arnal C., Surra J. C., Acín S., Carnicer R., Osada J., Roche H. M. (2006) Selective effect of conjugated linoleic acid isomers on atherosclerotic lesion development in apolipoprotein E knockout mice. Atherosclerosis 189, 318–327 [DOI] [PubMed] [Google Scholar]
- 15. Arbones-Mainar J. M., Johnson L. A., Altenburg M. K., Kim H.-S., Maeda N. (2010) Impaired adipogenic response to thiazolidinediones in mice expressing human apolipoproteinE4. FASEB J. 24, 3809–3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeKroon R. M., Osorio C., Robinette J. B., Mocanu M., Winnik W. M., Alzate O. (2011) Simultaneous detection of changes in protein expression and oxidative modification as a function of age and APOE genotype. J. Proteome Res. 10, 1632–1644 [DOI] [PubMed] [Google Scholar]
- 17. Osorio C., Sullivan P. M., He D. N., Mace B. E., Ervin J. F., Strittmatter W. J., Alzate O. (2007) Mortalin is regulated by APOE in hippocampus of AD patients and by human APOE in TR mice. Neurobiol. Aging 28, 1853–1862 [DOI] [PubMed] [Google Scholar]
- 18. Hayer A., Stoeber M., Bissig C., Helenius A. (2010) Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic 11, 361–382 [DOI] [PubMed] [Google Scholar]
- 19. Sánchez E., Lobo T., Fox J. L., Zeviani M., Winge D. R., Fernández-Vizarra E. (2013) LYRM7/MZM1L is a UQCRFS1 chaperone involved in the last steps of mitochondrial complex III assembly in human cells. Biochim. Biophys. Acta Bioenerg. 1827, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramirez-Zacarias J. L., Castro-Munozledo F., Kuri-Harcuch W. (1992) Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil Red O. Histochemistry 97, 493–497 [DOI] [PubMed] [Google Scholar]
- 21. Krishan A. (1975) Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol. 66, 188–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson L. A., Arbones-Mainar J. M., Fox R. G., Pendse A. A., Altenburg M. K., Kim H.-S., Maeda N. (2011) Apolipoprotein E4 exaggerates diabetic dyslipidemia and atherosclerosis in mice lacking the LDL receptor. Diabetes 60, 2285–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moreno-Navarrete J. M., Ortega F., Sabater M., Ricart W., Fernández-Real J. M. (2010) Telomere length of subcutaneous adipose tissue cells is shorter in obese and formerly obese subjects. Int. J. Obes. 34, 1345–1348 [DOI] [PubMed] [Google Scholar]
- 24. Moreno-Navarrete J. M., Petrov P., Serrano M., Ortega F., García-Ruiz E., Oliver P., Ribot J., Ricart W., Palou A., Bonet M. L., Fernández-Real J. M. (2013) Decreased RB1 mRNA, protein, and activity reflect obesity-induced altered adipogenic capacity in human adipose tissue. Diabetes 62, 1923–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Virtue S., Vidal-Puig A. (2008) It's not how fat you are, it's what you do with it that counts. PLoS Biol. 6, e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sakoda H., Ogihara T., Anai M., Funaki M., Inukai K., Katagiri H., Fukushima Y., Onishi Y., Ono H., Fujishiro M., Kikuchi M., Oka Y., Asano T. (2000) Dexamethasone-induced insulin resistance in 3T3-L1 adipocytes is due to inhibition of glucose transport rather than insulin signal transduction. Diabetes 49, 1700–1708 [DOI] [PubMed] [Google Scholar]
- 27. Farmer S. R. (2006) Transcriptional control of adipocyte formation. Cell Metab. 4, 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abu-Elheiga L., Jayakumar A., Baldini A., Chirala S. S., Wakil S. J. (1995) Human acetyl-CoA carboxylase: characterization, molecular cloning, and evidence for two isoforms. Proc. Natl. Acad. Sci. U.S.A. 92, 4011–4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ost A., Ortegren U., Gustavsson J., Nystrom F. H., Strålfors P. (2005) Triacylglycerol is synthesized in a specific subclass of caveolae in primary adipocytes. J. Biol. Chem. 280, 5–8 [DOI] [PubMed] [Google Scholar]
- 30. Aboulaich N., Ortegren U., Vener A. V., Strålfors P. (2006) Association and insulin regulated translocation of hormone-sensitive lipase with PTRF. Biochem. Biophys. Res. Commun. 350, 657–661 [DOI] [PubMed] [Google Scholar]
- 31. Hill M. M., Bastiani M., Luetterforst R., Kirkham M., Kirkham A., Nixon S. J., Walser P., Abankwa D., Oorschot V. M., Martin S., Hancock J. F., Parton R. G. (2008) PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zimmermann R., Strauss J. G., Haemmerle G., Schoiswohl G., Birner-Gruenberger R., Riederer M., Lass A., Neuberger G., Eisenhaber F., Hermetter A., Zechner R. (2004) Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306, 1383–1386 [DOI] [PubMed] [Google Scholar]
- 33. Lord C. C., Betters J. L., Ivanova P. T., Milne S. B., Myers D. S., Madenspacher J., Thomas G., Chung S., Liu M., Davis M. A., Lee R. G., Crooke R. M., Graham M. J., Parks J. S., Brasaemle D. L., Fessler M. B., Brown H. A., Brown J. M. (2012) CGI-58/ABHD5-derived signaling lipids regulate systemic inflammation and insulin action. Diabetes 61, 355–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H., Hu L., Dalen K., Dorward H., Marcinkiewicz A., Russell D., Gong D., Londos C., Yamaguchi T., Holm C., Rizzo M. A., Brasaemle D., Sztalryd C. (2009) Activation of hormone-sensitive lipase requires two steps, protein phosphorylation and binding to the PAT-1 domain of lipid droplet coat proteins. J. Biol. Chem. 284, 32116–32125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J. Y., Liu K., Zhou S., Tillison K., Wu Y., Smas C. M. (2008) Assessment of fat-specific protein 27 in the adipocyte lineage suggests a dual role for FSP27 in adipocyte metabolism and cell death. Am. J. Physiol. Endocrinol. Metab. 294, E654–E667 [DOI] [PubMed] [Google Scholar]
- 36. Volonte D., Galbiati F. (2011) Polymerase I and transcript release factor (PTRF)/cavin-1, a novel regulator of stress-induced premature senescence. J. Biol. Chem. 286, 28657–28661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jansa P., Mason S. W., Hoffmann-Rohrer U., Grummt I. (1998) Cloning and functional characterization of PTRF, a novel protein which induces dissociation of paused ternary transcription complexes. EMBO J. 17, 2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aboulaich N., Vainonen J. P., Strålfors P., Vener A. V. (2004) Vectorial proteomics reveal targeting, phosphorylation and specific fragmentation of polymerase I and transcript release factor (PTRF) at the surface of caveolae in human adipocytes. Biochem. J. 383, 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kozak L. P., Newman S., Chao P.-M., Mendoza T., Koza R. A. (2010) The early nutritional environment of mice determines the capacity for adipose tissue expansion by modulating genes of caveolae structure. PLoS ONE 5, e11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pilch P. F., Liu L. (2011) Fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol. Metab. 22, 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aboulaich N., Chui P. C., Asara J. M., Flier J. S., Maratos-Flier E. (2011) Polymerase I and transcript release factor regulates lipolysis via a phosphorylation-dependent mechanism. Diabetes 60, 757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jernås M., Palming J., Sjöholm K., Jennische E., Svensson P.-A., Gabrielsson B. G., Levin M., Sjögren A., Rudemo M., Lystig T. C., Carlsson B., Carlsson L. M., Lönn M. (2006) Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB J. 20, 1540–1542 [DOI] [PubMed] [Google Scholar]
- 43. Arner E., Westermark P. O., Spalding K. L., Britton T., Ryden M., Frisen J., Bernard S., Arner P. (2010) Adipocyte turnover: relevance to human adipose tissue morphology. Diabetes 59, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu L., Brown D., McKee M., Lebrasseur N. K., Yang D., Albrecht K. H., Ravid K., Pilch P. F. (2008) Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 8, 310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Razani B., Combs T. P., Wang X. B., Frank P. G., Park D. S., Russell R. G., Li M., Tang B., Jelicks L. A., Scherer P. E., Lisanti M. P. (2002) Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J. Biol. Chem. 277, 8635–8647 [DOI] [PubMed] [Google Scholar]
- 46. Gustavsson J., Parpal S., Karlsson M., Ramsing C., Thorn H., Borg M., Lindroth M., Peterson K. H., Magnusson K. E., Strâlfors P. (1999) Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 13, 1961–1971 [PubMed] [Google Scholar]
- 47. Kim C. A., Delépine M., Boutet E., El Mourabit H., Le Lay S., Meier M., Nemani M., Bridel E., Leite C. C., Bertola D. R., Semple R. K., O'Rahilly S., Dugail I., Capeau J., Lathrop M., Magré J. (2008) Association of a homozygous nonsense caveolin-1 mutation with berardinelli-seip congenital lipodystrophy. J. Clin. Endocrinol. Metab. 93, 1129–1134 [DOI] [PubMed] [Google Scholar]
- 48. Cao H., Alston L., Ruschman J., Hegele R. A. (2008) Heterozygous CAV1 frameshift mutations (MIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids Heal. Dis. 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. González-Muñoz E., López-Iglesias C., Calvo M., Palacín M., Zorzano A., Camps M. (2009) Caveolin-1 loss of function accelerates glucose transporter 4 and insulin receptor degradation in 3T3-L1 adipocytes. Endocrinology 150, 3493–3502 [DOI] [PubMed] [Google Scholar]
- 50. Park J. S., Kim H. Y., Kim H. W., Chae G. N., Oh H. T., Park J. Y., Shim H., Seo M., Shin E. Y., Kim E. G., Park S. C., Kwak S. J. (2005) Increased caveolin-1, a cause for the declined adipogenic potential of senescent human mesenchymal stem cells. Mech. Ageing Dev. 126, 551–559 [DOI] [PubMed] [Google Scholar]
- 51. Haupt Y., Maya R., Kazaz A., Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387(6630), 296–299 [DOI] [PubMed] [Google Scholar]
- 52. Bitar M. S., Abdel-Halim S. M., Al-Mulla F. (2013) Caveolin-1/PTRF upregulation constitutes a mechanism for mediating p53-induced cellular senescence: implications for evidence-based therapy of delayed wound healing in diabetes. Am. J. Physiol. Endocrinol. Metab. 305, E951963. [DOI] [PubMed] [Google Scholar]
- 53. Bai L., Deng X., Li J., Wang M., Li Q., An W., A D., Cong Y. S. (2011) Regulation of cellular senescence by the essential caveolar component PTRF/Cavin-1. Cell Res. 21, 1088–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Molchadsky A., Ezra O., Amendola P. G., Krantz D., Kogan-Sakin I., Buganim Y., Rivlin N., Goldfinger N., Folgiero V., Falcioni R., Sarig R., Rotter V. (2013) p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell Death Differ. 20, 774–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hallenborg P., Feddersen S., Francoz S., Murano I., Sundekilde U., Petersen R. K., Akimov V., Olson M. V., Lozano G., Cinti S., Gjertsen B. T., Madsen L., Marine J. C., Blagoev B., Kristiansen K. (2012) Mdm2 controls CREB-dependent transactivation and initiation of adipocyte differentiation. Cell Death Differ. 19, 1381–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cong Y.-S., Fan E., Wang E. (2006) Simultaneous proteomic profiling of four different growth states of human fibroblasts, using amine-reactive isobaric tagging reagents and tandem mass spectrometry. Mech. Ageing Dev. 127, 332–343 [DOI] [PubMed] [Google Scholar]
- 57. Chin L., Artandi S. E., Shen Q., Tam A., Lee S.-L., Gottlieb G. J., Greider C. W., DePinho R. A. (1999) p53 Deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97, 527–538 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.