

Abstract
Dysfunctions in Wnt signaling increase β-catenin stability and are associated with cancers, including colorectal cancer. In addition, β-catenin degradation is decreased by nutrient-dependent O-GlcNAcylation. Human colon tumors and colons from mice fed high-carbohydrate diets exhibited higher amounts of β-catenin and O-GlcNAc relative to healthy tissues and mice fed a standard diet, respectively. Administration of the O-GlcNAcase inhibitor thiamet G to mice also increased colonic expression of β-catenin. By ETD-MS/MS, we identified 4 O-GlcNAcylation sites at the N terminus of β-catenin (S23/T40/T41/T112). Furthermore, mutation of serine and threonine residues within the D box of β-catenin reduced O-GlcNAcylation by 75%. Interestingly, elevating O-GlcNAcylation in human colon cell lines drastically reduced phosphorylation at T41, a key residue of the D box responsible for β-catenin stability. Analyses of β-catenin O-GlcNAcylation mutants reinforced T41 as the most crucial residue that controls the β-catenin degradation rate. Finally, inhibiting O-GlcNAcylation decreased the β-catenin/α-catenin interaction necessary for mucosa integrity, whereas O-GlcNAcase silencing improved this interaction. These results suggest that O-GlcNAcylation regulates not only the stability of β-catenin, but also affects its localization at the level of adherens junctions. Accordingly, we propose that O-GlcNAcylation of β-catenin is a missing link between the glucose metabolism deregulation observed in metabolic disorders and the development of cancer.—Olivier-Van Stichelen, S., Dehennaut, V., Buzy, A., Zachayus, J.-L., Guinez, C., Mir, A.-M., El Yazidi-Belkoura, I., Copin, M.-C., Boureme, D., Loyaux, D., Ferrara, P., Lefebvre, T. O-GlcNAcylation stabilizes β-catenin through direct competition with phosphorylation at threonine 41.
Keywords: Wnt signaling, cancer, ETD-MS/MS
Wnt signaling is an evolutionarily conserved pathway involved in biological processes, including embryogenesis and intestinal tissue renewal (1). Cancer development is closely associated with deregulation of Wnt signaling and its key player β-catenin (2). β-Catenin is primarily found at adherens junctions in complex with E-cadherin and α-catenin and ensures epithelial integrity (3). Excess β-catenin is targeted to the proteasome by an efficient phosphorylation/ubiquitinylation-mediated degradation system (4, 5). First, a destruction complex including the tumor suppressors adenomatous polyposis coli (APC) and axin, as well as the kinases GSK3β and CK1α, sequestrates β-catenin (6). β-Catenin sequestration leads to the phosphorylation of the destruction box (D box) at S33, S37, T41, and S45; to the ubiquitination of β-catenin at K19 and K49; and to its proteasomal degradation. Intact β-catenin migrates into the nucleus and drives the expression of genes crucial for cell cycle progression (7–10).
Genetic alterations in Wnt signaling are frequently found in leukemia, lymphoma, and medulloblastoma and in gastric, hepatocellular, breast and colorectal cancers (CRCs) and include mutations in the APC, axin, TCF4, LRP5, and β-catenin genes (8). In addition, Wnt signaling is hyperactivated by a miR-301a-dependent PTEN-targeting mechanism in breast cancer (11). Direct mutations of β-catenin are found in hepatocellular carcinoma and CRC at 33 and 10%, respectively, increasing the protein's stability and therefore its oncogenic properties (12, 13). Diet, lifestyle, and metabolic pathologies also increase the risk of cancer, with a 2-fold increase of CRC in patients with type 2 diabetes (14–16). To understand the links between nutrition and CRC, we focus on β-catenin and its dynamic posttranslational modification (PTM) by O-linked N-acetylglucosamine (O-GlcNAc; ref. 17).
O-GlcNAc transferase (OGT) adds a single N-acetylglucosamine group to target proteins using the activated donor UDP-GlcNAc, the end product of the hexosamine biosynthesis pathway (HBP; ref. 18). A second enzyme, O-GlcNAcase (OGA), dynamically removes the GlcNAc residue. The UDP-GlcNAc concentration, and consequently O-GlcNAcylation, correlates closely with the nutrient status of the cell, and the nucleotide-sugar is therefore regarded as a nutrient sensor. We previously demonstrated that colons from unfed-state mice force-fed glucose or glucosamine have higher levels of both β-catenin expression and O-GlcNAcylation (19). Moreover, we observed higher levels of β-catenin, O-GlcNAcylation, and OGT in 2 CRC cell lines in comparison with a fetal colon cell line. These results led us to hypothesize that diet and metabolic disorders predispose for CRC by promoting an increase in β-catenin expression through up-regulation of HBP and O-GlcNAcylation. We demonstrated that modulation of the HBP flux by glucosamine stimulation yields increased β-catenin level in MCF7 cells, whereas its level is decreased on inhibition of the rate-limiting enzyme glutamine:fructose-6-phosphate amidotransferase (GFAT). In addition, simply manipulating the O-GlcNAcylation status of the cell directly correlates with the stability of β-catenin, suggesting that glycosylation protects it from degradation (19). Here, we describe one of the mechanisms by which O-GlcNAcylation may reduce β-catenin proteasomal susceptibility.
MATERIALS AND METHODS
Human tumor tissues
Colon tumor tissues and matching tumor-adjacent normal tissues from the same patient were obtained from the Tumor Bank, Regional Reference Center in Cancer [Centre Hospitalier Régional Universitaire (CHRU) of Lille, Lille, France; agreement CSTMT076]. Patient data are presented in Table 1.
Table 1.
Information on tissues used in the study and medical history of the related patients
| Patient and ref. no. | Tissue | Tumor tissue (%) | Healthy tissue (%) | Necrosis (%) | Cells (%) | Stroma (%) | pT | pN | Nodes removed | Nodes affected | Medical history |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 20 | 0 | Indeterminate (1.75 m; 85 kg); slight overweight | ||||||||
| 15417 | Tumor | 100 | 0 | 1 | 50 | 50 | 3 | 0 | |||
| 15419 | Healthy | 0 | 100 | 3 | 0 | ||||||
| 2 | 10 | 0 | Diabetes, hypertension | ||||||||
| 9269 | Tumor | 100 | 0 | <5 | 70 | 30 | 3 | 0 | |||
| 9271 | Healthy | 0 | 100 | 3 | 0 | ||||||
| 3 | 12 | 0 | Hypertension, diabetes, temporary ischemic attack | ||||||||
| 8453 | Tumor | 90 | 10 | 0 | 60 | 40 | 3 | 0 | |||
| 8458 | Healthy | 0 | 100 | 3 | 0 | ||||||
| 4 | 29 | 0 | Hypertension, dyslipidemia | ||||||||
| 14998 | Tumor | 90 | 10 | <5 | 80 | 20 | 4 | 0 | |||
| 15001 | Healthy | 0 | 100 | 4 | 0 |
pT, primary tumor; pN, regional lymph nodes.
Cell culture
HEK293T, MCF7, HT29, and HCT116 cells were maintained in Dulbecco's modified Eagle's medium (DMEM), and CCD841CoN cells were maintained in Eagle's minimum essential medium (EMEM). All the cell lines were maintained in medium supplemented with 10% (v/v) fetal calf serum (heat inactivated for HT29 and HCT116 cells), 2 mM l-glutamine, 5 IU/ml penicillin, and 50 μg/ml streptomycin at 37°C, in a 5% (v/v) CO2-enriched, humidified atmosphere.
Plasmids and transfection
Wild-type (WT) and tetramutant (TM; S33A, S37A, T41A, S45A) pCS2+-β-catenin-2XFLAG (provided by Dr. C. Liu, University of Kentucky, Lexington, KY, USA) are described elsewhere (4, 5). To generate the β-catenin O-GlcNAcylation mutants, we used the following primer sequences: S23A-β-catenin, sense oligonucleotide 5′-CGAAAGGCAGCAGTGGCCCACTGGCAGCAGCAG-3′/antisense oligonucleotide 5′-CTGCTGCTGCCAGTGGGCCACTGCTGCCTTTCG-3′; T40A-β-catenin, sense oligonucleotide 5′-ATTCATTCTGGAGCAGCCACCACAGCACCA-3′/antisense oligonucleotide 5′-TGGTGCTGTGGTGGCTGCTCCAGAATGAAT-3′; T41A-β-catenin, sense oligonucleotide 5′-ATTCTGGAGCAACCGCCACAGCACCATCTTT-3′/antisense oligonucleotide 5′-AAAGATGGTGCTGTGGCGGTTGCTCCAGAAT-3′; and T112A-β-catenin, sense oligonucleotide 5′-CAGATTCCATCCGCACAATTTGACTCT-3′/antisense oligonucleotide 5′-AGAGTCAAATTGTGCGGATGGAATCTG-3′. To generate the TM 4M β-catenin (S23A/T40A/T41A/T112A), we first generated the T40A/T41-β-catenin mutant using sense oligonucleotide 5′-ATTCTGGAGCAGCCGCCACAGCACCATCTTT-3′/antisense oligonucleotide 5′-AAAGATGGTGCTGTGGCGGCTGCTCCAGAAT-3′. Then, the S23A/T40/T41-β-catenin and the S23A/T40A/T41A/T112A-β-catenin (4M) were simultaneously generated by using the oligonucleotides mentioned above.
The HA-ubiquitin expression vector was provided by Dr. C. Couturier (Biology Institute of Lille, Lille, France).
MCF7 or HEK293T cells were transfected with 2.5 μg of DNA and 10 μl of Lipofectamine (Lipo2000; Invitrogen, Cergy Pontoise, France) in 10 ml of DMEM in 100 mm diameter dishes.
Small interfering RNA (siRNA)
With the exception of CCD841CoN cells, which were reverse transfected for 72 h, all cells were reverse transfected for 48 h with RNAiMax (Invitrogen), according to the manufacturer's instructions, using 10 nM of a pool of 4 siRNAs targeting OGA (siGenome Smart Pool, Human MGEA5, M-012805-01; Dharmacon, Lafayette, CO, USA), 10 nM siRNA targeting OGT (20), or a scrambled control (siCtrl) sequence (19).
Small molecule supplements
Proteasome inhibitor MG132 (Sigma-Aldrich, Saint-Quentin Fallavier, France) was used at a concentration of 1 μM. Glucosamine was used at a concentration of 5 mM. The OGT and OGA inhibitors, Ac5S-GlcNAc and NButGT, respectively (provided by Pr. D. Vocadlo, Simon Fraser University, Burnaby, BC, Canada), were used at a concentration of 100 μM. For thiamet G use, see In vivo Experiments below.
Immunoprecipitation and coimmunoprecipitation assays
Cells were washed with 10 ml cold phosphate-buffered saline (PBS), then lysed on ice with either IP lysis buffer [10 mM Tris/HCl, 150 mM NaCl, 1% Triton X-100 (v/v), 0.5% sodium deoxycholate (w/v), 0.1% sodium dodecyl sulfate (w/v), and protease inhibitors; pH 7.4] or octyl lysis buffer [25 mM Tris HCl, 150 mM NaCl, 1.7% (w/v) octyl glucoside, 0.084% (w/v) sodium fluoride, 0.44% (w/v) sodium pyrophosphate, 0.018% (w/v) sodium orthovanadate, 1 μM PUGNAc, and protease inhibitors; pH 7.5] for immunoprecipitation or co-IP lysis buffer [20 mM Tris/HCl, 150 mM NaCl, 0.5% Nonidet P-40 (v/v) and protease inhibitors; pH 8.0] for coimmunoprecipitation. The cell extracts were then centrifuged at 20,000 g for 10 min at 4°C. The supernatants were precleared with Sepharose-labeled protein A/G for 1 h. The beads were discarded after a 1 min centrifugation at 2500 g, and the supernatants were incubated with 2 μg of either mouse monoclonal anti-FLAG antibody (M2; Sigma-Aldrich) or rabbit polyclonal anti-β-catenin (H102; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rocked at 4°C overnight. Controls for immunoprecipitation specificities were performed with rabbit IgG (Santa Cruz Biotechnology). Antibody-bound proteins were recovered by rocking samples with 20 μl of Sepharose-labeled protein A/G (GE Healthcare, Buc, France) for 1 h at 4°C. For immunoprecipitation, the beads were gently centrifuged for 1 min at 2500 g and washed as follows: IP lysis buffer; IP lysis buffer supplemented with 500 mM NaCl; IP lysis buffer/TNE (10 mM Tris/HCl, 150 mM NaCl, and 1 mM EDTA; pH 7.4; v/v); and finally TNE alone. For coimmunoprecipitation, the beads were washed 4 times with co-IP lysis buffer. Bound proteins were eluted by boiling in Laemmli buffer.
Western blot analysis
Proteins were resolved on 8 or 10% SDS-PAGE under reducing conditions and transferred to nitrocellulose (GE Healthcare). Equal loading and transfer efficiency were confirmed by Ponceau red staining. The membranes were blocked for 45 min with 5% (w/v) non-fatty-acid milk in Tris-buffered saline (TBS)-Tween buffer [15 mM Tris/HCl, 140 mM NaCl, and 0.05% Tween20 (v/v); pH 8.0]. The membranes were incubated overnight at 4°C with the appropriate antibodies (described below). The membranes were then washed 3 times with TBS-Tween for 10 min and incubated with either an anti-rabbit or an anti-mouse horseradish peroxidase–labeled secondary antibody at a dilution of 1:10,000 for 1 h. Finally, the blots were washed with TBS-Tween 3 times for 10 min each, and signal was detected by enhanced chemiluminescence (GE Healthcare). Mouse monoclonal anti-O-GlcNAc (RL2; VWR International, Fontenay-sous-Bois, France), mouse monoclonal anti-phosphoT41-β-catenin (C8616; Sigma-Aldrich), rabbit polyclonal anti-phospho-T41/S45-β-catenin (9565; Cell Signaling Technology, Leiden, The Netherlands), rabbit polyclonal anti-phospho-S33/S37/T41-β-catenin (9561; Cell Signaling Technology), and rabbit polyclonal anti-HA (Santa Cruz Biotechnology) were used at a final dilution of 1:1000. Rabbit polyclonal anti-OGT (AL28) and chicken anti-OGA (345) (both provided by Prof. G. W. Hart, Johns Hopkins University, Baltimore, MD, USA), rabbit polyclonal anti α-catenin (H297; Santa Cruz Biotechnology), and rabbit polyclonal anti-β-catenin (H102; Santa Cruz Biotechnology) were used at a dilution of 1:2000. Mouse monoclonal anti-FLAG (M2; Sigma-Aldrich) and rabbit polyclonal anti-GAPDH (Abcam, Cambridge, UK) were used at a dilution of 1:5000.
Cell fractionation
The Proteoextract subcellular proteome extraction kit (Merck, Darmstadt, Germany) was used for cellular fractionation, as recommended by the manufacturer.
In vivo experiments
Eight-week-old male C57BL/6J mice were purchased from Charles River (Saint-Germain sur l'Arbresle, France) and adapted to the environment for 1 wk before the experiments. The mice were maintained in a 12 h light-dark cycle with water and a standard diet (SD; 65% carbohydrate, 11% fat, and 24% protein; SAFE, Augy, France) or a high-carbohydrate diet (HCD; 75% carbohydrate, 3% fat, and 22% protein; SAFE) for 9 wk. Procedures were performed according to the French guidelines for the care of experimental animals. Colons were collected, washed in PBS, and lysed in 2 ml of Nonidet P-40 lysis buffer [10 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P-40 (v/v), 0.018% Na3VO4, 1 μM PUGNAc, and protease inhibitors; pH 7.4]. An UltraTurrax disperser (Sigma-Aldrich) was used for tissue disruption. Samples were incubated with gentle agitation for 2 h at 4°C, after which the soluble fraction was obtained after centrifugation at 20,000 g for 30 min at 4°C.
For chronic thiamet G (Sigma-Aldrich) treatment, mice were intraperitoneally injected daily with thiamet G (20 mg/kg/d in PBS) or with PBS for 15 d. Before euthanasia, mice were denied access to food for 24 h.
Oral glucose tolerance test (OGTT)
To test glucose tolerance, mice were force-fed glucose (1 g glucose/kg) after overnight food withdrawal. Glycemia was measured every 15 min for 2 h with an Accu-Chek Performa apparatus (Roche, Basel Switzerland) before the animals were euthanized.
Preparation of β-catenin for mass spectrometry
HT29 cells and samples from 4 patients with CRC were lysed in octyl lysis buffer. The lysate was first precleared with protein-A-Sepharose, and β-catenin was isolated by immunoprecipitation. The Coomassie-stained band corresponding to β-catenin was excised, reduced with DTT, alkylated with iodoacetamide, and in-gel digested with trypsin (Promega France, Charbonnieres, France; ref. 21). Peptides were extracted with 50 mM NH4HCO3 and 50% CH3CN in 0.2% HCOOH and analyzed by nano-liquid chromatography–tandem mass spectrometry (LC-MS/MS) after partial evaporation in a Speed-vac concentrator (Thermo Fisher Scientific, Villebon-sur-Yvette, France), to remove acetonitrile.
Nano-LC-MS/MS analysis
LC-MS/MS experiments were performed on a nanoAcquity UPLC system (Waters, Milford, Ma, USA), connected to an LTQ Orbitrap Velos ETD mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray source. β-Catenin trypsin digest was loaded onto a precolumn (20 mm × 180 μm i.d.; Symmetry C18; Waters) and washed with solvent A (0.2% HCOOH) at 20 μl/min for 5 min. The peptides were then eluted on a C18 reverse-phase nanoflow column (200 mm × 75 μm i.d.; BEH130 C18; Waters) with a linear gradient of 7–40% solvent B [H2O/CH3CN/HCOOH, 10/90/0.2 (v/v/v)] for 120 min, 30–90% solvent B for 20 min, and 90% solvent B for 5 min, at a flow rate of 200 nl/min. The mass spectrometer was first operated in the data-dependent mode, to automatically switch between MS and MS/MS acquisition. Survey full-scan MS spectra (m/z 300–1700) were acquired in the Orbitrap with a resolution of 60,000 at m/z 400. The LTQ Orbitrap Velos ETD performed a full MS scan followed by 5 data-dependent electron-transfer dissociation–tandem mass spectrometry (ETD-MS/MS) scans with detection of the ETD fragment ions in the linear ion trap. Target values were 1e6 for full FTMS scans and 1e4 for ion trap MSn scans, with a maximum injection time of 200 ms. The anion target value was 3e5. ETD activation time was 100 ms. Supplemental activation was used for all ETD MSn scans. Dynamic exclusion was also enabled with an exclusion time of 180 s, with 1 repeat count and 180 s repeat duration. Additional targeted ETD-MS/MS experiments were performed, but only on precursors identified as O-GlcNAc-modified during the previous data-dependent experiments. ETD-MS/MS scans were acquired for the total duration of the peptide chromatographic peak. These MS/MS scans were then averaged, to obtain the final ETD-MS/MS spectrum.
LC-MS/MS data processing
LC-MS/MS data, acquired with Xcalibur 2.1.0.1140 software (Thermo Fisher Scientific), were processed with Visual Basic software (Microsoft, Redmond, WA, USA) developed with XRawfile libraries (Thermo Fisher Scientific). The program generates an MS/MS peak list (MGF file) that is used for database searching. This MGF file contains the exact parent mass and the retention time associated with each LTQ-MS/MS spectrum.
Database searching
An internal MASCOT 2.1 server (Matrix Science, Boston, MA, USA; http://www.matrixscience.com/) using the Swiss-Prot database was used to identify peptides and their PTMs.
The PTM search parameters used were an increase of +57.02146 Da on cysteine residues (fixed carboxyamidomethylation), +15.99491 Da addition on methionine residues (dynamic oxidation), +42.010565 on protein N-terminal residues (dynamic N-terminal acetylation), +79.966331 on serine and threonine residues (dynamic phosphorylation), and +203.079373 on serine/threonine residues (dynamic O-GlcNAcylation). The precursor mass tolerance was set to 5 ppm, and the fragment ion tolerance was set to 0.5 Da. The number of missed cleavage sites for trypsin was set to 3. Identified O-GlcNAc-modified peptide sequences and the exact site of modification were confirmed by manual interpretation of the targeted ETD-MS/MS spectra.
RESULTS
β-Catenin, OGT, and O-GlcNAcylation levels are higher in colon tumor tissues than in tumor-adjacent normal tissues
Human colon tumor tissues and tumor-adjacent normal tissues from 4 patients (see Table 1 for information about the tumor samples) were isolated, and levels of β-catenin, O-GlcNAc, and OGT were analyzed by Western blot (Fig. 1). Levels of β-catenin, O-GlcNAc, and OGT are elevated in tumor tissues compared to levels in tumor-adjacent normal tissue, and no mutation in β-catenin sequence was detected in the patient samples (data not shown). In addition, our previous work suggested that metabolic disorders, characterized by an up-regulation of HBP, high O-GlcNAcylation, and changes in β-catenin expression, correlate with an increase in occurrence of CRC (19). With these data, we hypothesized that, using cellular and mouse models, we could better characterize the mechanism by which OGT, O-GlcNAcylation, and β-catenin levels correlate in CRC.
Figure 1.
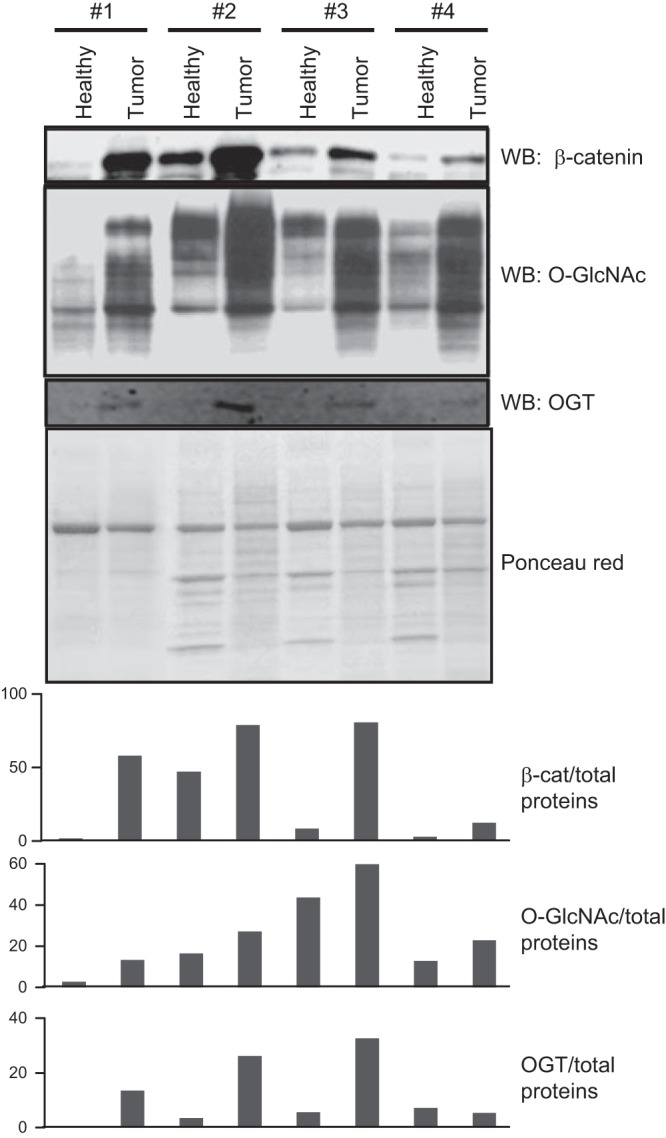
Human colorectal tumor tissue exhibits higher levels of β-catenin, O-GlcNAcylation, and OGT than does healthy tissue. Colon tumor and matching tumor-adjacent normal tissues from the same patient were analyzed by Western blot for levels of β-catenin, O-GlcNAcylation, and OGT contents. Ponceau red staining of the whole extracts confirmed equal loading (40 μg proteins/lane).
HCD or OGA inhibition enhance O-GlcNAcylation and β-catenin expression in vivo
To further characterize the relationship between O-GlcNAc and β-catenin in CRC, we profiled C57BL6 mice fed an HCD (Fig. 2A). At 9 wk after the beginning of the HCD, the mice were denied access to food overnight, and an OGTT was performed. Compared to the mice fed an SD, those fed an HCD had a slight (nonsignificant) increase in blood glucose. Colons of the unfed mice were collected and homogenized, and lysates were analyzed by Western blot. In the mice fed an HCD, we observed higher levels of β-catenin expression and O-GlcNAc in comparison to those fed an SD. In addition, when Thiamet-G, a potent OGA inhibitor, was injected into the peritoneum daily for 2 wk, colonic β-catenin levels increased (Fig. 2B), whereas blood glucose levels were not affected. These observations reinforce the hypothesis that perturbation of O-GlcNAc levels through an HCD or enzyme inhibition correlates with an elevation of β-catenin expression.
Figure 2.

Mice fed an HCD or injected with thiamet G exhibit increased β-catenin and O-GlcNAc levels. A) Colon homogenates from mice fed an HCD or an SD for 9 wk were analyzed by Western blot for β-catenin, O-GlcNAc, and GAPDH (loading control) levels. OGTTs were performed, and blood glucose levels were determined for the HCD- and SD-fed mice by overnight food withdrawal after glucose force-feeding (differences are statistically nonsignificant). B) Colon homogenates from mice treated with the OGA inhibitor thiamet G for 2 wk were analyzed as in panel A. Anti-O-GlcNAc antibody condition was treated with free N-acetylglucosamine to show specificity. Glycemic levels were measured for the same animals with no significant differences observed. A, B) Average ± sd ratios of β-catenin/GAPDH and O-GlcNAc/GAPDH are shown. *P < 0.05, **P < 0.01, ***P < 0.001.
T41 O-GlcNAcylation and phosphorylation influence degradation of β-catenin
In MCF7 cells, which are free of Wnt signaling mutations, O-GlcNAcylation interferes with β-catenin proteasomal degradation (19). To better understand the mechanism responsible for β-catenin stabilization by O-GlcNAcylation, we transfected MCF7 cells with a FLAG-tagged β-catenin, an HA-tagged ubiquitin, and siRNA was used to down-regulate OGA expression (Fig. 3A). After the cells were treated with the proteasome inhibitor MG132 to stabilize the ubiquitinated forms of β-catenin, the β-catenin was immunoprecipitated. Western blots were probed for ubiquitin via the HA tag. Results showed that siOGA (or NButGT, data not shown) reduced the level of ubiquitinated β-catenin.
Figure 3.

O-GlcNAcylation occurs in the D box of β-catenin and reduces its ubiquitinylation. A) MCF7 cells were cotransfected with vectors encoding FLAG-β-catenin and ubiquitin-HA and treated with siOGA and MG132. β-Catenin immunoprecipitates were analyzed by Western blot with anti-FLAG and anti-HA antibodies. B) Schematic representation of the β-catenin structure. The N-terminal region contains a D box, in which phosphorylation of 4 specific residues drives the ubiquitinylation and the subsequent proteasomal degradation of β-catenin. CK1α first phosphorylates S45, and then GSK3β successively modifies T41, S37, and S33. To investigate the O-GlcNAcylation of the D box, we used a mutant form of β-catenin in which these 4 phosphorylated residues were mutated into alanine (TM). The central region of β-catenin contains 12 imperfect sequence repeats of 42 aa (the armadillo repeats) that are essential in the interaction with many partners, including cadherins, APC, and TCF/LEF. The C-terminal region displays the transactivator function required for activation of target genes. C) MCF7 cells were transfected with FLAG-β-catenin (WT), TM-FLAG-β-catenin (TM), or an empty vector (Mock). At 48 h after transfection, the cells were lysed, and the lysates were analyzed by Western blot with anti-FLAG and -actin (loading control) antibodies. D) MCF7 cells were transfected with the vectors described above. At 36 h after transfection, the cells were treated with or without 5 mM glucosamine for 16 h and lysed, and β-catenin was immunoprecipitated with an anti-FLAG antibody. Immunoprecipitates were analyzed by Western blot with anti-O-GlcNAc and anti-FLAG antibodies. Average ± sd ratios of O-GlcNAcylated-β-catenin/total β-catenin are shown. **P < 0.01.
Ubiquitination and subsequent targeting of β-catenin to the proteasome is controlled by phosphorylation of key residues (S33/S37/T41/S45) located within its D box (Fig. 3B). The priming site of phosphorylation resides at S45 and is driven by CK1α. This phosphorylation is followed by the phosphorylation of T41 and finally S37 and S33 by GSK3β. Then, the E3 ubiquitin ligase β-TrCp binds to phospho-S33 and phospho-S37 (5), leading to the ubiquitination of the K19 and K49 moieties (22), after which β-catenin is targeted for degradation (23). To determine whether O-GlcNAcylation competes with phosphorylation to modify the β-catenin D box, we examined the level of O-GlcNAcylation on a TM form of β-catenin in which S33, S37, T41, and S45 were replaced by alanine (Fig. 3B). As previously demonstrated, this mutant is more stable than the WT β-catenin (ref. 4 and Fig. 3C). After transfection with WT or TM FLAG β-catenin, MCF7 cells were treated with MG132, to freeze β-catenin degradation, and glucosamine, to increase UDP-GlcNAc production while bypassing the rate-limiting enzyme of the HBP, GFAT. Equal amounts of β-catenin were immunopurified, and O-GlcNAc levels were analyzed by Western blot (Fig. 3D). Addition of glucosamine enhanced the O-GlcNAcylation of WT β-catenin but not the glycosylation of TM, suggesting that β-catenin is O-GlcNAcylated at one or several of the 4 phosphorylation sites that drive the ubiquitination of β-catenin.
To determine the site at which β-catenin is O-GlcNAcylated in a biologically relevant cancer cell line, HT29, we used ETD-MS/MS. Along with nano-LC/MS/MS, which profiles different peptide retention times, ETD-MS/MS allowed the production of specific c- and z-type fragment ions without the loss of labile PTM (Fig. 4A). ETD-MS/MS analyses of peaks corresponding to modified peptides revealed 4 O-GlcNAcylation sites located at S23, T40, T41, and T112 (Fig. 4B and Supplemental Figs. S1–S4). In addition, 2 novel and 7 previously described phosphorylation sites were mapped. The O-GlcNAcylation and phosphorylation sites identified in this study are depicted in Fig. 5 and listed in Table 2.
Figure 4.

β-Catenin is O-GlcNAcylated at S23, T40, T41, and T112 in HT29 cells. A) Nano-LC-ETD MS/MS analysis of an in-gel β-catenin trypsin digest. a) Total ion current (TIC) chromatogram, m/z 300–1700. b) Reconstructed ion chromatogram (RIC) of the [M+4H]4+ ion, m/z 772.1298, corresponding to the unmodified peptide, residues 20–49 of β-catenin. c) RIC of the [M+4H]4+ ion, m/z 822.8996, corresponding to 3 differentially O-GlcNAc-modified peptides, residues 20–49 of β-catenin. d) RIC of the [M+3H]3+ ion, m/z 1068.1693, corresponding to the unmodified peptide, residues 96–124 of β-catenin. e) RIC of the [M+3H]3+ ion, m/z 1135.8624, corresponding to the O-GlcNAc modified peptide, residues 96–124 of β-catenin. NL, normalized level. B) Sequencing by ETD-MS/MS of precursor ions corresponding to the O-GlcNAc residues S23, T40, T41, and T112 in β-catenin. a) ETD-MS/MS spectrum of a tryptic O-GlcNAc-modified peptide precursor ion at m/z 822.8996 [M+4H]4+ from β-catenin. MS/MS scans were acquired for the total duration of the chromatographic peak at a retention time of 69.4 min, as shown in panel Ac. Thirty MS/MS spectra were averaged to obtain this spectrum. The peptide AAVS(GlcNAc)HWQQQSYLDSGIHSGATTTAPSLSGK (residues 20–49 of β-catenin) contained 1 site of O-GlcNAc modification at S23. The c ion series, c4 to c13 (c4 being modified), and the z2+ ion series, z172+ to z292+ (z272+ being modified), demonstrate that S23 is O-GlcNAcylated. Fragments carrying the O-GlcNAc moiety are indicated by an asterisk. b) ETD-MS/MS spectrum of a tryptic O-GlcNAc modified peptide precursor ion at m/z 822.900 [M+4H]4+ from β-catenin. MS/MS scans were acquired for the total duration of the chromatographic peak at a retention time of 66.2 min, as shown in panel Ac. Thirty MS/MS spectra were averaged to obtain this spectrum. The peptide AAVSHWQQQSYLDSGIHSGATT(GlcNAc)TAPSLSGK (residues 20–49 of β-catenin) contained 1 site of O-GlcNAc modification at T41. The z ion series, z3 to z17 (z9 being modified); the y ion series, y3 to y10 (y9 being modified); and the c2+ ion series, c172+ to c292+ (c222+ being modified) demonstrate that T41 is O-GlcNAcylated. Fragments carrying the O-GlcNAc moiety are indicated by an asterisk. c) ETD-MS/MS spectrum of a tryptic O-GlcNAc modified peptide precursor ion at m/z 822.900 [M+4H]4+ from β-catenin. MS/MS scans were acquired for the total duration of the chromatographic peak at a retention time of 68.6 min, as shown in panel Ac. Thirty MS/MS spectra were averaged to obtain this spectrum. The peptide AAVSHWQQQSYLDSGIHSGAT(GlcNAc)TTAPSLSGK (residues 20–49 of β-catenin) contained 1 site of O-GlcNAc modification at T40. The z ion series, z4 to z17 (z10 being modified), and the c2+ ion series, c172+ to c292+ (c212+ being modified), demonstrates that T40 is O-GlcNAcylated. Fragments carrying the O-GlcNAc moiety are indicated by an asterisk. d) ETD MS/MS spectrum of a tryptic O-GlcNAc modified peptide precursor ion at m/z 1135.8624 [M+3H]3+ from β-catenin. MS/MS scans were acquired for the total duration of the chromatographic peak at a retention time of 103.3 min, as shown in panel Ae. Thirty MS/MS spectra were averaged to obtain this spectrum. The peptide AAMFPETLDEGMQIPST(GlcNAc)QFDAAHPTNVQR (residues 96–124 of β-catenin) contained 1 site of O-GlcNAc modification at T112. The z ion series, z4 to z14 (z13 being modified), demonstrates that T112 is O-GlcNAcylated. Fragments carrying the O-GlcNAc moiety are indicated by an asterisk. Horizontal brackets: charge reduced ions and fragment.
Figure 5.
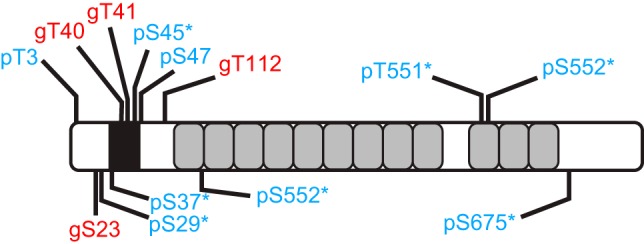
O-GlcNAcylation (red) and phosphorylation (blue) sites mapped in the study. Asterisks indicate that the corresponding phosphorylation sites are described in the literature. Black box: D box; gray boxes: armadillo repeats.
Table 2.
List of the O-GlcNAcylation and phosphorylation sites mapped in the study
| Residues | Sequence | Modified residue | Mascot ion score |
|---|---|---|---|
| 2-19 | AT(Pho)QADLMELDMAMEPDRK | 3 | 46 |
| 20-49 | AAVS(GlcNAc)HWQQQSYLDSGIHSGATTTAPSLSGK | 23 | 36 |
| AAVSHWQQQSYLDSGIHSGAT(GlcNAc)TTAPSLSGK | 40 | M | |
| AAVSHWQQQSYLDSGIHSGATT(GlcNAc)TAPSLSGK | 41 | M | |
| AAVSHWQQQS(Pho)YLDSGIHSGATTTAPSLSGK | 29 | M | |
| AAVSHWQQQYLDSGIHS(Pho)GATTTAPSLSGK | 37 | M | |
| AAVSHWQQQSYLDSGIHSGATTTAPS(Pho)LSGK | 45 | 76 | |
| AAVSHWQQQSYLDSGIHSGATTTAPSLS(Pho)GK | 47 | M | |
| 96-124 | AAMFPETLDEGMQIPST(GlcNAc)QFDAAHPTNVQR | 112 | 38 |
| 186-200 | HAIMRS(Pho)PQMVSAIVR(T) | 191 | 39 |
| 191-200 | S(Pho)PQMVSAIVR(T) | 191 | 48 |
| 550-565 | RT(Pho)SMGGTQQQFVEGVR(M) | 551 | 82 |
| 550-565 | RTS(Pho)MGGTQQQFVEGVR(M) | 552 | 107 |
| 551-565 | T(Pho)SMGGTQQQFVEGVR(M) | 551 | 98 |
| 551-565 | TS(Pho)MGGTQQQFVEGVR(M) | 552 | 93 |
| 672-684 | KRLS(Pho)VELTSSLFR(T) | 675 | 75 |
| 673-684 | RLS(Pho)VELTSSLFR(T) | 675 | 73 |
Phosphorylation (Pho) and O-GlcNAcylation (GlcNAc) sites are underscored.
We next investigated whether the O-GlcNAcylation of β-catenin influences its phosphorylation status (Fig. 6). First, we tested increasing glucose amounts and a condition in which glucosamine was used on 3 colon cell lines: CCD841CoN, a normal fetal cell line, and HT29 and HCT116, 2 colon cancer cell lines (Fig. 6A). Equal amounts of endogenous β-catenin were immunoprecipitated and analyzed according to their O-GlcNAc content and their phosphorylation status at T41/S45. We observed that these conditions increased the O-GlcNAcylation of β-catenin in the HT29 and HCT116 cells and that glucosamine (and glucose for HT29) treatment correlated with a decrease in phosphorylation at T41/S45. A longer exposure was necessary to detect the T41 phosphorylation of β-catenin in the HCT116 cells, as S45 is deleted in this cell line. Whereas the CCD841CoN cells showed a decreased in their T41/S45 phosphorylation status, the O-GlcNAcylation of β-catenin was below the detection limit, which is not surprising, as normal cells have lower levels of O-GlcNAc than cancer cells have (19, 24–26). Knockdown of OGA interfered with the phosphorylation of β-catenin in the HT29 and HCT116 cells, whereas no effect was observed in the CCD841CoN cells (Fig. 6B), consistent with the lack of detection of β-catenin O-GlcNAcylation in this cell line in contrast with HT29 and HCT116 (Fig. 6A). Last, a decrease in pT41-β-catenin was observed in HEK293 cells transfected with WT FLAG-β-catenin and treated with NButGT (Fig. 6C). Together, our results support a reciprocal relationship between phosphorylation and Ο-GlcNAcylation at T41 of β-catenin.
Figure 6.

O-GlcNAcylation of β-catenin D box competes with its phosphorylation. A) CD841CoN, HT29, and HCT116 cells were cultured in the presence of increasing concentrations of glucose (Glc) or with glucosamine (GlcNH2). Whole-cell lysates were probed for O-GlcNAc level and phosphorylation of β-catenin at T41 and S45. Equal amounts of immunopurified β-catenin were loaded and analyzed according to their O-GlcNAc levels by immunoblot. B) CD841CoN, HT29, and HCT116 cells were transfected with either siOGA or siCtrl and incubated with MG132. β-Catenin, phospho-β-catenin, O-GlcNAcylation, OGA, and GAPDH (loading control) levels were determined by immunoblot. C) HEK293 cells were transfected with WT FLAG-β-catenin and incubated with MG132, with or without NButGT. Cell lysates were immunopurified with an anti-FLAG antibody, and the immunoprecipitates were analyzed with a phospho-T41-dependent antibody.
O-GlcNAcylation at S23, T40, and T112 is not involved in β-catenin ubiquitinylation and stability
We generated single-point mutants for all O-GlcNAc-modified sites of β-catenin, as well as another TM (4M: S23A/T40A/T41A/T112A; Fig. 7A). We investigated the protein's expression (Fig. 7B), subcellular localization (Fig. 7C), and ubiquitinylation (Fig. 7D) in HEK293T cells. We observed a higher expression of the T41A mutant (and consequently of the mutant 4M) compared with WT β-catenin (Fig. 7B). However, we found no difference in S23A and a slight increase in T40A and T112A. Subcellular fractionation showed that the T41A single-point mutant and 4M accumulate in the cytoplasm, suggesting stabilization of the protein (Fig. 7C, F1). Cotransfection of the different β-catenin mutants with HA-ubiquitin demonstrated that T41A and 4M were less ubiquitinated than WT β-catenin, whereas we observed a faint decrease in ubiquitinylation of the S23A, T40A, and T112A mutants in comparison with WT β-catenin (Fig. 7D). Overall, these results support that O-GlcNAcylation and phosphorylation at T41 rather than modification at S23, T40, or T112 regulates β-catenin ubiquitinylation and stability.
Figure 7.

O-GlcNAcylation at T41, but not at S23, T40, or T112, regulates β-catenin ubiquitinylation and stability. A) Schematic representation of β-catenin showing the O-GlcNAcylation sites mapped in the study. Single-point mutants and a tetramutant of β-catenin (4M: S23A/T40A/T41A/T112A) were generated. B) HEK293T cells were transfected either with a WT or mutant FLAG-β-catenin or with an empty vector (mock). At 36 h after transfection, the cells were treated with or without 1 μM MG132 for 16 h. Cell lysates were analyzed by Western blot for β-catenin using the anti-FLAG tag and anti-GAPDH antibodies (loading control). C) Cells were transfected as described in panel B. At 48 h after transfection, a subcellular fractionation was performed, and distribution of FLAG-β-catenin in each fraction (F1, cytosol; F2, membranes; F3, nucleus; and F4, cytoskeleton) was investigated by Western blot analysis. Antibodies directed against GAPDH (F1), E-cadherin (F2 and F4), H2B (F3), and CK8 (F4) were used to ensure the purity of each fraction. D) Cells were cotransfected with the different β-catenin constructs and with a ubiquitin-HA-expressing vector. At 36 h after transfection, the cells were treated with 1 μM MG132 for 16 h and lysed, and immunoprecipitation was performed with an anti-FLAG antibody. β-Catenin immunoprecipitates were analyzed by Western blot for β-catenin (anti-FLAG) and ubiquitin (anti-HA). Asterisk indicates a nonspecific band. IgG HC, immunoglobulin G heavy chain; Ub-β-cat, ubiquitinated β-catenin.
O-GlcNAcylation interferes in the α-catenin–β-catenin interaction
Among the 4 O-GlcNAcylation sites that we identified, only T41 (known also to be a phospho-site) appeared to be involved in β-catenin stabilization, suggesting that O-GlcNAcylation regulates other functions of β-catenin. T112 is located in the β-catenin–α-catenin interaction domain, and it has been proposed that this residue is phosphorylated by CK2 to increase the affinity between β- and α-catenins (27). We hypothesized that O-GlcNAcylation would affect the β-catenin interaction at the adherens junctions by changing this interaction. To answer this question, we inhibited O-GlcNAcylation by treatment of MCF7 cells with Ac-5SGlcNAc, an inhibitor of OGT (28), and tested the interaction of β-catenin with α-catenin and E-cadherin by coimmunoprecipitation (Fig. 8A). We observed that Ac-5SGlcNAc had no effect on the interaction between β-catenin and E-cadherin. On the other hand, decreased O-GlcNAcylation clearly diminished the interaction between the α- and β-catenins. Such a decrease was also observed when OGT was silenced with siRNA (Fig. 8B), whereas knockdown of the expression of OGA enhanced the interaction between the 2 catenins (Fig. 8C). However, coimmunoprecipitation experiments between the different mutants of O-GlcNAcylation of β-catenin (described in Fig. 7A) and α-catenin revealed no significant difference in comparison with the WT protein (Fig. 9).
Figure 8.

Perturbation of O-GlcNAc level disturbs the interaction between β-catenin and α-catenin. A) MCF7 cells were treated with the potent OGT inhibitor Ac5S-GlcNAc at a concentration of 100 μM for 16 h or with DMSO as a vehicle control and with MG132 at 1 μM. The cells were lysed, and coimmunoprecipitation was performed with an anti-β-catenin antibody. Nonimmune rabbit IgG was used as a negative control. Immunoprecipitates and 1% of whole-cell lysates (input) were analyzed by Western blot with the indicated antibodies. (Note that the anti-O-GlcNAc antibody was used with and without free N-acetylglucosamine.) B) MCF7 cells were transfected with a nontarget siRNA (siCtrl) or with an OGT siRNA (siOGT). The cells were treated with 1 μM MG132 for 16 h. At 72 h after transfection, the cells were lysed, and coimmunoprecipitation and Western blot analysis were performed as in panel A, C) The same procedure as in panel B was performed, except that an OGA siRNA (siOGA) was used.
Figure 9.
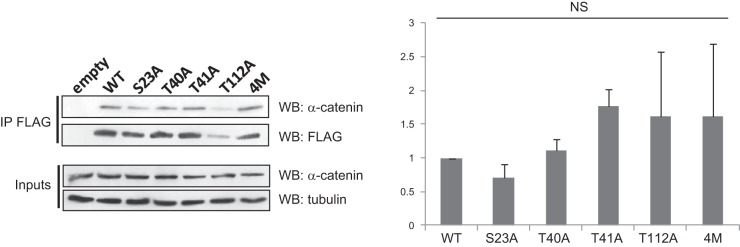
O-GlcNAcylation of β-catenin at S23, T40, T41, and T112 does not affect its interaction with α-catenin. HEK293T cells were transfected with WT or mutant FLAG-β-catenin or an empty vector (Mock). At 36 h after transfection, the cells were treated with 1 μM MG132 for 16 h. The cells were lysed, and β-catenin was immunoprecipitated with the FLAG Tag. Immunoprecipitated β-catenin and 1% of the whole-cell lysate (inputs) were analyzed by Western blot with the indicated antibodies. Densitometric values of 3 distinct experiments are represented. NS, not significant.
DISCUSSION
Cancer is the second leading cause of death in Western societies, next to cardiovascular disease. Modern lifestyles, characterized by junk-food diets, low energy expenditure, and metabolic disorders, increase the risk of cancer (29). Intriguingly, several groups reported that O-GlcNAcylation and OGT are more elevated in cancer-derived cells (breast, ref. 24; pancreas, ref. 25; prostate, ref. 26; and colon, ref. 19; see also refs. 30, 31 for reviews), when compared to normal cells.
In this study, O-GlcNAcylation and OGT were highly expressed in tumors from patients with CRC, each displaying at least 1 problem related to metabolic disorder: diabetes, hypertension, overweight, or dyslipidemia (Table 1). O-GlcNAc levels are closely tied to those of the donor UDP-GlcNAc, which is implicated as a key player in many metabolic pathways (31, 32). We proposed that there is a key relationship between metabolic disorders and cancers that is governed by nutrient flux.
Cancer cells change their metabolism and increase glucose utilization through the Warburg effect (33). Indeed, an initial observation noted that the wing of a hen with Rous sarcoma produced more lactic acid than the normal wing (34, 35). It was therefore originally suggested that a deficiency in glucose metabolism occurs at the origin of cancer (36). More recently, the metabolism shift found in tumor cells was found to be a consequence rather than a cause of cancer, and oncogenic signaling pathways controlling HIF1 are responsible for this change (37). Interestingly, the use of both glucose and glutamine are up-regulated in cancer cells (38), and both nutrients are used to generate UDP-GlcNAc, the donor for the O-GlcNAcylation processes (31, 32). In cancer cells, increased glucose affects glycolysis, the HBP, and the pentose phosphate pathway (PPP). Indeed, regarding the PPP, it has been recently demonstrated that O-GlcNAcylation of the glycolysis rate-limiting enzyme PFK1 at S529 interferes with the binding of the metabolic sensor fru-2,6-bis-phosphate (39). Glucose is then diverted to generate 6-phospho-gluconate, a precursor of pentoses needed for nucleic acid synthesis during active cell division. Thus, cancer cells consume their glucose differently; nonoxidative consumption of glucose is sufficient to generate enough ATP to proliferate, and above all, it allows the production of nucleic acids, amino acids, glycans, and lipids, to increase the biomass. We suggest that HBP activation contributes to hexosamine synthesis for cellular structure construction as well as regulation of metabolic flux, signaling pathways, and cell homeostasis processes. β-Catenin, the central component of Wnt signaling and an O-GlcNAcylated protein, perfectly fits this hypothesis.
Three of the 4 O-GlcNAc sites identified on β-catenin are located within the protein flexible domain (S23/T40/T41; Fig. 7A). This domain concentrates the phosphorylation and ubiquitination sites that regulate the fate of β-catenin. First, CK1α modifies S45 and subsequently T41, S37, and S33 are modified by GSK3β. APC then prevents the dephosphorylation of the D box (40). The E3 ubiquitin ligase β-TrCp binds to phospho-S33 and phospho-S37 (5) and ubiquitinates of K29 and K49 (22), thus targeting β-catenin to the proteasomal pathway (23). S23 is mutated in hepatocellular cancers (41) and phosphorylated by GSK3β (42), but its function is not well understood, although it has been suggested that it takes part in embryogenesis and carcinogenesis. We hypothesized that the proximity of S23 with K19 suggests that the S23 O-GlcNAcylation interferes with the K19 ubiquitination, but we found only a slight decrease in the ubiquitination of S23A-β-catenin when compared to WT (Fig. 7D). Last, it has been demonstrated that the β-catenin protein homologue plakoglobin is O-GlcNAcylated at T14, equivalent to β-catenin S23 (43). The O-GlcNAcylation of plakoglobin at T14 prevents its proteasomal degradation emphasizing the importance of β-catenin S23 O-GlcNAcylation (Fig. 7).
The fourth O-GlcNAcylation site was identified as T112 in the interaction domain with α-catenin. It has been proposed that phosphorylated T112 yields an increase in affinity between β- and α-catenins (27). Although perturbing the O-GlcNAcylation processes interfered with the α-/β-catenin interaction (Fig. 8), none of the β-catenin mutants exhibited a drastic change in the α-catenin interaction shown by immunoprecipitation (Fig. 9).
When mutated, the GSK3β substrate T41, but not T40, causes the most changes in β-catenin ubiquitination, expression, and cytosolic accumulation (Fig. 7). Phospho-modified T41 is a checkpoint allowing the subsequent phosphorylation of S33 and S37. The proteasome machinery recognizes phospho-modified S33 and S37, as required for β-catenin degradation. Logically, S33, S37, T41, and S45 are frequently mutated in cancers, leading to an uncontrolled stabilization of β-catenin and an increase in its oncogenic features. By competing with phosphorylation on T41, O-GlcNAcylation stabilizes β-catenin in the same manner and may increase its oncogenic properties as well. Interestingly, metabolic disorders, such as type 2 diabetes, correlate with elevated levels of CRC (14–16). We propose that increased levels of glucose (by nutrient excess, unhealthy diet, or metabolic disorders) cause or exacerbate cancer emergence, especially CRC. Indeed, elevation of CRC incidence may occur due to O-GlcNAcylation of WT nonmutated β-catenin. Our study provides a mechanistic explanation of how nutrition and cancer are linked and support that T41 is a critical residue for β-catenin regulation.
Supplementary Material
Acknowledgments
The authors thank Prof. D. Vocadlo (Simon Fraser University, Burnaby, BC, Canada), who provided NButGT and Ac-5SGlcNAc; Dr. C. Couturier (Biology Institute of Lille, France), who provided the HA-ubiquitin expression; Dr. C. Liu (University of Kentucky, Lexington, KY, USA), who provided pCS2+β-catenin-2XFlag (WT and TM); the Site de Recherche Intégrée sur le Cancer ONCOLille, and the Tumor Bank [Regional Reference Center in Cancer, Centre Hospitalier Régional Universitaire (CHRU)] of Lille (Lille, France) for providing colon tumor tissues and matching tumor-adjacent normal tissues; and Michelle Bond [National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), U.S. National Institutes of Health, Bethesda, MD, USA] for editing the English in the manuscript and for helpful comments.
This work was supported by the Ligue Contre le Cancer/Comité du Nord, the Association pour la Recherche sur le Cancer, the Region Nord-Pas de Calais (Cancer Regional Program), Lille 1 University, and the Centre National de la Recherche Scientifique. V.D. is the recipient of a postdoctoral fellowship from the Ligue Contre le Cancer.
The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- APC
- adenomatous polyposis coli
- CRC
- colorectal cancer
- D box
- destruction box
- DMEM
- Dulbecco's modified Eagle's medium
- EDT-MS/MS
- electron-transfer dissociation–tandem mass spectrometry
- GFAT
- glutamine:fructose-6-phosphate amidotransferase
- HBP
- hexosamine biosynthesis pathway
- HCD
- high-carbohydrate diet
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- OGA
- O-linked N-acetylglucosaminase
- O-GlcNAc
- O-linked N-acetylglucosamine
- OGT
- O-linked N-acetylglucosamine transferase
- OGTT
- oral glucose tolerance test
- PBS
- phosphate-buffered saline
- PPP
- pentose phosphate pathway
- PTM
- posttranslational modification
- SD
- standard diet
- siCtrl
- scrambled control
- siRNA
- small interfering RNA
- TM
- tetramutant
- WT
- wild type
REFERENCES
- 1. Clevers H. (2006) Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 2. Reya T., Clevers H. (2005) Wnt signalling in stem cells and cancer. Nature 434, 843–850 [DOI] [PubMed] [Google Scholar]
- 3. Meng W., Takeichi M. (2009) Adherens junction: molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 1, a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu C., Kato Y., Zhang Z., Do V. M., Yankner B. A., He X. (1999) Beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc. Natl. Acad. Sci. U. S. A. 96, 6273–6278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu C., Li Y., Semenov M., Han C., Baeg G. H., Tan Y., Zhang Z., Lin X., He X. (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 [DOI] [PubMed] [Google Scholar]
- 6. Kimelman D., Xu W. (2006) Beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene 25, 7482–7491 [DOI] [PubMed] [Google Scholar]
- 7. He T. C., Sparks A. B., Rago C., Hermeking H., Zawel L., da Costa L. T., Morin P. J., Vogelstein B., Kinzler K. W. (1998) Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512 [DOI] [PubMed] [Google Scholar]
- 8. Polakis P. (2012) Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 4, a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shtutman M., Zhurinsky J., Simcha I., Albanese C., D'Amico M., Pestell R., Ben-Ze'ev A. (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. U. S. A. 96, 5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daniels D. L., Weis W. I. (2005) Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat. Struct. Mol. Biol. 12, 364–371 [DOI] [PubMed] [Google Scholar]
- 11. Ma F., Zhang J., Zhong L., Wang L., Liu Y., Wang Y., Peng L., Guo B. (2014) Upregulated microRNA-301a in breast cancer promotes tumor metastasis by targeting PTEN and activating Wnt/β-catenin signaling. Gene 535, 191–197 [DOI] [PubMed] [Google Scholar]
- 12. De La Coste A., Romagnolo B., Billuart P., Renard C. A., Buendia M. A., Soubrane O., Fabre M., Chelly J., Beldjord C., Kahn A., Perret C. (1998) Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. U. S. A. 95, 8847–8851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morin P. J., Sparks A. B., Korinek V., Barker N., Clevers H., Vogelstein B., Kinzler K. W. (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275, 1787–1790 [DOI] [PubMed] [Google Scholar]
- 14. Khaw K. T., Wareham N., Bingham S., Luben R., Welch A., Day N. (2004) Preliminary communication: glycated hemoglobin, diabetes, and incident colorectal cancer in men and women: a prospective analysis from the European prospective investigation into cancer-Norfolk study. Cancer Epidemiol. Biomarkers Prev. 13, 915–919 [PubMed] [Google Scholar]
- 15. Ahmed R. L., Schmitz K. H., Anderson K. E., Rosamond W. D., Folsom A. R. (2006) The metabolic syndrome and risk of incident colorectal cancer. Cancer 107, 28–36 [DOI] [PubMed] [Google Scholar]
- 16. Pais R., Silaghi H., Silaghi A. C., Rusu M. L., Dumitrascu D. L. (2009) Metabolic syndrome and risk of subsequent colorectal cancer. World J. Gastroenterol. 15, 5141–5148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lefebvre T., Baert F., Bodart J. F., Flament S., Michalski J. C., Vilain J. P. (2004) Modulation of O-GlcNAc glycosylation during Xenopus oocyte maturation. J. Cell Biochem. 93, 999–1010 [DOI] [PubMed] [Google Scholar]
- 18. Marshall S., Bacote V., Traxinger R. R. (1991) Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system: role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 266, 4706–4712 [PubMed] [Google Scholar]
- 19. Olivier-Van Stichelen S., Guinez C., Mir A. M., Perez-Cervera Y., Liu C., Michalski J. C., Lefebvre T. (2012) The hexosamine biosynthetic pathway and O-GlcNAcylation drive the expression of β-catenin and cell proliferation. Am. J. Physiol. Endocrinol. Metab. 302, E417–424 [DOI] [PubMed] [Google Scholar]
- 20. Guinez C., Mir A. M., Dehennaut V., Cacan R., Harduin-Lepers A., Michalski J. C., Lefebvre T. (2008) Protein ubiquitinylation is modulated by O-GlcNAc glycosylation. FASEB J. 22, 2901–2911 [DOI] [PubMed] [Google Scholar]
- 21. Shevchenko A., Chernushevish I., Ens W., Standing K. G., Thomson B., Wilm M., Mann M. (1997) Rapid ‘de novo’ peptide sequencing by a combination of nanoelectrospray, isotopic labeling and a quadrupole/time-of-flight mass spectrometer. Rapid Comm. Mass. Spectrom. 11, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 22. Winer I. S., Bommer G. T., Gonik N., Fearon E. R. (2006) Lysine residues Lys-19 and Lys-49 of beta-catenin regulate its levels and function in T cell factor transcriptional activation and neoplastic transformation. J. Biol. Chem. 281, 26181–26187 [DOI] [PubMed] [Google Scholar]
- 23. Hart M., Concordet J. P., Lassot I., Albert I., del los Santos R., Durand H., Perret C., Rubinfeld B., Margottin F., Benarous R., Polakis P. (1999) The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr. Biol. 9, 207–210 [DOI] [PubMed] [Google Scholar]
- 24. Caldwell S. A., Jackson S. R., Shahriari K. S., Lynch T. P., Sethi G., Walker S., Vosseller K., Reginato M. J. (2010) Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 29, 2831–2842 [DOI] [PubMed] [Google Scholar]
- 25. Lynch T. P., Ferrer C. M., Jackson S. R., Shahriari K. S., Vosseller K., Reginato M. J. (2012) Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J. Biol. Chem. 287, 11070–11081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma Z., Vocadlo D. J., Vosseller K. (2013) Hyper O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-κB activity in pancreatic cancer cells. J. Biol. Chem. 288, 15121–15130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bek S., Kemler R. (2002) Protein kinase CKII regulates the interaction of beta-catenin with alpha-catenin and its protein stability. J. Cell Sci. 115, 4743–4753 [DOI] [PubMed] [Google Scholar]
- 28. Gloster T. M., Zandberg W. F., Heinonen J. E., Shen D. L., Deng L., Vocadlo D. J. (2011) Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat. Chem. Biol. 7, 174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dossus L., Kaaks R. (2008) Nutrition, metabolic factors and cancer risk. Best Pract. Res. Clin. Endocrinol. Metab. 22, 551–571 [DOI] [PubMed] [Google Scholar]
- 30. Fardini Y., Dehennaut V., Lefebvre T., Issad T. (2013) O-GlcNAcylation: a new cancer hallmark? Front. Endocrinol. 4, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma Z., Vosseller K. (2013) O-GlcNAc in cancer biology. Amino Acids 45, 719–733 [DOI] [PubMed] [Google Scholar]
- 32. Hanover J. A., Krause M. W., Love D. C. (2010) The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim. Biophys. Acta 1800, 80–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warburg O., Wind F., Negelein E. (1927) The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cori C. F., Cori G. T. (1925) The carbohydrate metabolism of tumors: I, the free sugar, lactic acid, and glycogen content of malignant tumors. J. Biol. Chem. 64, 11–22 [Google Scholar]
- 35. Cori C. F., Cori G. T. (1925) The carbohydrate metabolism of tumors: II, changes in the sugar, lactic acid, and CO2-combining power of blood passing through a tumor. J. Biol. Chem. 65, 397–405 [Google Scholar]
- 36. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 37. Shaw R. J. (2006) Glucose metabolism and cancer. Curr. Opin. Cell Biol. 18, 598–608 [DOI] [PubMed] [Google Scholar]
- 38. Deberardinis R. J., Sayed N., Ditsworth D., Thompson C. B. (2008) Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 18, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yi W., Clark P. M., Mason D. E., Keenan M. C., Hill C., Goddard W.A., 3rd, Peters E. C., Driggers E. M., Hsieh-Wilson L. C. (2012) Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337, 975–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Su Y., Fu C., Ishikawa S., Stella A., Kojima M., Shitoh K., Schreiber E. M., Day B. W., Liu B. (2008) APC is essential for targeting phosphorylated beta-catenin to the SCFbeta-TrCP ubiquitin ligase. Mol. Cell 32, 652–661 [DOI] [PubMed] [Google Scholar]
- 41. Legoix P., Bluteau O., Bayer J., Perret C., Balabaud C., Belghiti J., Franco D., Thomas G., Laurent-Puig P., Zucman-Rossi J. (1999) Beta-catenin mutations in hepatocellular carcinoma correlate with a low rate of loss of heterozygosity. Oncogene 18, 4044–4046 [DOI] [PubMed] [Google Scholar]
- 42. Van Noort M., van de Wetering M., Clevers H. (2002) Identification of two novel regulated serines in the N terminus of beta-catenin. Exp. Cell Res. 276, 264–272 [DOI] [PubMed] [Google Scholar]
- 43. Hatsell S., Medina L., Merola J., Haltiwanger R., Cowin P. (2003) Plakoglobin is O-glycosylated close to the N-terminal destruction box. J. Biol. Chem. 278, 37745–37752 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.