

Abstract
Hyperhomocysteinemia (HHcy) is prevalent in patients with hypertension and is an independent risk factor for aortic pathologies. HHcy is known to cause an imbalance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), leading to the accumulation of collagen in the aorta and resulting in stiffness and development of hypertension. Although the exact mechanism of extracellular matrix (ECM) remodeling is unclear, emerging evidence implicates epigenetic regulation involving DNA methylation. Our purpose was to investigate whether 5-aza-2′-deoxycytidine (Aza), a DNA methyltransferase (DNMT1) inhibitor, reduces high blood pressure (BP) by regulating aortic ECM remodeling in HHcy. Wild-type and cystathionine β-synthase (CBS)+/− HHcy mice were treated with Aza (0.5 mg/kg body weight). In HHcy mice, Aza treatment normalized the plasma homocysteine (Hcy) level and BP. Thoracic and abdominal aorta ultrasound revealed a reduction in the resistive index and wall-to-lumen ratio. Vascular response to phenylephrine, acetylcholine, and sodium nitroprusside improved after Aza in HHcy mice. Histology showed a marked reduction in collagen deposition in the aorta. Aza treatment decreased the expression of DNMT1, MMP9, TIMP1, and S-adenosyl homocysteine hydrolase (SAHH) and upregulated methylene tetrahydrofolate reductase (MTHFR). We conclude that reduction of DNA methylation by Aza in HHcy reduces adverse aortic remodeling to mitigate hypertension.—Narayanan, N., Pushpakumar, S. B., Givvimani, S., Kundu, S., Metreveli, N., James, D., Bratcher, A. P., Tyagi, S. C. Epigenetic regulation of aortic remodeling in hyperhomocysteinemia.
Keywords: DNA methylation, extracellular matrix, 5-aza-2′-deoxycytidine
Aortic disease is one of the most prevalent causes of increased mortality in the world (1–3). Several risk factors, including hyperglycemia, high serum cholesterol, and an elevated level of homocysteine (Hcy), known as hyperhomocysteinemia (HHcy), are associated with aortic aneurysm and atherosclerosis (4, 5). The vascular pathology associated with HHcy is characterized by excess extracellular matrix (ECM) turnover, causing increased deposition of collagen, leading to vessel stiffness (6, 7). Under physiological conditions, ECM homeostasis is maintained by the equilibrium between matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs). A previous study has demonstrated that HHcy increases the expression and activity of MMP9 and causes matrix degradation and accumulation of collagen in the ECM (8).
Arterial hypertension is associated with endothelial dysfunction (9). HHcy causes autooxidation of sulfhydryl groups, promoting reactive oxygen species production, which uncouples endothelial nitric oxide (NO) reaction to reduce NO synthesis and bioavailability (10–12). A reduction in NO signaling further increases superoxide generation, forming a vicious cycle, and impairs endothelial function. The functional consequence of endothelial dysfunction impairs vasodilation resulting in arterial hypertension (13, 14).
Hcy is a non-protein-coding amino acid synthesized by demethylation of methionine. The methyl group, which is excised during the synthesis of Hcy, is used in various methylation reactions involving DNA, proteins, amino acids, and Hcy (15). DNA methylation is one of the most intensely studied epigenetic mechanisms in the development of aortic diseases. There are 3 DNA methyltransferases (DNMTs) involved in DNA methylation: DNMT1 is involved in maintenance methylation, and DNMT3a and -3b catalyze de novo methylation (16). Current research is focusing on the use of DNMT inhibitors in several disease conditions. Decitabine or 5-aza-2′-deoxycytidine (Aza), a DNMT1 inhibitor, has been approved by the U.S. Food and Drug Administration (FDA) for treatment of myelodysplastic syndrome (MDS). Other inhibitors, such as Vidaza (5-aza cytidine) are currently in phase 2 and 3 cancer trials (17).
The purpose of the present study was to investigate the role of DNA methylation in aortic ECM remodeling and vascular dysfunction in HHcy-associated hypertension. We hypothesized that increased levels of Hcy and DNMT1 result in adverse ECM remodeling and endothelial dysfunction, leading to arterial hypertension. We also examined whether the DNMT1 inhibitor Aza could modulate ECM metabolism enzymes to mitigate hypertension. We report that Aza treatment in HHcy mice protects the aorta by regulating the epigenetic mechanism of genes involved in ECM metabolism.
MATERIALS AND METHODS
Antibodies and reagents
Monoclonal antibodies DNMT1, methylenetetrahydrofolate reductase (MTHFR; mouse) MMP9, TIMP1, and Hcy (rabbit) were purchased from Abcam (Cambridge, MA, USA), and the mouse polyclonal antibody S-adenosyl homocysteine hydrolase (SAHH) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The following secondary fluorescent antibodies were used: Texas Red anti-rabbit and Alexa Fluor 488 anti-mouse (Invitrogen, Carlsbad, CA, USA).
Animal models
Male C57BL/6J (wild-type; WT) and cystathionine β-synthase heterozygous knockout (CBS+/−; B6129P2) mice, aged 8–12 wk were obtained from Jackson Laboratories (Bar Harbor, ME, USA). The disruption of the CBS gene in the heterozygous model results in mild HHcy. All mice were fed standard chow (LabDiet 5010; LabDiet, St. Louis, MO, USA) and water ad libitum. The WT and CBS+/− mice were treated with Aza (Sigma-Aldrich, St. Louis, MO, USA) for 4 wk by intraperitoneal injection (0.5 mg/kg body weight; 3 consecutive times every week). Control animals received saline injections only. The animals were divided into 4 groups: WT, WT treated with Aza (WT+Aza), CBS+/−, and CBS+/− treated with Aza (CBS+/−+Aza). At least 4 animals (n≥4) were used in each group. All procedures were performed according to U.S. National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee of the University of Louisville School of Medicine.
Blood pressure (BP) measurements
BP was measured in conscious mice by the noninvasive tail cuff method (CODA; Kent Scientific, Torrington, CT, USA). The animals were allowed to acclimatize in the restraining chambers on a warm platform for short periods (20–30 min) for a few days before data were recorded. Under standard conditions (room temperature, lighting, and quiet surroundings), weekly BP was recorded before and after Aza treatment commenced, including systolic, diastolic, and mean pressures.
Ultrasound
With the mice under isoflurane anesthesia, ultrasound of the aorta was performed with the Vevo 2100 system (Visual Sonics, Toronto, ON, Canada), before and after Aza treatment. The thoracic and abdominal areas were depilated, and the animal was placed supine on a warm platform (37°C). A Vevo MS550D (22–55 MHz) transducer (Viual Sonics) was used to image the thoracic and abdominal aorta. Cross-sectional images of the aorta in B mode were used to measure wall thickness and lumen diameter to obtain the wall-to-lumen ratio. Peak systolic velocities (PSVs) and end-diastolic velocities (EDVs) were measured in pulsed-wave Doppler mode. The resistive index (RI) was calculated as RI = (PSV − EDV)/PSV.
Plasma Hcy levels
Vascular reactivity studies with aortic rings
Aortic ring preparation
Thoracic aortas were extracted from euthanized mice and immersed in Krebs solution (pH 7.4, 37°C) containing (in mM) 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 12.5 NaHCO3, and 10.9 d-glucose. Aortic rings were prepared by cutting 2-mm-wide segments and mounting them with 2 tungsten wire triangles (0.002 inch diameter; Scientific Instruments Services, Ringoes, NJ, USA) in a tissue myobath (Radnoti, Monrovia, CA, USA) that was continuously aerated with 95% O2-5% CO2. The rings were stretched to develop 0.5 g optimal resting tension and equilibrated for an hour to obtain baseline measurements with a DigiMed Tissue Force Analyzer (Micromed, Louisville, KY, USA; ref. 21).
Response to phenylephrine (Phe), acetylcholine (Ach), and sodium nitroprusside (SNP)
After a baseline recording was acquired, vasoconstriction was studied by adding different concentrations of Phe from 10−6 to 10−2 M to the organ bath to make a final concentration of 10−9 to 10−5 M, respectively. Once maximal constriction was reached at 10−5 M Phe, Ach was added to the organ bath in a similar manner, in gradually increasing concentrations, to detect endothelium-dependent vasorelaxation. The tissue rings were washed with fresh Krebs solution and preconstricted with a maximum dose of Phe, followed by 10−5 M SNP, to study endothelium-independent vasorelaxation. The tissue responses were recorded for 10 min for each drug concentration with DMSI-410 1.8.27 software (Micromed).
Cryosectioning
Aortic tissue was cryopreserved in Peel-A-Way disposable plastic tissue-embedding molds (Polysciences, Inc., Warrington, PA, USA) containing tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC, USA). The tissues were stored at −80°C until use. Serial sections of 7 μm thickness were made on a Cryocut (CM 1850; Leica Microsystems, Buffalo Grove, IL, USA). The cryosections were placed on Superfrost Plus microscope slides (Thermo Fisher Scientific, Pittsburgh, PA, USA), air dried, and stored at −80°C until further use.
Collagen staining
To measure the deposition of collagen in the aortic wall, we stained the sections with a Masson trichrome kit (Richard Allan Scientific, Kalamazoo, MI, USA), according to the manufacturer's instructions. Collagen deposition stained blue. Images were captured on a light microscope (Olympus FluoView1000; B&B Microscope Ltd., Pittsburgh, PA, USA). Sections were stained separately to differentiate type I and III collagen with a Picrosirius red staining kit (Polysciences, Inc.; ref. 22). Images were captured under a polarized light filter (Olympus FluoView1000; B&B Microscope).
Immunohistochemistry
Immunofluorescence staining was performed on 7-μm-thick aortic sections with anti-MTHFR, -MMP9, -TIMP1, -SAHH, -DNMT1, and -Hcy antibodies. Images were captured by confocal microscope (Olympus FluoView1000; B&B Microscope) and analyzed by Image Proplus 7.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Quantification of 5-methylcytosine (5-mC)
Genomic DNA was extracted from the aorta, and the amount of 5-mC was measured by ELISA, according to the manufacturer's instructions (Zymo Research Corp., Irvine, CA, USA). Values are presented as means ± sem as a percentage of 5-mC.
Statistical analysis
Statistical analysis was performed with Primer of Biostatistics 7.0 (McGraw-Hill, New York, NY, USA). One-way analysis of variance (ANOVA) followed by Bonferroni correction was used for comparison between the experimental groups. Differences were considered significant when P < 0.05. Values are presented as means ± sem (n≥4).
RESULTS
Effect of Aza on physiological parameters
The body and heart weights of the CBS+/− (HHcy) mice were lower than in the WT groups (Fig. 1A, B). A significant increase was noted after Aza treatment. The plasma Hcy level (Fig. 1C) and baseline systolic, diastolic, and mean BP (Fig. 2A–C) in the CBS+/− mice were higher than those in the WT groups. After 4 wk of Aza treatment, a significant decrease was observed in both Hcy level and BP parameters.
Figure 1.

Physiological parameters. A, B) Body (A) and heart (B) weights of 10-wk-old male WT and CBS+/− mice (n=6). C) Plasma Hcy levels from all groups (n=4). *P < 0.05 vs. WT and WT + Aza; †P < 0. 05 vs. CBS.
Figure 2.

BP was measured by the tail cuff method. Line graphs represent systolic BP (A), diastolic BP (B), and mean BP (C). Values are expressed as means ± se (n=4). *P < 0.05 vs. WT and WT + Aza; †P < 0.05 vs. CBS.
Wall-to-lumen ratio and RI
HHcy is known to cause aortic vessel remodeling. To analyze the structural changes in the aorta, we measured the lumen diameter and wall thickness of the ascending aorta and lumen diameter of the abdominal aorta. The wall-to-lumen ratio of the ascending aorta in the CBS+/− mice increased compared with that in the WT groups (Fig. 3A, B). Similarly, the diameter of the abdominal aorta in the CBS+/− mice decreased significantly more than in the WT and WT+Aza mice (Fig. 4A, B). After Aza treatment, the wall-to-lumen ratio of the ascending aorta decreased, and the lumen diameter of the abdominal aorta increased in the CBS+/− mice and was similar to that in the WT groups. The RI is a measure of the resistance offered by the vessel to blood flow. The RI of the ascending (Fig. 3C, D) and abdominal (Fig. 4C, D) aorta increased in the CBS+/− mice compared to that in the WT groups and decreased significantly after Aza treatment.
Figure 3.

A) B-mode ultrasound image of ascending aorta. Wall thickness (mm) and lumen diameter (mm) were measured by Vevo 2100 (Visual Sonics Vevo Ultra Imaging System). B) Wall-to-lumen ratio of the ascending aorta. C) Pulse wave mode images of ascending aorta. D) RI provides an estimate of the resistance offered by the ascending aorta and can be calculated as (PSV − EDV)/EDV. Bars represent means ± sem (n=4). *P < 0.05 vs. WT and WT + Aza; †P < 0.05 vs. CBS.
Figure 4.

A) B-mode ultrasound image of abdominal aorta. Lumen diameter (mm) was measured by Vevo 2100 (Visual Sonics Vevo Ultra Imaging System). B) Lumen diameter of the abdominal aorta. C) Pulse wave mode images of abdominal aorta. D) RI provides an estimate of the resistance offered by the abdominal aorta and can be calculated as (PSV − EDV)/EDV. Bars represent means ± sem (n=4). *P < 0.05 vs. WT and WT + Aza; †P < 0.05 vs. CBS.
Aortic response to Phe, Ach, and SNP
To evaluate the effect of Aza on aortic function, we measured the response of aortic rings from the experimental groups to vasoconstriction and vasorelaxation by applying Phe and Ach, respectively, in a dose-dependent manner. Aorta from CBS+/− mice was nearly 3- and 7-fold less responsive to Phe (Fig. 5A) and the vasodilator Ach (Fig. 5B), respectively, compared to the responses in the WT groups. Similarly, endothelium-independent relaxation in response to the maximum concentration of SNP decreased by 3-fold (Fig. 5C) in the CBS+/− mice compared to that in the WT groups. Aza treatment restored the aortic response to Phe, Ach, and SNP to levels similar to those in the WT groups.
Figure 5.

Aorta was extracted from experimental mice (WT, WT+Aza, CBS, and CBS+Aza) according to the protocol described in Materials and Methods. Aortic rings were mounted in a myobath containing Krebs solution. The rings were treated with Phe (A) and Ach (B) in a dose-dependent manner and SNP (C), as described in Materials and Methods (means±se; n = 4). *P < 0.05 vs. WT and WT+Aza; †P < 0.05 vs. CBS.
Effect of Aza on collagen deposition
Collagen deposition was quantified in the aorta as an indication of vascular stiffness. The WT groups, without or with Aza, showed normal blue intensity, whereas increased intensity was observed in the CBS+/− mice, suggesting increased collagen deposition in the adventitia (Fig. 6A, B). Aza treatment reduced total collagen content by 2.4-fold compared to that in the untreated control. Under a polarized light filter, the Picrosirius red stain shows type I collagen as yellow and type III as green (Fig. 7A). Type I and III collagen from the WT mice without Aza treatment was considered normal and was used for further analysis. Aortas from the WT + Aza group did not differ from the control aortas (Fig. 7A–C). In contrast, the CBS+/− mice showed a 3.8-fold increase in type I and a 4.5-fold increase in type III collagen compared with levels in the untreated WT mice (Fig. 7B, C). After treatment with Aza in the CBS+/− mice, type I collagen decreased by 2-fold and type III by 1.5-fold compared with levels in the untreated CBS+/− mice (Fig. 7B, C).
Figure 6.
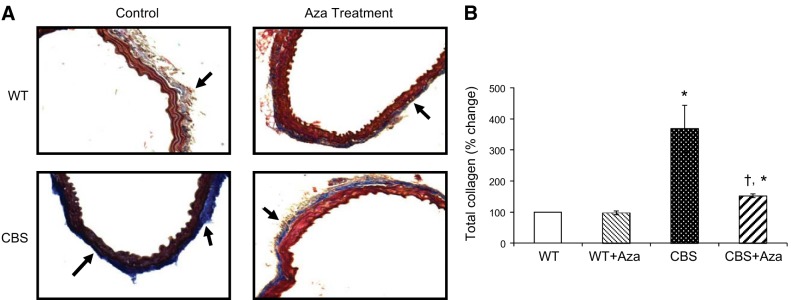
Mason trichrome staining. A) Collagen is stained dark blue. (B) Mean ± sem percentage intensity of blue staining in the aorta (n=4). Original image, ×20. *P < 0.05 vs. WT and WT + Aza; †P < 0.05 vs. CBS.
Figure 7.

A) Picrosirius red staining for type I and III collagen (yellow and green represent type I and type III collagen, respectively). B, C) Mean ± sem percentage intensity of yellow (B) and green (C) in aorta. Original image, ×20. *P < 0.05 vs. WT and WT + Aza; †P < 0.05 vs. CBS.
DNMT1 inhibition decreases ECM remodeling and Hcy synthesis and triggers Hcy remethylation
To examine the effects of Hcy and Aza treatment on the expression of proteins involved in Hcy metabolism, we measured the expression of MTHFR, SAHH, and Hcy by immunohistochemistry. There was an 8-fold increase in Hcy and a 2-fold increase in SAHH expression (Fig. 8A, B) in the CBS+/− mice when compared to that in the WT groups. MTHFR expression was decreased by 1.5-fold in the CBS+/− mice (Fig. 8A, B) compared with that in the WT groups. These results suggest an up-regulation of Hcy synthesis and accumulation and down-regulation of the Hcy remethylation pathway in the CBS+/− mice. Aza treatment decreased the expression of Hcy and SAHH and increased MTHFR in the CBS+/− mice which were similar to the WT controls. To evaluate the expression of ECM remodeling genes, we measured the expression of MMP9 and TIMP1. There was a 3-fold increase in MMP9 and 2.6-fold increase in TIMP1 in the CBS+/− mice (Fig. 8C, D), compared to that in the WT control. Following Aza treatment, their levels declined and were similar to those in the WT groups.
Figure 8.

Immunohistochemistry. A, C) Protein expression of SAHH, Hcy, and MTHFR (A) and MMP9 and TIMP1 (C) was measured. Expression of MTHFR and SAHH is shown as green fluorescence; Hcy, MMP9, and TIMP1 as red fluorescence. B, D) Bar graph representations of the immunohistochemistry images. The y axis represents the percentage change in mean ± sem intensity (n=4). Original image: Hcy, ×20; SAHH, MTHFR, MMP9, and TIMP1, ×10. *P < 0.05 vs. WT, WT + Aza, and CBS + Aza; †P < 0.05 vs. WT, WT + Aza, and CBS+Aza; ψP < 0.05 vs. WT and WT + Aza; ‡P < 0.05 vs. WT and WT + Aza.
Global DNA methylation in HHcy
To better understand the effect of HHcy on maintenance methylation, we measured the expression of DNMT1 in all the groups. There was a 3-fold increase in DNMT1 expression in the CBS+/− mice compared with expression in the WT controls, which was improved by Aza treatment (Fig. 9). To determine the overall methylation levels in aorta during HHcy, we measured 5-mC from the extracted DNA. As shown in Fig. 10, we observed a significant increase in methylation levels in the CBS+/− mice compared to those in the WT groups. Following treatment with Aza, there was a significant reduction in the CBS+/− group compared with the levels in the control.
Figure 9.

Immunohistochemistry. A) Protein expression of DNMT1 is shown as green fluorescence. B) Bar graph representation of DNMT1 expression. The y axis represents the percentage change in mean ± sem intensity (n=4). Original image: ×10. *P < 0.05 vs. WT, WT + Aza, and CBS + Aza.
Figure 10.
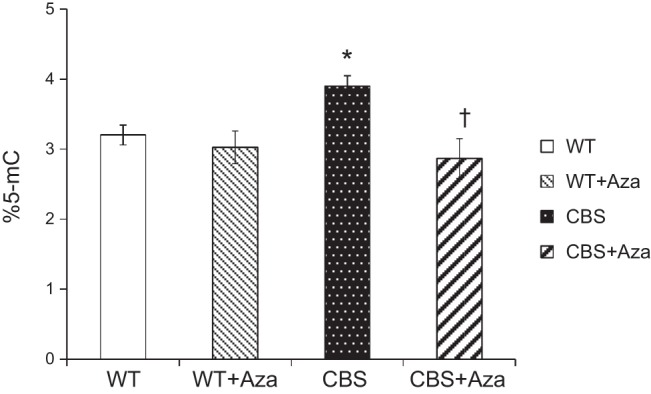
Overall methylation analysis was measured using ELISA. Bar graphs represent mean ± sem percentage of 5-mC (n=4). *P < 0.05 vs. WT and WT + Aza; †P < 0.05 vs. CBS.
DISCUSSION
HHcy plays a critical role in the development of various aortic diseases (23–26). HHcy induces the expression of MMPs involved in ECM metabolism, promoting aortic remodeling resulting in arterial hypertension (7). Epigenetic mechanisms such as DNA methylation are known to control the expression of ECM components (27). Although various studies report an aberrant DNA methylation pattern in the early stages of atherosclerosis (28) and aortic aneurysm (29), the role of DNA hypermethylation in aortic remodeling and arterial hypertension in HHcy remains unclear. In the recent years, epigenetic inhibitors have been used as therapeutic agents in various cancer drug trials (17). Our study provides new insights into the mechanism and the use of epigenetic inhibitors as therapeutic options in hypertension-associated aortic pathologies.
In the present study, we used CBS+/− mice as an HHcy model. In a previous study, Gupta et al. (30) demonstrated that CBS-deficient mice have a decreased fat mass caused by a reduction in lipogenesis. Our observation of reduced body weight in our CBS+/− mice supports this earlier finding. Plasma Hcy level is an important predictor of pulse pressure in hypertensive subjects (31). Lim and Cassano (32) reported that a 5 μM increase in plasma Hcy caused an increase in systolic and diastolic blood pressure by 0.7/0.5 mmHg in men and 1.2/0.7 mmHg in women. In the present study, increased plasma Hcy in CBS+/− mice was associated with elevated systolic, diastolic, and mean blood pressures. Evidence from earlier studies suggests that lowering plasma Hcy level by supplemental vitamins like folic acid, B6, and B12 is effective in delaying the formation of atherosclerotic plaques (33). Our results show that Aza lowered plasma Hcy in CBS+/− mice to normal levels. During HHcy, the conversion of S-adenosyl methionine to S-adenosyl homocysteine liberates excess methyl groups that are used in methylating substrates including DNA and proteins. Indeed, hypermethylation of gene promoters has been described in hypertension. For example, the sulfatase 1 (SULF1) gene, an endosulfatase, has been reported to be hypermethylated in hypertensive subjects (34). In this study, Aza treatment in hypertensive CBS+/− mice restored systolic, diastolic, and mean blood pressures to normal levels, suggesting an important role for hypermethylation in HHcy-associated hypertension.
Excessive accumulation of ECM proteins in HHcy causes hypertrophic vascular remodeling by increasing the wall thickness and decreasing lumen diameter (35, 36). In the present study, ultrasound examination revealed an increase in the wall-to-lumen ratio of the ascending aorta and a decrease in the lumen diameter of the abdominal aorta in the CBS+/− mice. In an earlier study from our lab, treatment of CBS+/− mice with SAHH inhibitor, deazaadenosine (DZA), decreased systolic blood pressure without a change in the aortic wall-to-lumen ratio within the first week (7). This result suggests that the reduction in systolic blood pressure is secondary structural remodeling (7). In the present study, Aza treatment decreased BP at 3 wk, suggesting a significant role for the epigenetic mechanism in aortic remodeling during HHcy-induced hypertension. The reduction in blood pressure (systolic, diastolic, and mean) was associated with a reduction in the wall-to-lumen ratio of the ascending aorta and an increase in the lumen diameter of the abdominal aorta. The RI of the aorta was measured as an indication of resistance to blood flow. In an earlier study, a positive correlation was reported for carotid artery RI and atherosclerosis (37). In our study, increased RI in the CBS+/− suggested vascular stiffness and was associated with increased MMP9 and TIMP1 expression. The increase in TIMP1 appears to be compensatory to increased MMP9. Consistent with our findings, increased MMP9 and TIMP1 have been reported in pathologies such as neonatal encephalopathy (38) and aortic aneurysm (39). In our study, Aza treatment reduced collagen deposition and also decreased MMP9 and TIMP1 expression in HHcy mice. The reduction in the wall-to-lumen ratio and RI by Aza treatment suggests mitigation of aortic ECM remodeling.
Although previous studies reported no significant changes in smooth muscle contraction (by Phe; ref. 40) or endothelium independent relaxation (by SNP; ref. 41) between WT and HHcy mice, we saw a significant decrease in the aortic response to Phe, Ach, and SNP in HHcy mice. Consistent with our findings, Tyagi et al. (42) reported that HHcy mice showed a decreased response to a vasoconstrictor (endothelin-1) compared to that of control animals. In the present study, the impairment of vessel function suggests endothelial and smooth muscle dysfunction and vascular stiffness. Aza treatment improved vascular response to Phe, Ach, and SNP in HHcy and also reduced vessel stiffness, as indicated by reduction of collagen deposition. These observations suggest that DNMT1 inhibition improves aortic function during HHcy, thereby mitigating hypertension.
Hcy is derived from demethylation of dietary methionine. SAHH plays a major role in the synthesis of Hcy from S-adenosyl homocysteine. After synthesis, Hcy has 2 fates: transsulfuration to cysteine aided by CBS and remethylation to methionine by MTHFR in the folate metabolism pathway (15). A prior study showed that, in HHcy, betaine Hcy methyltransferase (BHMT) and formation of 5-methyltetrahydrofolate by MTHFR are inhibited (43). The fate of Hcy in the body is controlled by a balance between the remethylation and transsulfuration pathways. During consumption of a high-methionine diet, Hcy catabolism is increased, favoring transsulfuration over remethylation. In contrast, with a low-methionine diet, the remethylation pathway is favored compared to transsulfuration (15). Since CBS is a key enzyme in Hcy metabolism, genetic mutations in CBS (44) and dietary deficiency in amino acids, such as methionine or taurine and vitamins (folate and B12), can affect the plasma Hcy concentrations. In a recent study by Tang et al. (45), when WT and transgenic mice expressing human CBS were fed a low-methionine diet, the protein levels of CBS in the liver and the activity were decreased to conserve methionine levels in the body. In the transsulfuration pathway, Hcy is catabolized to yield taurine, which reduces total serum cholesterol by lowering very-low-density lipoprotein (VLDL) and low-density lipoprotein (LDL) (46). When CBS+/− mice were fed a taurine-deficient diet, there was an up-regulation of the CBS monoallele, along with a reduction in total serum cholesterol levels. These studies suggest the role of enzymes involved in Hcy metabolism, such as CBS, SAHH, and MTHFR, in maintaining plasma Hcy levels. In the present study, immunohistochemical analysis revealed up-regulation of SAHH and Hcy expression and down-regulation of MTHFR in aorta of HHcy mice. On treatment with Aza, there was a reduction in the SAHH expression and increase in MTHFR, suggesting increased remethylation to reduce plasma Hcy levels.
Previous reports in the literature link HHcy to both global hypermethylation (47) and hypomethylation (48). Zhang et al. (49) showed that the effect of Hcy on methylation patterns of gene promoters is dependent on Hcy concentration. HHcy is known to inhibit the activity of dimethylarginine dimethylaminohydrolase (DDAH2), an enzyme involved in the NO synthase pathway that causes endothelial dysfunction. Another study reported hypermethylation of DDAH2 promoters in HHcy (49). Also, an earlier study in our lab (50) showed increased DNMT1 expression in HHcy mice, suggesting hypermethylation. In the present study, we confirmed global hypermethylation by quantitating the percentage of 5-mC in HHcy mice. Treatment with Aza decreased the expression of DNMT1 and also reduced global methylation levels.
An approach to alleviating HHcy-mediated aortic pathology is to lower plasma Hcy levels. Although folic acid supplementation decreases the Hcy level and prevents disease progression, it does not reverse the existing HHcy-induced aortic pathology, termed the Hcy memory effect (51). The present study for the first time provides evidence that an epigenetic DNMT inhibitor can be a potential therapeutic agent in aortic diseases, independently or in combination with other therapies such as folic acid supplementation.
Summary
HHcy promotes pathological aortic remodeling and endothelial dysfunction, leading to arterial hypertension. Inhibition of DNMT1 using Aza ameliorates Hcy level and reduces high blood pressure (Fig. 11). Aza appears to mediate its action by modifying the expression of enzymes involved in Hcy metabolism and ECM proteins to reduce vascular stiffness, thus improving vascular function.
Figure 11.
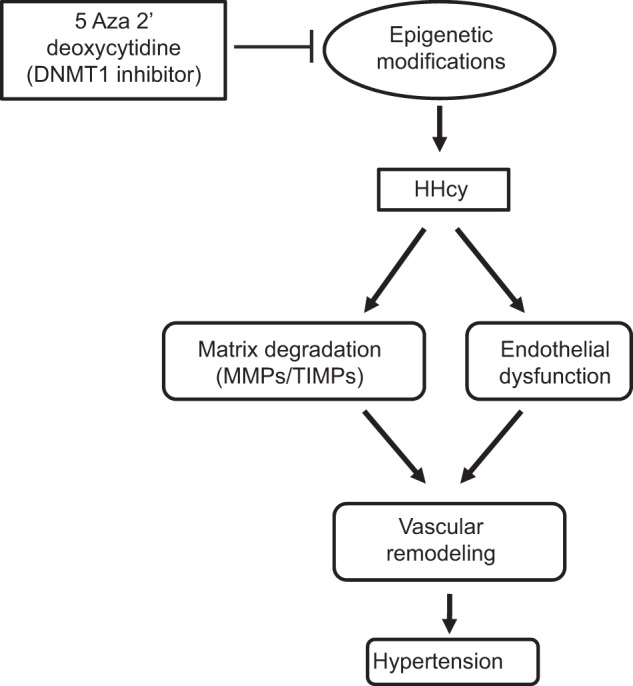
Schematic representation of epigenetic regulation of aortic remodeling in hyperhomocysteinemia.
Limitations of the study
Hypertension-induced aortic remodeling is a complex process involving enzymes that synthesize ECM components and their regulators, such as MMPs and TIMPs. This study mainly focused on the effect of the epigenetic inhibitor, Aza on ECM remodeling proteins (MMP9, TIMP1, and collagen) and Hcy metabolism enzymes (SAHH and MTHFR) in the aorta. However, as the enzymes involved in Hcy metabolism are located in the liver, further studies are needed to quantitate the specific effect of Aza on these individual enzymes. Although immunohistochemistry is a reliable method of quantitating protein expression, Western blot analysis would have been a better method; however, those experiments would require a very large tissue sample, especially of aorta. Although this study provides evidence of impairment in vascular function in HHcy mice and its improvement with Aza, understanding the mechanism by which HHcy affects promoter methylation of individual genes involved in NO synthase pathway requires elucidation.
Acknowledgments
This work was supported by U.S. National Institutes of Health grants R01HL074185-10 and NS-084823.
The authors declare no conflicts of interest.
Footnotes
- 5-mC
- 5-methylcytosine
- Ach
- acetylcholine
- ANOVA
- analysis of variance
- Aza
- 5-aza-2′-deoxycytidine
- BP
- blood pressure
- CBS
- cystathionine β-synthase
- DDAH2
- dimethylarginine dimethylaminohydrolase
- DNMT
- DNA methyltransferase
- ECM
- extracellular matrix
- EDV
- end diastolic velocity
- Hcy
- homocysteine
- HHcy
- hyperhomocysteinemia
- MMP
- matrix metalloproteinase
- MTHFR
- methylene tetrahydrofolate reductase
- Phe
- phenylephrine
- PSV
- peak systolic velocity
- RI
- resistive index
- SAHH
- S-adenosyl homocysteine hydrolase
- SNP
- sodium nitroprusside
- TIMP
- tissue inhibitor of metalloproteinase
- WT
- wild-type
REFERENCES
- 1. Jain D., Fishman E. K., Argani P., Shah A. S., Halushka M. K. (2011) Unexpected sclerosing mediastinitis involving the ascending aorta in the setting of a multifocal fibrosclerotic disorder. Pathol. Res. Pract. 207, 60–62 [DOI] [PubMed] [Google Scholar]
- 2. Puranik R., Chow C. K., Duflou J. A., Kilborn M. J., McGuire M. A. (2005) Sudden death in the young. Heart Rhythm 2, 1277–1282 [DOI] [PubMed] [Google Scholar]
- 3. Nishimura R. A. (2002) Cardiology patient pages: aortic valve disease. Circulation 106, 770–772 [DOI] [PubMed] [Google Scholar]
- 4. Moroz P., Le M. T., Norman P. E. (2007) Homocysteine and abdominal aortic aneurysms. ANZ J. Surg. 77, 329–332 [DOI] [PubMed] [Google Scholar]
- 5. Guthikonda S., Haynes W. G. (2006) Homocysteine: role and implications in atherosclerosis. Curr. Atherosclerosis Rep. 8, 100–106 [DOI] [PubMed] [Google Scholar]
- 6. Sundstrom J., Sullivan L., D'Agostino R. B., Jacques P. F., Selhub J., Rosenberg I. H., Wilson P. W., Levy D., Vasan R. S. (2003) Plasma homocysteine, hypertension incidence, and blood pressure tracking: the Framingham Heart Study. Hypertension 42, 1100–1105 [DOI] [PubMed] [Google Scholar]
- 7. Ovechkin A. V., Tyagi N., Sen U., Lominadze D., Steed M. M., Moshal K. S., Tyagi S. C. (2006) 3-Deazaadenosine mitigates arterial remodeling and hypertension in hyperhomocysteinemic mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L905–L911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tyagi S. C., Matsubara L., Weber K. T. (1993) Direct extraction and estimation of collagenase(s) activity by zymography in microquantities of rat myocardium and uterus. Clin. Biochem. 26, 191–198 [DOI] [PubMed] [Google Scholar]
- 9. Mareckova Z., Heller S., Horky K. (1999) [Endothelial dysfunction and arterial hypertension]. Vnitr. Lek. 45, 232–237 [PubMed] [Google Scholar]
- 10. Li N., Yi F. X., Rute E., Zhang D. X., Slocum G. R., Zou A. P. (2002) Effects of homocysteine on intracellular nitric oxide and superoxide levels in the renal arterial endothelium. Am J. Physiol. Heart Circ. Physiol. 283, H1237–H1243 [DOI] [PubMed] [Google Scholar]
- 11. Ungvari Z., Csiszar A., Bagi Z., Koller A. (2002) Impaired nitric oxide-mediated flow-induced coronary dilation in hyperhomocysteinemia: morphological and functional evidence for increased peroxynitrite formation. Am. J. Pathol. 161, 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Topal G., Brunet A., Millanvoye E., Boucher J. L., Rendu F., Devynck M. A., David-Dufilho M. (2004) Homocysteine induces oxidative stress by uncoupling of NO synthase activity through reduction of tetrahydrobiopterin. Free Radic. Biol. Med. 36, 1532–1541 [DOI] [PubMed] [Google Scholar]
- 13. Sander M., Chavoshan B., Victor R. G. (1999) A large blood pressure-raising effect of nitric oxide synthase inhibition in humans. Hypertension 33, 937–942 [DOI] [PubMed] [Google Scholar]
- 14. Juonala M., Viikari J. S., Ronnemaa T., Helenius H., Taittonen L., Raitakari O. T. (2006) Elevated blood pressure in adolescent boys predicts endothelial dysfunction: the cardiovascular risk in young Finns study. Hypertension 48, 424–430 [DOI] [PubMed] [Google Scholar]
- 15. Selhub J. (1999) Homocysteine metabolism. Annu. Rev. Nutr. 19, 217–246 [DOI] [PubMed] [Google Scholar]
- 16. Mizuno S., Chijiwa T., Okamura T., Akashi K., Fukumaki Y., Niho Y., Sasaki H. (2001) Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 97, 1172–1179 [DOI] [PubMed] [Google Scholar]
- 17. Heyn H., Esteller M. (2012) DNA methylation profiling in the clinic: applications and challenges. Nat. Rev. Genet. 13, 679–692 [DOI] [PubMed] [Google Scholar]
- 18. Amarnath K., Amarnath V., Amarnath K., Valentine H. L., Valentine W. M. (2003) A specific HPLC-UV method for the determination of cysteine and related aminothiols in biological samples. Talanta 60, 1229–1238 [DOI] [PubMed] [Google Scholar]
- 19. Sen U., Basu P., Abe O. A., Givvimani S., Tyagi N., Metreveli N., Shah K. S., Passmore J. C., Tyagi S. C. (2009) Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am. J. Physiol. Renal Physiol 297, F410–F419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sen U., Tyagi N., Kumar M., Moshal K. S., Rodriguez W. E., Tyagi S. C. (2007) Cystathionine-beta-synthase gene transfer and 3-deazaadenosine ameliorate inflammatory response in endothelial cells. Am. J. Physiol. Cell Physiol. 293, C1779–1787 [DOI] [PubMed] [Google Scholar]
- 21. Sen U., Rodriguez W. E., Tyagi N., Kumar M., Kundu S., Tyagi S. C. (2008) Ciglitazone, a PPARgamma agonist, ameliorates diabetic nephropathy in part through homocysteine clearance. Am. J. Physiol. Endocrinol Metab. 295, E1205–E1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pushpakumar S., Kundu S., Pryor T., Givvimani S., Lederer E., Tyagi S. C., Sen U. (2013) Angiotensin-II induced hypertension and renovascular remodelling in tissue inhibitor of metalloproteinase 2 knockout mice. J. Hypertension 31, 2270–2281; discussion 2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giusti B., Marcucci R., Lapini I., Sestini I., Lenti M., Yacoub M., Pepe G. (2004) Role of hyperhomocysteinemia in aortic disease. Cell. Mol. Biol. 50, 945–952 [PubMed] [Google Scholar]
- 24. Takagi H., Umemoto T. (2005) Homocysteinemia is a risk factor for aortic dissection. Med. Hypotheses 64, 1007–1010 [DOI] [PubMed] [Google Scholar]
- 25. Tribouilloy C. M., Peltier M., Iannetta Peltier M. C., Trojette F., Andrejak M., Lesbre J. P. (2000) Plasma homocysteine and severity of thoracic aortic atherosclerosis. Chest 118, 1685–1689 [DOI] [PubMed] [Google Scholar]
- 26. Novaro G. M., Aronow H. D., Mayer-Sabik E., Griffin B. P. (2004) Plasma homocysteine and calcific aortic valve disease. Heart 90, 802–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chicoine E., Esteve P. O., Robledo O., Van Themsche C., Potworowski E. F., St-Pierre Y. (2002) Evidence for the role of promoter methylation in the regulation of MMP-9 gene expression. Biochem. Biophys. Res. Commun. 297, 765–772 [DOI] [PubMed] [Google Scholar]
- 28. Zaina S., Lindholm M. W., Lund G. (2005) Nutrition and aberrant DNA methylation patterns in atherosclerosis: more than just hyperhomocysteinemia? J. Nutr. 135, 5–8 [DOI] [PubMed] [Google Scholar]
- 29. Krishna S. M., Dear A. E., Norman P. E., Golledge J. (2010) Genetic and epigenetic mechanisms and their possible role in abdominal aortic aneurysm. Atherosclerosis 212, 16–29 [DOI] [PubMed] [Google Scholar]
- 30. Gupta S., Kruger W. D. (2011) Cystathionine beta-synthase deficiency causes fat loss in mice. PloS One 6, e27598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsai J. C., Kuo H. T., Chiu Y. W., Hwang S. J., Chuang H. Y., Chang J. M., Chen H. C., Lai Y. H. (2005) Correlation of plasma homocysteine level with arterial stiffness and pulse pressure in hemodialysis patients. Atherosclerosis 182, 121–127 [DOI] [PubMed] [Google Scholar]
- 32. Lim U., Cassano P. A. (2002) Homocysteine and blood pressure in the Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiol. 156, 1105–1113 [DOI] [PubMed] [Google Scholar]
- 33. Vermeulen E. G., Stehouwer C. D., Twisk J. W., van den Berg M., de Jong S. C., Mackaay A. J., van Campen C. M., Visser F. C., Jakobs C. A., Bulterjis E. J., Rauwerda J. A. (2000) Effect of homocysteine-lowering treatment with folic acid plus vitamin B6 on progression of subclinical atherosclerosis: a randomised, placebo-controlled trial. Lancet 355, 517–522 [DOI] [PubMed] [Google Scholar]
- 34. Wang X., Falkner B., Zhu H., Shi H., Su S., Xu X., Sharma A. K., Dong Y., Treiber F., Gutin B., Harshfield G., Snieder H. (2013) A genome-wide methylation study on essential hypertension in young African American males. PloS One 8, e53938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reddy H. K., Koshy S. K., Wasson S., Quan E. E., Pagni S., Roberts A. M., Joshua I. G., Tyagi S. C. (2006) Adaptive-outward and maladaptive-inward arterial remodeling measured by intravascular ultrasound in hyperhomocysteinemia and diabetes. J. Cardiovasc. Pharmacol. Ther. 11, 65–76 [DOI] [PubMed] [Google Scholar]
- 36. Basu P., Qipshidze N., Tyagi S. C., Sen U. (2011) Remodeling in vein expresses arterial phenotype in hyperhomocysteinemia. Int. J. Physiol. Pathophysiol. Pharmacol. 3, 266–279 [PMC free article] [PubMed] [Google Scholar]
- 37. Frauchiger B., Schmid H. P., Roedel C., Moosmann P., Staub D. (2001) Comparison of carotid arterial resistive indices with intima-media thickness as sonographic markers of atherosclerosis. Stroke 32, 836–841 [DOI] [PubMed] [Google Scholar]
- 38. Bednarek N., Svedin P., Garnotel R., Favrais G., Loron G., Schwendiman L., Hagberg H., Morville P., Mallard C., Gressens P. (2012) Increased MMP-9 and TIMP-1 in mouse neonatal brain and plasma and in human neonatal plasma after hypoxia-ischemia: a potential marker of neonatal encephalopathy. Pediatr. Res. 71, 63–70 [DOI] [PubMed] [Google Scholar]
- 39. Koullias G. J., Ravichandran P., Korkolis D. P., Rimm D. L., Elefteriades J. A. (2004) Increased tissue microarray matrix metalloproteinase expression favors proteolysis in thoracic aortic aneurysms and dissections. Ann. Thorac. Surg. 78, 2106–2110; discussion 2110–2101 [DOI] [PubMed] [Google Scholar]
- 40. Powers R. W., Gandley R. E., Lykins D. L., Roberts J. M. (2004) Moderate hyperhomocysteinemia decreases endothelial-dependent vasorelaxation in pregnant but not nonpregnant mice. Hypertension 44, 327–333 [DOI] [PubMed] [Google Scholar]
- 41. Hucks D., Thuraisingham R. C., Raftery M. J., Yaqoob M. M. (2004) Homocysteine induced impairment of nitric oxide-dependent vasorelaxation is reversible by the superoxide dismutase mimetic TEMPOL. Nephrol. Dial. Transplant. 19, 1999–2005 [DOI] [PubMed] [Google Scholar]
- 42. Tyagi N., Qipshidze N., Sen U., Rodriguez W., Ovechkin A., Tyagi S. C. (2011) Cystathionine beta synthase gene dose dependent vascular remodeling in murine model of hyperhomocysteinemia. Int. J. Physiol. Pathophysiol. Pharmacol. 3, 210–222 [PMC free article] [PubMed] [Google Scholar]
- 43. Finkelstein J. D., Martin J. J. (1984) Methionine metabolism in mammals: distribution of homocysteine between competing pathways. J. Biol. Chem. 259, 9508–9513 [PubMed] [Google Scholar]
- 44. Kraus J. P., Janosik M., Kozich V., Mandell R., Shih V., Sperandeo M. P., Sebastio G., de Franchis R., Andria G., Kluijtmans L. A., Blom H., Boers G. H., Gordon R. B., Kamoun P., Tsai M. Y., Kruger W. D., Koch H. G., Ohura T., Gaustadnes M. (1999) Cystathionine beta-synthase mutations in homocystinuria. Hum. Mutat. 13, 362–375 [DOI] [PubMed] [Google Scholar]
- 45. Tang B., Mustafa A., Gupta S., Melnyk S., James S. J., Kruger W. D. (2010) Methionine-deficient diet induces post-transcriptional downregulation of cystathionine beta-synthase. Nutrition 26, 1170–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen W., Guo J. X., Chang P. (2012) The effect of taurine on cholesterol metabolism. Mol. Nutr. Food Res. 56, 681–690 [DOI] [PubMed] [Google Scholar]
- 47. Bonsch D., Lenz B., Reulbach U., Kornhuber J., Bleich S. (2004) Homocysteine associated genomic DNA hypermethylation in patients with chronic alcoholism. J. Neural Transm. 111, 1611–1616 [DOI] [PubMed] [Google Scholar]
- 48. Rampersaud G. C., Kauwell G. P., Hutson A. D., Cerda J. J., Bailey L. B. (2000) Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am. J. Clin. Nutr. 72, 998–1003 [DOI] [PubMed] [Google Scholar]
- 49. Zhang J. G., Liu J. X., Li Z. H., Wang L. Z., Jiang Y. D., Wang S. R. (2007) Dysfunction of endothelial NO system originated from homocysteine-induced aberrant methylation pattern in promoter region of DDAH2 gene. Chin. Med. J (Engl.) 120, 2132–2137 [PubMed] [Google Scholar]
- 50. Narayanan N., Tyagi N., Shah A., Pagni S., Tyagi S. C. (2013) Hyperhomocysteinemia during aortic aneurysm, a plausible role of epigenetics. Int. J. Physiol. Pathophysiol. Pharmacol. 5, 32–42 [PMC free article] [PubMed] [Google Scholar]
- 51. Krishna S. M., Dear A., Craig J. M., Norman P. E., Golledge J. (2013) The potential role of homocysteine mediated DNA methylation and associated epigenetic changes in abdominal aortic aneurysm formation. Atherosclerosis 228, 295–305 [DOI] [PubMed] [Google Scholar]