

Abstract
Selenoprotein P (Sepp1) and its receptor, apolipoprotein E receptor 2 (apoER2), account for brain retaining selenium better than other tissues. The primary sources of Sepp1 in plasma and brain are hepatocytes and astrocytes, respectively. ApoER2 is expressed in varying amounts by tissues; within the brain it is expressed primarily by neurons. Knockout of Sepp1 or apoER2 lowers brain selenium from ∼120 to ∼50 ng/g and leads to severe neurodegeneration and death in mild selenium deficiency. Interactions of Sepp1 and apoER2 that protect against this injury have not been characterized. We studied Sepp1, apoER2, and brain selenium in knockout mice. Immunocytochemistry showed that apoER2 mediates Sepp1 uptake at the blood-brain barrier. When Sepp1−/− or apoER2−/− mice developed severe neurodegeneration caused by mild selenium deficiency, brain selenium was ∼35 ng/g. In extreme selenium deficiency, however, brain selenium of ∼12 ng/g was tolerated when both Sepp1 and apoER2 were intact in the brain. These findings indicate that tandem Sepp1-apoER2 interactions supply selenium for maintenance of brain neurons. One interaction is at the blood-brain barrier, and the other is within the brain. We postulate that Sepp1 inside the blood-brain barrier is taken up by neurons via apoER2, concentrating brain selenium in them.—Burk, R. F., Hill, K. E., Motley, A. K., Winfrey, V. P., Kurokawa, S., Mitchell, S. L., Zhang, W. Selenoprotein P and apolipoprotein E receptor-2 interact at the blood-brain barrier and also within the brain to maintain an essential selenium pool that protects against neurodegeneration.
Keywords: choroid plexus, megalin
The brain is a privileged organ with respect to selenium metabolism (1, 2). Under conditions of dietary selenium deficiency so severe that whole-body selenium concentration fell to 13% of its level in selenium-replete mice, brain selenium concentration remained 56% of its selenium-replete value (3). This finding indicates that there is a mechanism that maintains brain selenium at the expense of other tissues. Reports in the past decade have implicated selenoprotein P (Sepp1) as a component of the mechanism supplying selenium to tissues, including to the brain (4, 5).
Sepp1 is a selenium-rich protein that is secreted by the liver into the plasma but is also produced by other tissues, albeit in lesser quantities (6). Its N-terminal domain contains one selenocysteine that has redox activity (7), and its smaller C-terminal domain, containing 9 selenocysteines, endows it with a selenium transport function (8). When mice with deletion of Sepp1 became available, it was determined that loss of Sepp1 depressed tissue selenium levels, with brain and testis affected more than other tissues (4). Sepp1−/− male mice were infertile and both males and females developed brain injury with severe neurological dysfunction when fed a low-selenium diet. Feeding a high-selenium diet to Sepp1−/− mice prevented the neurological dysfunction, however, suggesting that a small-molecule form of selenium (as yet uncharacterized) can supply selenium to the brain when the element is abundant (4, 5).
Immunocytochemistry (ICC) of the testis revealed vesicles containing Sepp1 in Sertoli cells, leading to the discovery that apolipoprotein E receptor 2 (apoER2), a member of the low-density lipoprotein receptor family, facilitates endocytosis of Sepp1, thereby supplying the testis with selenium for spermatogenesis (9). Extrahepatic tissues express varying amounts of apoER2, and endocytosis of Sepp1 mediated by this receptor supplies their cells with selenium (10). ApoER2−/− mice have depressed brain selenium, and feeding them a selenium-deficient diet causes neurodegeneration and neurological dysfunction similar to that observed in Sepp1−/− mice fed the same diet (11). Thus, it has become clear that both Sepp1 and apoER2 are necessary to maintain brain selenium and brain neuron viability under selenium-deficient conditions.
Insertion of a transgene that directed hepatic expression of human SEPP1 into Sepp1−/− mice delayed, but did not prevent, severe neurological dysfunction when the mice were fed a low-selenium diet (12). That finding suggests that some SEPP1 selenium was transferred across the blood-brain barrier but that liver expression of SEPP1 provided only partial protection against neurodegeneration. A role for apoER2 was not addressed in that report because the work was carried out before the role of apoER2 in Sepp1 uptake had been described.
Sepp1 is synthesized within the brain because its mRNA is expressed there in a glial pattern (13), and primary rat astrocytes secrete it (14). Within the brain, apoER2 has been detected exclusively in neurons (15). Its presence at the blood-brain barrier has been established in the choroid plexus (13) but not in brain capillaries. No direct demonstration of a Sepp1-apoER2 interaction at the blood-brain barrier has been reported.
These observations raise the possibility that Sepp1 and apoER2 interact both at the blood-brain barrier and within the brain to supply neurons with selenium. Such tandem interactions might explain the better retention of selenium by brain than by other tissues that acquire selenium from the systemic circulation by a single apoER2-mediated endocytosis of Sepp1. This report presents results supporting such a two-level process for supplying selenium to brain neurons.
MATERIALS AND METHODS
Reagents
Digoxigenin-labeled nucleotides and alkaline phosphatase-conjugated antidigoxigenin antibody were purchased from Roche Diagnostics (Indianapolis, IN, USA). Oligonucleotides were obtained from the Molecular Cell Biology Resources Core at Vanderbilt University Medical Center. High-capacity cDNA reverse transcriptase kit and Power SYBR Green PCR master mix were purchased from Applied Biosystems (Foster City, CA, USA). RNeasy mini kits were purchased from Qiagen (Valencia, CA, USA). All other chemicals were of reagent grade.
Animal husbandry
Colonies of Sepp1+/− and apoER2+/− mice congenic with C57BL/6 mice were maintained and bred to produce Sepp1−/− and apoER2−/− mice and their wild-type littermates for experiments (16). Sepp1C/C/alb-cre+/− mice were bred as described previously (6), and megalin+/− breeding pairs were maintained as described previously (7). The mice received food and tap water ad libitum.
Experimental diets were formulated by Harlan-Teklad (Madison, WI, USA) to our specifications (17). The diets were Torula yeast-based and contained supplemental selenium as sodium selenite. The basal (selenium-deficient) form of this experimental diet was assayed, and it contained <0.01 mg Se/kg. Sodium selenite was added to this diet during mixing to give added selenium concentrations of 0.25 mg/kg (selenium-adequate diet) and 4.0 mg/kg (high-selenium diet, but not toxic). The Vanderbilt University Institutional Animal Care and Use Committee approved the protocol of these studies.
Fetal experiments
Timed-mating experiments to produce d 18 fetuses were accomplished as described previously (16). For experiments in which selenium content was determined or RNA was quantified, d 18 pregnant dams, neonates, or adult male mice were killed by isoflurane inhalation. Fetuses were removed from the uterus and a 0.5-cm portion of tail was taken from each fetus for isolation of genomic DNA and genotyping by PCR. Brains were removed and stored at −80°C until they were assayed for selenium content or until RNA was isolated.
For ICC experiments, d 18 pregnant dams or adult male mice were killed by isoflurane inhalation. The uterus was removed and placed in ice-cold phosphate-buffered saline. Individual fetuses were removed. Brains were harvested and processed for ICC as described before (16). A 0.5-cm portion of the fetal tail or ear clipping was removed for genotyping.
Anti-Sepp1 monoclonal antibody 9S4 that binds to the N-terminal domain was used for detection of Sepp1. Anti-apoER2 polyclonal antibody 2561, a gift of Dr. Joachim Herz (University of Texas Southwestern Medical School, Dallas, TX, USA), was used to detect apoER2 (18). ICC was carried out as described previously (16).
Effects of selenium deficiency
To assess the effect of selenium deficiency on brain selenium, mice were fed selenium-deficient diet beginning at weaning. Control mice were fed a diet supplemented with 0.25 mg Se/kg diet. At the desired time, the mice were anesthetized with isoflurane, and blood was removed from the inferior vena cava with a syringe and needle. The brain was removed and stored at −80°C until it was assayed for selenium content or RNA was isolated from it. To assess the effect of selenium deficiency on neurological dysfunction, weanling male Sepp1c/c, Sepp1c/c/alb-cre+/−, Sepp1U40S/U40S, apoER2+/+, apoER2−/−, Sepp1+/+, and Sepp1−/− mice were fed selenium-deficient or selenium-adequate diet. The mice were observed daily for gait abnormality and obvious neurological impairment. They were weighed every 4 wk or more frequently.
Biochemical measurements
Selenium was measured using a modification of the fluorometric assay of Koh and Benson (19, 20). Frozen brain tissue was pulverized under liquid nitrogen and treated with TRIzol reagent for RT-PCR. Total RNA was isolated and used to prepare cDNA, as described previously (6). Relative quantitation of RNA levels was determined by comparative CT reactions (ΔΔCT analysis), as described previously (6). The apoER2 primers used were forward AGATGGGCTCAACAGTCACC and reverse AGTGGGCGATCATAGTTGCT.
Statistics
Statistical comparisons between groups were made on an iMac (Apple, Cupertino, CA, USA) using Prism 4 for Macintosh 4.0c software (GraphPad Software, La Jolla, CA, USA). Student's t test and Tukey's multiple-comparison test after 1-way ANOVA were used to compare groups. Groups were considered to be significantly different when P < 0.05.
RESULTS
ApoER2 mediates Sepp1 uptake at the blood-brain barrier
Because deletion of either Sepp1 or apoER2 lowers brain selenium content (4, 11), we sought a Sepp1-apoER2 interaction at the blood-brain barrier—in brain capillaries and in the choroid plexus—as the potential mechanism of selenium transport from the systemic circulation into the brain. The brain of the d 18 fetus was studied with ICC for evidence of apoER2-mediated uptake of Sepp1. Sepp1 was observed in a punctate pattern in capillary endothelial cells, suggesting its presence there in vesicles (Fig. 1A). ApoER2 was observed to be present in the same cells (Fig. 1B), and merging those two images demonstrated that Sepp1 and apoER2 were present in the same compartment (Fig. 1C). No Sepp1 was observed in apoER2−/− capillaries (Fig. 1D). These findings are compatible with apoER2-mediated endocytosis of Sepp1 by fetal brain capillary cells.
Figure 1.

Sepp1 binds to apoER2 in d 18 fetal brain capillaries (Cap) and choroid plexus (CP). Sepp1 is stained red, apoER2 is stained green, and nuclei are stained blue. A–C) Views of the same fetal brain cortex section. Section stained for Sepp1 (A), section stained for apoER2 (B), and a merge of A and B (C). D) Brain cortex of an apoER2−/− mouse stained for Sepp1. E, F) CP in a lateral ventricle and the adjacent ventricle wall stained for Sepp1. Section from a wild-type mouse (E) and section from an apoER2−/− mouse (F). CD 31 staining was used to verify the identity of capillaries (not shown).
Fetal choroid plexus epithelial cells also contained Sepp1 in vesicles (Fig. 1E). Sepp1 was not detected in the choroid plexus in apoER2−/− mice (Fig. 1F), indicating that the presence of apoER2 facilitates uptake of Sepp1 by the fetal choroid plexus. Thus, apoER2-mediated binding and, presumably, uptake of Sepp1 occurred in both components of the fetal blood-brain barrier.
ICC did not detect Sepp1 in the brain capillaries of adult mice fed a selenium-adequate diet, and Sepp1 was only occasionally detected in the choroid plexus (not shown). For this reason, adult mice were fed a high-selenium diet and were studied. ICC consistently demonstrated Sepp1 in the choroid plexus of mice fed a high-selenium diet but never in their brain capillaries (Fig. 2A). ApoER2 was also detected in the choroid plexus (Fig. 2B). In mice with deletion of apoER2, neither Sepp1 nor apoER2 was detected in the choroid plexus (Fig. 2C, D). Thus, apoER2 mediates uptake of Sepp1 by choroid plexus cells in adult brain, but ICC provided no evidence of Sepp1 uptake in adult brain capillaries.
Figure 2.

Sepp1 and apoER2 in adult brain and choroid plexus (CP) from apoER2+/+ and apoER2−/− mice fed a diet supplemented with 4 mg Se/kg. Nuclei are stained blue in all panels. A) Sepp1 staining (red) in wild-type brain with CP in the third ventricle and ependymal cells (Ep) indicated. B) ApoER2 staining (red) in wild-type brain with CP in a lateral ventricle and Ep indicated. C) Sepp1 staining (red) of apoER2−/− brain with CP in the third ventricle and Ep indicated. D) ApoER2 staining (red) of apoER2−/− brain with CP in a lateral ventricle and Ep indicated.
Because apoER2-mediated uptake of Sepp1 was more robust at the fetal blood-brain barrier than at the adult blood-brain barrier, we assessed apoER2 expression in fetal and adult brains by RT-PCR. Figure 3A shows that fetal brain apoER2 mRNA was >9 times greater than adult brain apoER2 mRNA. A possible reason for this difference is that the growing fetal brain has a greater need than adult brain for apoER2 ligands. This finding raised the question of whether selenium deficiency would increase brain expression of apoER2 to foster increased acquisition of selenium (as Sepp1). Figure 3B shows that selenium deficiency did not increase adult brain apoER2 mRNA. This finding indicates that total adult brain apoER2 expression is not regulated by selenium nutritional status, even though it is regulated by other physiological factors related to developmental stage (Fig. 3A). Because apoER2 is expressed elsewhere in the brain in addition to the blood-brain barrier, it remains possible that selenium deficiency affects its expression selectively at the blood-brain barrier with compensatory change of expression in another compartment.
Figure 3.

Effects of developmental status (A) and selenium status (B) on apoER2 mRNA in mouse brain. Values are expressed as means + sd; n = 3. A) Comparison of apoER2 mRNA between adult and d 18 fetus brains. Values are significantly different (P<0.05) by Student's t test. B) Comparison of selenium-deficient and control diet effects on adult brain apoER2 mRNA. Mice were fed the respective diets for 12 wk from weaning. Values are not significantly different (P>0.05) by Student's t test.
Because of the difference in apoER2 expression between fetal and adult brains, we examined the effect of apoER2 knockout on brain selenium in the fetus, the neonate, and the adult (Fig. 4). Brain selenium concentration in apoER2−/− d 18 fetuses was 75% of that in apoER2+/+ d 18 fetuses. Within a month of birth, however, the effect of apoER2 deletion had increased, with brain selenium concentration in apoER2−/− mice falling to 39%. These findings are consistent with the Sepp1-apoER2 mechanism being more important in maintaining brain selenium in the adult mouse than in the mouse fetus. A potential cause of this difference is that immaturity of the blood-brain barrier in the fetus may allow selenium forms to enter the brain directly, lessening the need for apoER2-mediated Sepp1 uptake at the blood-brain barrier.
Figure 4.

Effect of apoER2 on mouse fetus and pup brain selenium concentrations. Once weaned, mice were fed a selenium-adequate diet. Values are means + sd, n = 4–17. Pairs of values at each time point are all significantly different (P<0.05) by Student's t test.
Megalin, another member of the low-density lipoprotein receptor family, facilitates endocytosis of filtered Sepp1 forms by renal proximal convoluted tubule (PCT) cells (21). Megalin is also present in the brain, in ependymal and choroid plexus cells (22, 23). Even though Sepp1 was not seen to be associated with ependymal cells (Figs. 1E and 2A) and deletion of apoER2 abolished Sepp1 association with the choroid plexus (Figs. 1F and 2C), we assessed the effect of megalin deletion on Sepp1 in d 18 fetal brain using ICC. Figure 5A depicts Sepp1 in megalin+/+ brain, and Fig. 5B depicts Sepp1 in the megalin−/− brain. Deletion of megalin did not abolish the presence of Sepp1 in either capillary or choroid plexus cells. Also, megalin deletion did not affect brain selenium concentration in adult mice fed selenium-adequate diet (see Fig. 2A in ref. 7). These experiments provide no evidence for a megalin-Sepp1 interaction in the brain but neither do they rule one out.
Figure 5.
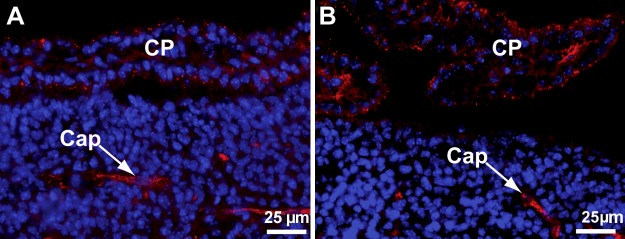
Deletion of megalin does not abolish binding of Sepp1 (red) to choroid plexus (CP) in a lateral ventricle or in brain capillaries (Cap). A) Brain section from a d 18 megalin+/+ fetus. B) Brain section from a d 18 megalin−/− fetus.
Sepp1 expression within the brain protects against severe neurodegeneration associated with low brain selenium
When weanling mice with deletion of Sepp1 or apoER2 are fed a selenium-deficient diet, within several weeks they develop severe neurodegeneration that causes neurological dysfunction and death (4, 11). This devastating condition has been attributed to a decrease in brain selenium that leads to injury of neurons (24). However, brain selenium levels have not been reported in mice developing this injury. We performed an experiment to determine those selenium levels.
Figure 6 presents brain selenium concentrations of Sepp1−/− mice and of apoER2−/− mice fed a selenium-deficient diet on the day that they developed severe neurological dysfunction (bold arrows). The primary comparisons with affected mice were mice of the same genotype that had been fed selenium-adequate diet and, consequently, had not developed neurological dysfunction. A 19% difference in brain selenium level was associated with development of severe neurological dysfunction when Sepp1 had been deleted, and a 44% difference was associated with development of the severe dysfunction when apoER2 had been deleted (Fig. 6). Thus, development of severe neurological dysfunction in mice with either Sepp1 or apoER2 deleted occurred when brain selenium concentration fell to ∼35 ng/g. Two other control groups were studied for each experimental group. The additional controls, without gene deletion, confirmed that feeding selenium-deficient diet to littermate wild-type mice caused relatively small decreases in their much higher brain selenium levels (Fig. 6). None of the control mice developed observable neurological dysfunction.
Figure 6.

Brain selenium concentrations in Sepp1−/− and apoER2−/− mice on developing selenium deficiency-induced severe neurological dysfunction (indicated by bold arrows). One mouse from each of the 3 control groups was studied with each experimental mouse. Sepp1−/− mice fed a selenium-deficient diet (n=6) developed signs of severe neurological dysfunction (ataxia, hyperactivity, and/or inability to right) from 8 to 14 d after weaning; apoER2−/− mice fed a selenium-deficient diet (n=4) developed signs of severe neurological dysfunction from 19 to 23 d after weaning. Values are expressed as means + sd. *P < 0.05; Student's t test.
To further evaluate the effect of brain selenium on severe neurological dysfunction, we produced extremely severe selenium deficiency in mice by feeding selenium-deficient diet to mice with conditional knockout of Sepp1 in hepatocytes (Sepp1C/C/alb-cre+/− mice). In the liver, Sepp1 synthesis competes for metabolically available selenium with synthesis of selenium excretory metabolites. Consequently, deletion of hepatocyte Sepp1 leads to increased selenium excretion, which exacerbates dietary selenium deficiency (6). In addition, it lowers plasma Sepp1 concentrations, decreasing selenium supply to extrahepatic tissues, including the brain. By feeding selenium-deficient diet to weanling Sepp1C/C/alb-cre+/− mice, we produced mice with extremely severe selenium deficiency without loss of brain Sepp1 or apoER2. The deficiency became so severe by 8 wk after weaning that the mice failed to gain more weight (Fig. 7A). Beginning 22 wk after weaning, the Sepp1C/C/alb-cre+/− mice developed crossing of the hind limbs when they were lifted by the tail and weakness of those limbs that progressively worsened and resulted in the hind limbs making broad-based swimming motions and in dragging of the rear portion of the body when the mice ambulated (for illustration see Fig. 11 in ref. 6). However, the qualitatively different syndrome of severe neurological dysfunction, as had been observed in Sepp1−/− and apoER2−/− mice fed a selenium-deficient diet (24), did not occur in the selenium-deficient Sepp1C/C/alb-cre+/− mice, and none of them died during the 40 wk of the experiment.
Figure 7.

Effects of selenium deficiency on the weight of mice with selective deletion of Sepp1 in hepatocytes (A) and mice with mutation of the redox-active selenocysteine of Sepp1 to serine (B). All mice were fed a selenium-deficient diet from the time of weaning. Values are means + sd (n=5/group in A and n=4–8 in B). A) Weights of the 2 groups at each time point beginning at 12 wk were different (P<0.05) by Student's t test. B) Weights of the 2 groups at each time point were not significantly different by Student's t test.
Figure 8 shows that brain selenium concentration decreased with time in the Sepp1C/C/alb-cre+/− mice fed a selenium-deficient diet, reaching a low of 12 ± 4 ng/g (n=5) at 40 wk, approximately one-third of the concentration that triggered severe neurological dysfunction in Sepp1−/− mice or apoER2−/− mice fed the same diet. Brain selenium concentration in the selenium-deficient Sepp1C/C mice that served as controls for the Sepp1C/C/alb-cre+/− mice (Fig. 7A) was 62 ± 3 ng/g (n=5) at 40 wk, illustrating the severe exacerbation of the selenium deficiency caused by selective knockout of Sepp1 in hepatocytes. Also, severe neurological dysfunction had appeared in the global gene knockout mice within 4 wk of weaning, while the milder hind-limb dysfunction that occurred in the Sepp1C/C/alb-cre+/− mice was not observed until 22 wk after weaning. Thus, when both Sepp1 and apoER2 were present in the brain, a much lower brain selenium concentration was tolerated without severe neurological dysfunction than when either protein was not expressed in the brain. This finding is compatible with a Sepp1-apoER2 interaction within the brain concentrating the very limited brain selenium in a compartment that protected against the severe neurodegeneration.
Figure 8.

Expression of Sepp1 in brain protects against severe neurological dysfunction caused by feeding selenium-deficient diet. Sepp1−/− mice (same group as in Fig. 6) developed severe neurological dysfunction at 1–2 wk, but none of the Sepp1−/−/alb-cre+/− mice (weight curves shown in Fig. 7A) developed severe neurological dysfunction. Sepp1−/−/alb-cre+/− mice fed the selenium-deficient diet for 12 wk beginning at weaning had no neurological signs, but those fed the diet for 28 and 40 wk had weakness of the hind limbs, first observed at 22 wk. Values are expressed as means + sd; n = 4–6. *P < 0.05; Tukey's multiple-comparison test after 1-way ANOVA.
Disruption of the peroxidase activity of Sepp1 does not cause severe neurological dysfunction
Sepp1 consists of an N-terminal domain that contains a selenocysteine residue in a thioredoxin-like redox motif and a C-terminal domain that contains 9 selenocysteine residues. The C-terminal domain is responsible for the selenium transport function of Sepp1 (8), and the N-terminal domain has peroxidase activity when coupled with thioredoxin reductase 1 (7).
Recently, we acquired mice with mutation of the redox-active selenocysteine residue to serine (Sepp1U40S mice). This mutation abolished the peroxidase activity of Sepp1 (7) but had no affect on tissue (including brain) selenium concentrations in mice fed a selenium-adequate diet (unpublished results). Sepp1U40S mice were fed a selenium-adequate diet for 4 wk beginning at weaning without developing obvious neurological signs.
Because feeding a selenium-deficient diet to Sepp1−/− mice caused severe neurodegeneration, we fed Sepp1U40S mice selenium-deficient diet to determine whether loss of Sepp1 redox function combined with selenium deficiency would cause severe neurological dysfunction. Figure 7B shows that Sepp1U40S mice fed a selenium-deficient diet for 36 wk gained weight, as well as wild-type mice fed the same diet. The mice showed no obvious neurological signs, and brain selenium concentration in them was maintained as well as in controls after both had been fed a selenium-deficient diet for 36 wk (50±5 ng/g, n=8, in Sepp1U40S mice; 44±4 ng/g, n=4, in C57BL/6 controls). Thus, loss of Sepp1 redox function did not cause severe neurological dysfunction or affect brain selenium concentration.
DISCUSSION
This report presents evidence that Sepp1 is taken up at the blood-brain barrier by an apoER2-dependent mechanism, transferring selenium from the systemic circulation to the brain. It also demonstrates that a much lower brain selenium concentration is tolerated without severe neurodegeneration when both Sepp1 and apoER2 are expressed in the brain than when either of them is not so expressed. This latter finding implies that a second brain Sepp1-apoER2 interaction, this one inside the blood-brain barrier, concentrates limited brain selenium in a compartment in which it protects against severe neurodegeneration.
Sepp1-apoER2 interaction at the blood-brain barrier
Fetal brain capillary endothelial cells take up Sepp1 by apoER2-mediated endocytosis, but such uptake was not detected in the adult brain (Figs. 1 and 2). This difference correlates with the greater expression of apoER2 mRNA in fetal brain than in adult brain (Fig. 3A).
Whole-brain apoER2 mRNA is not regulated by selenium status (Fig. 3B), suggesting that the uptake of Sepp1 by endothelial cells is not specifically regulated but is a “bystander” event, dependent on apoER2 regulation by factors other than the need for selenium. Supporting this argument is the fact that the blood-brain barrier is incomplete in the d 18 fetus, presumably allowing blood Sepp1 to have access to the brain interstitium at that stage of development and rendering the Sepp1-apoER2 uptake mechanism at the blood-brain barrier less necessary in the fetus than in the adult (Fig. 4). Thus, Sepp1 uptake by fetal brain capillaries serves to demonstrate that apoER2 can mediate Sepp1 endocytosis in brain capillaries. Failure to demonstrate uptake in adult brain capillaries (Fig. 2) does not mean that such uptake is not occurring at a physiologically significant level below the limit of detection by our ICC method. Tissues in which vesicles containing Sepp1 are detectable by ICC, e.g., testis and placenta, have higher apoER2 mRNA levels than tissues in which vesicles are not visible and are likely taking up larger amounts of Sepp1 than the “low-apoER2” tissues (10).
Fetal brain capillaries appear to take up Sepp1 in the manner of other types of cells that have access to the systemic circulation. However, brain capillary cells and testis Sertoli cells have a different function in selenium metabolism from that of other apoER2-expressing cells. Both of these types of cells constitute barriers between their organs and the systemic circulation. They take up substances that must be transferred to other cells within their organs, whereas cells in tissues without barriers between themselves and the blood, e.g., muscle cells (10), take up Sepp1 by apoER2-mediated endocytosis and digest it in lysosomes, so that its selenium can be used for synthesis of selenoproteins in the same cell.
The immediate fate of Sepp1 taken up by brain capillary cells is not known. However, the close physical association of endothelial cells with astrocytes, which synthesize and secrete Sepp1 (14), is consistent with the selenium taken up as Sepp1 by capillary cells being transferred by transcytosis or other means to astrocytes for further transport within the brain as newly synthesized Sepp1 (Scheme 1). In the testis, Sertoli cells have contacts with germ cells, the main consumers of selenium in that organ, and appear to transfer selenium taken up as Sepp1 from the systemic circulation to them. The mechanism of selenium transfer between the cells of Sepp1 uptake from the systemic circulation to cells within brain and testis is not known.
Scheme 1.

Scheme of postulated tandem Sepp1-apoER2 interactions for delivering selenium to brain neurons. The first interaction is at the blood-brain barrier and results in selenium from plasma Sepp1 being transported into the brain in an unknown form, represented by Se*. Se* provides selenium to astrocytes, and possibly to other brain cells, which use it to synthesize Sepp1 within the brain. Cells that express apoER2, primarily neurons, take up Sepp1 in the second interaction of Sepp1 and apoER2.
The role of the choroid plexus in acquiring selenium for the brain appears to be more complex than that of the brain capillaries. The choroid plexus produces cerebrospinal fluid (CSF), which contains Sepp1 (25, 26). CSF enters the brain parenchyma from the subarachnoid space via a paravascular space between penetrating arteries and astrocyte endfeet (27). Then, CSF contributes to the flow of fluid through the brain interstitium. The interstitial fluid exits the brain in “glymphatics” that are adjacent to draining veins. This physiological mechanism has been described only recently, and details of protein transport from the choroid plexus through the CSF to cells within the brain have not been reported. However, there is evidence that the choroid plexus transfers micronutrients from the blood to the brain (28).
The choroid plexus expresses mRNAs of both Sepp1 and apoER2 (13), and using ICC, we detected both proteins in it (Fig. 2A, B). Our observation that deletion of apoER2 renders Sepp1 undetectable in the choroid plexus (Figs. 1 and 2) suggests that Sepp1 is taken up from the blood by apoER2-mediated endocytosis. It is possible that Sepp1 taken up by choroid plexus epithelial cells is digested, freeing selenium for synthesis of Sepp1 (or another form of selenium) to be secreted into the CSF. It is not known whether the Sepp1 in CSF can reach the interstitial fluid in the brain or whether it has another fate. However, both brain capillaries and the choroid plexus appear to contribute to the transfer of selenium from the systemic circulation across the blood-brain barrier.
Megalin mediates endocytosis of filtered N-terminal Sepp1 forms by renal PCT cells (7). Megalin is also present on the choroid plexus and on ependymal cells and has been suggested to play a role in brain selenium metabolism (29). We did not detect Sepp1 in ependymal cells using ICC (Figs. 2 and 5). Neither did we detect an effect of megalin knockout on capillary or choroid plexus Sepp1 in fetal brain (Fig. 5). Megalin knockout did not affect brain selenium concentration in mice fed a diet containing 0.25 mg selenium/kg. However, it did exacerbate selenium deficiency, lowering selenium concentration in all tissues studied, including brain, presumably because N-terminal Sepp1 forms and other filtered selenium-containing proteins were lost in the urine (7).
The megalin-Sepp1 interaction appears to be highly variable. While megalin-mediated uptake of filtered N-terminal Sepp1 forms by renal PCT cells clearly occurs (7), we have been unable to detect megalin-Sepp1 interactions in the visceral yolk sac (16) or in the brain (Fig. 5). This variability might be related to Sepp1 being present in more than one form and/or to limitations of our detection methods. ApoER2 binds to the selenium-rich C-terminal domain of Sepp1 but not to the N-terminal domain (10, 30). Megalin, on the other hand, binds to the N-terminal domain of Sepp1 (7). In addition, megalin-mediated endocytosis can result after the ligand binds directly to megalin or after it binds to cubilin, the endocytosis of which depends on megalin (22). Thus, apoER2-mediated uptake of Sepp1 forms occurs in many tissues, but megalin-mediated uptake of Sepp1 forms has, thus far, been documented only in renal PCT cells.
Nature of the neurological dysfunction induced by selenium deficiency differs between mice with Sepp1 and apoER2 intact in brain and mice with either gene deleted
While the brain competes with other tissues for selenium through apoER2-mediated endocytosis of Sepp1 at the blood-brain barrier, it also has another Sepp1-dependent mechanism that prevents severe neurodegeneration. The resistance of mice expressing both Sepp1 and apoER2 in brain to severe neurological dysfunction in the face of extreme selenium deficiency is evidence for such an intrabrain mechanism. Work showing abundant expression of apoER2 by interneurons (31), a cell type with its viability dependent on selenoprotein expression (32), supports apoER2-mediated uptake of Sepp1 by neuronal cells as that mechanism.
ApoER2 has signaling properties when it binds the ligand reelin (33), and it has been suggested that apoER2 binding of Sepp1 might also provide a signal (34). It is unlikely that a signaling event can explain the severe neurological dysfunction observed with Sepp1 or apoER2 deletion, however, because dietary supplementation with inorganic selenium prevents the dysfunction.
A report that appeared 3 decades ago demonstrated that wild-type mice fed a selenium-deficient diet through 3 generations began to show neurological abnormalities (35). When lifted by the tail, those mice crossed their hind limbs rather than extending them, as do selenium-replete mice. This observation indicated that some, although slight, central nervous system dysfunction occurs in extreme selenium deficiency. When fed selenium-deficient diet, Sepp1C/C/alb-cre+/− mice developed more profound selenium deficiency (except in the liver) than had been reported before. We noted hind limb abnormalities that were similar to, but more severe than, those reported in extremely severe dietary selenium deficiency in wild-type mice (35). However, the severe neurological dysfunction and death that had been observed in mice with global deletion of Sepp1 or apoER2 and fed a selenium-deficient diet did not occur. Thus, the neurological dysfunction caused by extreme selenium deficiency alone is qualitatively different from the more severe neurological dysfunction caused by the simultaneous occurrence of mild selenium deficiency with either Sepp1 or apoER2 deletion. These two types of neurological dysfunction are also distinguished from one another by brain selenium concentration (Figs. 6 and 8). When either Sepp1 or its receptor apoER2 was not expressed in the brain, severe neurodegeneration occurred in brains with a selenium level ∼3 times greater than the level in mice with Sepp1 and apoER2 intact in the brain and suffering only the relatively mild neurological dysfunction. On the basis of this observation, we conclude that a Sepp1-apoER2 interaction protects brain neurons from severe injury despite whole-brain selenium being extremely low. Because brain apoER2 is predominantly expressed by neurons (15), we postulate that the Sepp1-apoER2 interaction within the brain serves to maintain neuron selenium at the expense of other brain cells (Scheme 1).
CONCLUSIONS
On the basis of the evidence presented in this report, we conclude that the superior ability of the brain to retain selenium in dietary selenium deficiency is due to a unique two-tier mechanism of Sepp1-apoER2 interactions (Scheme 1). The first interaction is at the blood-brain barrier where Sepp1 is taken up from the systemic circulation by brain capillary cells and choroid plexus epithelial cells. Those cells then release selenium into the brain in a form that has not yet been determined. We postulate that, within the brain, astrocytes, and possibly other glial cells, take up selenium and secrete it into brain interstitial fluid as Sepp1. Then cells within the brain that express apoER2, primarily neurons, acquire selenium by apoER2-mediated endocytosis of Sepp1. The selenium that is acquired supports synthesis of selenoproteins essential to the function and viability of the neurons.
This 2-level mechanism allows the brain to compete with other tissues for Sepp1 in the systemic circulation and, once selenium has been thus acquired, to retain it by intrabrain cycling as Sepp1. Moreover, because apoER2 is expressed within the brain principally by neurons, we propose that this mechanism concentrates brain selenium in neurons, protecting them from degeneration.
Many important details of brain selenium metabolism remain to be clarified: How is selenium transferred from the cells that take up Sepp1 at the blood-brain barrier to cells inside the brain? What is the role of small molecule forms of selenium in the brain? Is uptake of Sepp1 by apoER2 on neurons regulated? Our proposed outline of brain selenium transport (Scheme 1) provides a basis for future research to answer these and other questions. Because selenium is important for neuron function and viability, further research on its metabolism in the brain might shed light on neurodegenerative disorders.
Acknowledgments
The authors are grateful to Teri D. Stevenson for animal husbandry and to Dr. William E. Valentine for a critical reading of the manuscript and helpful suggestions for its improvement.
The work reported here was supported by U.S. National Institutes of Health grants R37 ES-002497, 5P30 DK-058404, and 5P30 ES-000267.
The authors declare no conflicts of interest.
Footnotes
- apoER2
- apolipoprotein E receptor 2
- CSF
- cerebrospinal fluid
- ICC
- immunocytochemistry
- PCT
- proximal convoluted tubule
- Sepp1
- selenoprotein P
REFERENCES
- 1. Behne D., Hilmert H., Scheid S., Gessner H., Elger W. (1988) Evidence for specific selenium target tissues and new biologically important selenoproteins. Biochim. Biophys. Acta 966, 12–21 [DOI] [PubMed] [Google Scholar]
- 2. Nakayama A., Hill K. E., Austin L. M., Motley A. K., Burk R. F. (2007) All regions of mouse brain are dependent on selenoprotein P for maintenance of selenium. J. Nutr. 137, 690–693 [DOI] [PubMed] [Google Scholar]
- 3. Burk R. F., Hill K. E. (2009) Selenoprotein P: expression, functions, and roles in mammals. Biochim. Biophys. Acta 1790, 1441–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Atkins J. F., Gesteland R. F., Burk R. F. (2003) Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 278, 13640–13646 [DOI] [PubMed] [Google Scholar]
- 5. Schomburg L., Schweizer U., Holtmann B., Flohé L., Sendtner M., Köhrle J. (2003) Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 370, 397–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hill K. E., Wu S., Motley A. K., Stevenson T. D., Winfrey V. P., Capecchi M. R., Atkins J. F., Burk R. F. (2012) Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J. Biol. Chem. 287, 40414–40424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kurokawa S., Eriksson S., Rose K. L., Wu S., Motley A. K., Hill S., Winfrey V. P., McDonald W. H., Capecchi M. R., Atkins J. F., Arnér E. S., Hill K. E., Burk R. F. (2014) Sepp1UF forms are N-terminal selenoprotein P truncations that have peroxidase activity when coupled with thioredoxin reductase-1. Free Radic. Biol. Med. 69, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hill K. E., Zhou J., Austin L. M., Motley A. K., Ham A. J., Olson G. E., Atkins J. F., Gesteland R. F., Burk R. F. (2007) The selenium-rich C-terminal domain of mouse selenoprotein P is necessary for supply of selenium to brain and testis but not for maintenance of whole-body selenium. J. Biol. Chem. 282, 10972–10980 [DOI] [PubMed] [Google Scholar]
- 9. Olson G. E., Winfrey V. P., Nagdas S. K., Hill K. E., Burk R. F. (2007) Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J. Biol. Chem. 282, 12290–12297 [DOI] [PubMed] [Google Scholar]
- 10. Kurokawa S., Hill K. E., McDonald W. H., Burk R. F. (2012) Long isoform mouse selenoprotein P (Sepp1) supplies rat myoblast L8 cells with selenium via endocytosis mediated by heparin binding properties and apolipoprotein E receptor-2 (apoER2). J. Biol. Chem. 287, 28717–28726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burk R. F., Hill K. E., Olson G. E., Weeber E. J., Motley A. K., Winfrey V. P., Austin L. M. (2007) Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. J. Neurosci. 27, 6207–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Renko K., Werner M., Renner-Muller I., Cooper T. G., Yeung C. H., Hollenbach B., Scharpf M., Köhrle J., Schomburg L., Schweizer U. (2008) Hepatic selenoprotein P (SePP) expression restores selenium transport and prevents infertility and motor-incoordination in Sepp-knockout mice. Biochem. J. 409, 741–749 [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y., Zhou Y., Schweizer U., Savaskan N. E., Hua D., Kipnis J., Hatfield D. L., Gladyshev V. N. (2008) Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J. Biol. Chem. 283, 2427–2438 [DOI] [PubMed] [Google Scholar]
- 14. Yang X., Hill K. E., Maguire M. J., Burk R. F. (2000) Synthesis and secretion of selenoprotein P by cultured rat astrocytes. Biochem. Biophys. Acta 1474, 390–396 [DOI] [PubMed] [Google Scholar]
- 15. Clatworthy A. E., Stockinger W., Christie R. H., Schneider W. J., Nimpf J., Hyman B. T., Rebeck G. W. (1999) Expression and alternate splicing of apolipoprotein E receptor 2 in brain. Neuroscience 90, 903–911 [DOI] [PubMed] [Google Scholar]
- 16. Burk R. F., Olson G. E., Hill K. E., Winfrey V. P., Motley A. K., Kurokawa S. (2013) Maternal-fetal transfer of selenium in the mouse. FASEB J. 27, 3249–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Burk R. F. (2004) Neurological dysfunction occurs in mice with targeted deletion of selenoprotein P gene. J. Nutr. 134, 157–161 [DOI] [PubMed] [Google Scholar]
- 18. Trotter J. H., Klein M., Jinwal U. K., Abisambra J. F., Dickey C. A., Tharkur J., Masiulis I., Ding J., Locke K. G., Rickman C. B., Birch D. G., Weeber E. J., Herz J. (2011) ApoER2 function in the establishment and maintenance of retinal synaptic connectivity. J. Neurosci. 31, 14413–14423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koh T. S., Benson T. H. (1983) Critical re-appraisal of fluorometric method for determination of selenium in biological materials. J. Assoc. Off. Anal. Chem. 66, 918–926 [PubMed] [Google Scholar]
- 20. Sheehan T. M. T., Gao M. (1990) Simplified fluorometric assay of total selenium in plasma and urine. Clin. Chem. 36, 2124–2126 [PubMed] [Google Scholar]
- 21. Olson G. E., Winfrey V. P., Hill K. E., Burk R. F. (2008) Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J. Biol. Chem. 283, 6854–6860 [DOI] [PubMed] [Google Scholar]
- 22. Christensen E. I., Birn H. (2002) Megalin and cubilin: multifunctional endocytic receptors. Nat. Rev. Mol. Cell Biol. 3, 256–266 [DOI] [PubMed] [Google Scholar]
- 23. Zheng G., Bachinsky D. R., Stamenkovic I., Strickland D. K., Brown D., Andres G., McCluskey R. T. (1994) Organ distribution in rats of two members of the low-density lipoprotein receptor gene family, gp330 and LRP/alpha 2MR, and the receptor-associated protein (RAP). J. Histochem. Cytochem. 42, 531–542 [DOI] [PubMed] [Google Scholar]
- 24. Valentine W. M., Abel T. W., Hill K. E., Austin L. M., Burk R. F. (2008) Neurodegeneration in mice resulting from loss of functional selenoprotein P or its receptor apolipoprotein E receptor 2. J. Neuropathol. Exp. Neurol. 67, 68–77 [DOI] [PubMed] [Google Scholar]
- 25. Michalke B., Berthele A. (2011) Contribution to selenium speciation in cerebrospinal fluid samples. J. Anal. At. Spectrom. 26, 165–170 [Google Scholar]
- 26. Scharpf M., Schweizer U., Arzberger T., Roggendorf W., Schomburg L., Köhrle J. (2007) Neuronal and ependymal expression of selenoprotein P in the human brain. J. Neural Transm. 114, 877–884 [DOI] [PubMed] [Google Scholar]
- 27. Iliff J. J., Wang M., Liao Y., Plogg B. A., Peng W., Gundersen G. A., Benveniste H., Vates G. E., Deane R., Goldman S. A., Nagelhus E. A., Nedergaard M. (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Science Transl. Med 4, 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spector R., Johanson C. (2006) Micronutrient and urate transport in choroid plexus and kidney: implications for drug therapy. Pharmaceut. Res. 23, 2515–2524 [DOI] [PubMed] [Google Scholar]
- 29. Chiu-Ugalde J., Theilig F., Behrends T., Drebes J., Sieland C., Subbarayal P., Köhrle J., Hammes A., Schomburg L., Schweizer U. (2010) Mutation of megalin leads to urinary loss of selenoprotein P and selenium deficiency in serum, liver, kidneys and brain. Biochem. J. 431, 103–111 [DOI] [PubMed] [Google Scholar]
- 30. Kurokawa S., Bellinger F. P., Hill K. E., Burk R. F., Berry M. J. (2014) Isoform-specific binding of selenoprotein P to the β-propeller domain of apolipoprotein E receptor 2 mediates selenium supply. J. Biol. Chem. 289, 9195–9207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pitts M. W., Raman A. V., Hashimoto A. C., Todorovic C., Nichols R. A., Berry M. J. (2012) Deletion of selenoprotein P results in impaired function of parvalbumin interneurons and alterations in fear learning and sensorimotor gating. Neuroscience 208, 58–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wirth E. K., Conrad M., Winterer J., Wozny C., Carlson B. A., Roth S., Schmitz D., Bornkamm G. W., Coppola V., Tessarollo L., Schomburg L., Köhrle J., Hatfield D. L., Schweizer U. (2010) Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J. 24, 844–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. May P., Herz J., Bock H. H. (2005) Molecular mechanisms of lipoprotein receptor signalling. Cell. Mol. Life Sci. 62, 2325–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peters M. M., Hill K. E., Burk R. F., Weeber E. J. (2006) Altered hippocampus synaptic function in selenoprotein P deficient mice. Mol. Neurodegener. 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wallace E., Calvin H., Cooper G. (1983) Progressive defects observed in mouse sperm during the course of three generations of selenium deficiency. Gamete Res. 7, 377–387 [Google Scholar]